

Abstract
Objective:
To investigate the associations of cerebral amyloidosis with concurrent cognitive performance and with longitudinal cognitive decline in asymptomatic and symptomatic stages of autosomal dominant Alzheimer disease (ADAD).
Methods:
Two hundred sixty-three participants enrolled in the Dominantly Inherited Alzheimer Network observational study underwent neuropsychological evaluation as well as PET scans with Pittsburgh compound B. One hundred twenty-one participants completed at least 1 follow-up neuropsychological evaluation. Four composite cognitive measures representing global cognition, episodic memory, language, and working memory were generated using z scores from a battery of 13 standard neuropsychological tests. General linear mixed-effects models were used to investigate the relationship between baseline cerebral amyloidosis and baseline cognitive performance and whether baseline cerebral amyloidosis predicts cognitive change over time (mean follow-up 2.32 years ± 0.92, range 0.89–4.19) after controlling for estimated years from expected symptom onset, APOE ε4 allelic status, and education.
Results:
In asymptomatic mutation carriers, amyloid burden was not associated with baseline cognitive functioning but was significantly predictive of longitudinal decline in episodic memory. In symptomatic mutation carriers, cerebral amyloidosis was correlated with worse baseline performance in multiple cognitive composites and predicted greater decline over time in global cognition, working memory, and Mini-Mental State Examination.
Conclusions:
Cerebral amyloidosis predicts longitudinal episodic memory decline in presymptomatic ADAD and multidomain cognitive decline in symptomatic ADAD. These findings imply that amyloidosis in the brain is an indicator of early cognitive decline and provides a useful outcome measure for early assessment and prevention treatment trials.
β-Amyloid (Aβ) is thought to be an initiating factor in the pathophysiologic process of Alzheimer disease (AD).1,2 However, the relationship between cognition, brain Aβ burden, and the future development of dementia is still unclear.
Postmortem neuropathologic examination has found inconsistent relationships between cognition and Aβ deposition.3–6 Studies using amyloid PET in cognitively normal elderly individuals and individuals with mild cognitive impairment (MCI) and AD dementia have found significant relationships between cognitive deficits and increased brain fibrillar amyloid using both cross-sectional7–12 and longitudinal data13–23; however, other studies have not shown amyloid and cognitive correlations.24–26
In autosomal dominant AD (ADAD), brain amyloid deposition is known to occur 15 years or more before the onset of clinical symptoms,27,28 and estimated years from expected symptom onset (EYO) calculated from family history data can provide an objective biomarker-independent estimate of an individual's relative point in the disease process. The predictable age of symptom onset and low rate of comorbidities in the younger ADAD individuals make them an ideal population in which to directly assess the amyloid–cognition relationship across the course of disease, although such relationships may differ from those in sporadic AD.
Using data from 263 participants in the Dominantly Inherited Alzheimer Network (DIAN) observational study, we performed cross-sectional and longitudinal analyses to investigate the associations of brain amyloid deposition with concurrent cognitive performance and with longitudinal cognitive decline.
METHODS
Participants.
Participants were enrolled in the DIAN observational study, an international study of families with ADAD-associated mutations in APP, PSEN1, or PSEN2.29
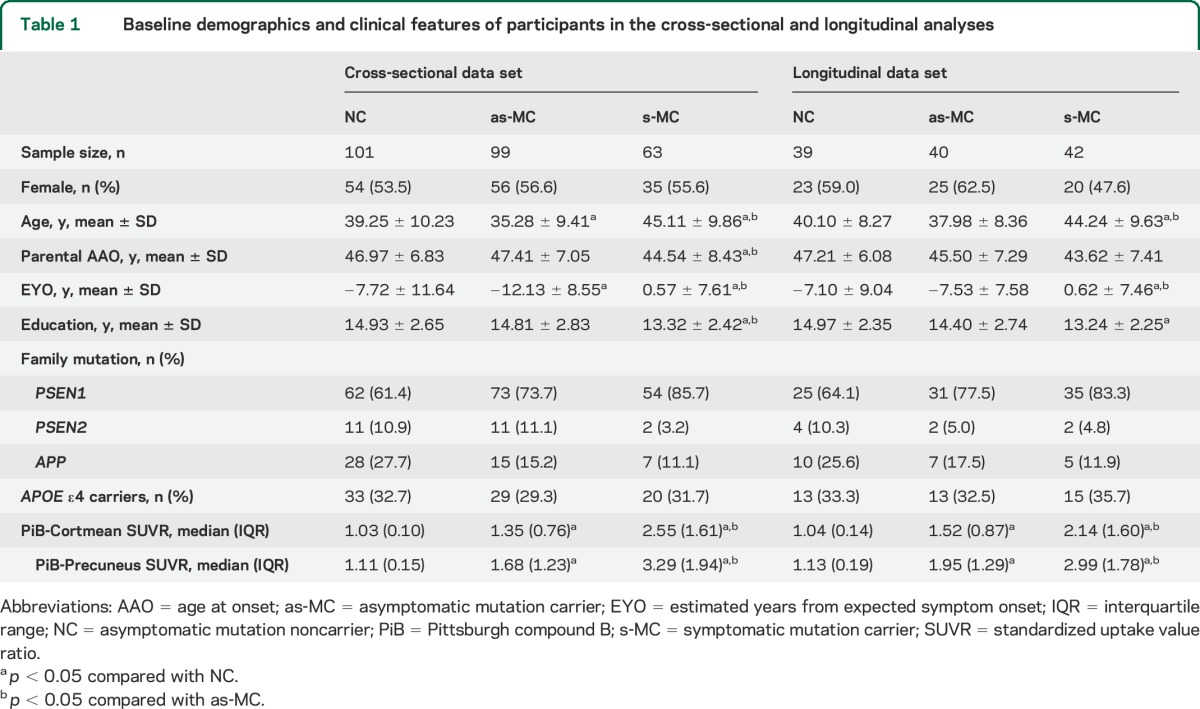
Based on mutation status and Clinical Dementia Rating (CDR) score,30 participants were classified as asymptomatic mutation noncarriers (NCs, CDR = 0), asymptomatic mutation carriers (as-MCs, CDR = 0), and symptomatic mutation carriers (s-MCs, CDR > 0). A small number of mutation noncarriers with CDR >0 in the cross-sectional data set (n = 7) and longitudinal dataset (n = 2) were excluded from analyses due to the potential presence of non-AD pathology. Cross-sectional data (table 1), including baseline neuropsychological tests and PET scans with Pittsburgh compound B (PiB), were obtained from 263 participants (101 NCs, 99 as-MCs, and 63 s-MCs) from 98 families carrying an ADAD mutation in PSEN1 (80.6%), PSEN2 (3.1%), or APP (16.3%). Longitudinal data (table 1) were obtained from a subset of 121 participants (39 NCs, 40 as-MCs, and 42 s-MCs) who completed at least 1 cognitive assessment at follow-up. The average follow-up time was 2.32 ± 0.92 years (range 0.89–4.19) and the average number of visits was 2.51 ± 0.76 (range 2–5). Baseline demographics between groups were compared using approximate t tests from the mixed models. Degrees of freedom were approximated by Satterthwaite method.
Table 1.
Baseline demographics and clinical features of participants in the cross-sectional and longitudinal analyses
Standard protocol approvals, registrations, and patient consents.
The study was approved by the local institutional review boards of each site. Participants provided written informed consent or assent with a proxy.
Clinical evaluation.
Per standard DIAN study protocols, each participant and a collateral source underwent semi-structured interviews collecting detailed demographics, medical history, and family history. Values for EYO were calculated as the difference between the participant's current age and the age at onset of his or her affected parent or first-degree relative, as previously described.28 Cognitive status was clinically assessed by global CDR score, with CDR 0 indicating normal cognitive function, CDR 0.5 both MCI and very mild dementia, CDR 1 mild dementia, CDR 2 moderate dementia, and CDR 3 severe dementia.
All participants completed a physical and neurologic examination. Mutations in APP, PSEN1, and PSEN2 and APOE ε4 carrier status were identified from DNA extracted from peripheral blood samples using methods described previously.31,32 Clinical evaluators remained blinded to the mutation status of each participant.
Neuropsychological assessments.
Three domain-specific cognitive composites and a global cognitive composite were calculated by averaging z scores from a battery of 13 standard paper-and-pencil neuropsychological tests, previously described in detail in this cohort.33 The structure of the cognitive composites was derived from an exploratory factor analysis conducted on a subset of participants at baseline. The Episodic Memory composite (EM) included Logical Memory Immediate Recall and Delayed Recall and Word List Immediate Recall and Delayed Recall. The Language Function composite (LF) was generated from Letter Fluency for the letters “F,” “A,” and “S,” Category Fluency for animals and vegetables, and the 30-item version of the Boston Naming Test. The Working Memory composite (WM) was generated from Digit Span (Forwards and Backwards), the Trail Making Test Parts A and B, and the Digit Symbol Coding test. The Global Cognitive composite (GC) included all 13 measures. Participants with missing data were excluded from analyses. In addition, scores from the Mini-Mental State Examination (MMSE) and CDR sum of boxes (CDR-SB) were analyzed individually.
PiB-PET imaging.
PiB-PET scans were performed to quantify cerebral fibrillar Aβ deposition within 6 months of baseline clinical and neuropsychological evaluations. The mean interval from assessment to imaging was 16.21 days, with a range from 0 to 158 days. As described previously,34,35 the PiB-PET data in the time frame from 40 to 70 minutes postinjection were analyzed by a region-of-interest (ROI) approach. For each FreeSurfer ROI, a regional spread function–based technique was used to correct for partial volume effects before regional image intensities were referenced to cerebellar gray matter to calculate a standardized uptake value ratio (SUVR).36 We examined the mean cortical SUVR (PiB-Cortmean) derived from an average across left and right lateral orbitofrontal, inferior parietal, precuneus, rostral middle frontal, superior frontal, superior temporal, and middle temporal regions. We additionally analyzed SUVR in the precuneus (PiB-Precuneus), known to be an area of early Aβ deposition.34,37 Analyses in which PiB SUVRs were calculated using the brainstem as a reference region were also conducted and the results are shown in tables e-1 and e-2 on the Neurology® Web site at Neurology.org.
Statistical analysis.
General linear mixed-effects models were used for both cross-sectional and longitudinal analyses. The cross-sectional analysis was conducted to investigate the association between baseline amyloidosis and baseline cognitive performance. The longitudinal analysis was conducted to assess whether baseline amyloidosis predicts subsequent longitudinal cognitive decline. These analyses included both PiB and EYO as fixed effects, as well as patient groups based on mutation and clinical status (NC, as-MC, s-MC), and all possible interactions. The family affiliation was treated as a random effect in these models. For longitudinal analyses, time (number of years since baseline cognitive test), as well as its interaction with patient groups (NC, as-MC, and s-MC), baseline PiB (centered at the mean), and/or EYO, and all possible 2- and 3-factor interaction terms were included as fixed effects in the models. Patients were also treated as a random effect in the longitudinal analyses. Additional analyses were also conducted to adjust for other potential covariates, including years of education and APOE ε4 status (positive or negative). We did not specifically analyze the effect of APOE ε4 status on the amyloid–cognition relationship due to the limitation of the sample size. SPSS 16.0 (SPSS Inc., Chicago, IL) and the PROC MIXED procedure in SAS 9.3 (SAS Institute, Cary, NC) were used to implement the analyses. Family and patient random effects were modeled with an unstructured covariance structure. Statistical significance was defined as p < 0.05.
RESULTS
Cross-sectional analyses.
as-MCs were younger than NCs, and s-MCs were older and closer to EYO than NCs and as-MCs (p < 0.05) (table 1). Mean years of education was lower in s-MCs than NCs and as-MCs (p < 0.05). Among s-MCs, 88.9% were CDR 0.5 or 1 (very mild and mild dementia), and only 7 participants were CDR 2 or 3 (moderate and severe dementia). PiB-Cortmean and PiB-Precuneus values were greater in as-MCs than NCs and greater in s-MCs than all other groups (p < 0.05). Sex and APOE ε4 status did not differ significantly among groups. PSEN1 mutation was the most common type of family mutation in all groups.
The relationships between PiB-PET values and cognitive performance in each group are shown in table 2 and figure 1. The β values presented in all tables represent the slope of the relationship between amyloid and cognition. The magnitude of β is in terms of an SD of the predicted value. For example, a β of −0.2 would indicate that a 1-unit change in the predictor (e.g., PiB-Cortmean) leads to 20% of an SD decrease in the predicted value (e.g., EM composite). In s-MCs, higher cerebral amyloidosis correlated with significantly worse performance on all cognitive composites except language. After controlling for EYO, years of education, and APOE ε4 status, higher PiB-Cortmean values correlated with lower scores in GC (estimated β = −0.167, p = 0.011), EM (estimated β = −0.158, p = 0.026), WM (estimated β = −0.237, p = 0.002), and MMSE (estimated β = −2.452, p < 0.001) and higher scores in CDR-SB (estimated β = 1.095, p < 0.001). PiB-Precuneus showed similar associations as PiB-Cortmean in s-MCs. There were no significant negative relationships between levels of amyloid deposition and baseline cognition for the as-MC and NC groups. Unexpectedly, the only effect for these groups was a slightly positive relationship with greater PiB-PET values associated with better LF scores in as-MCs (PiB-Cortmean: estimated β = 0.211, p = 0.029; PiB-Precuneus: estimated β = 0.167, p = 0.028).
Table 2.
Cross-sectional analysis: Associations between baseline PiB-PET and baseline cognitive performance
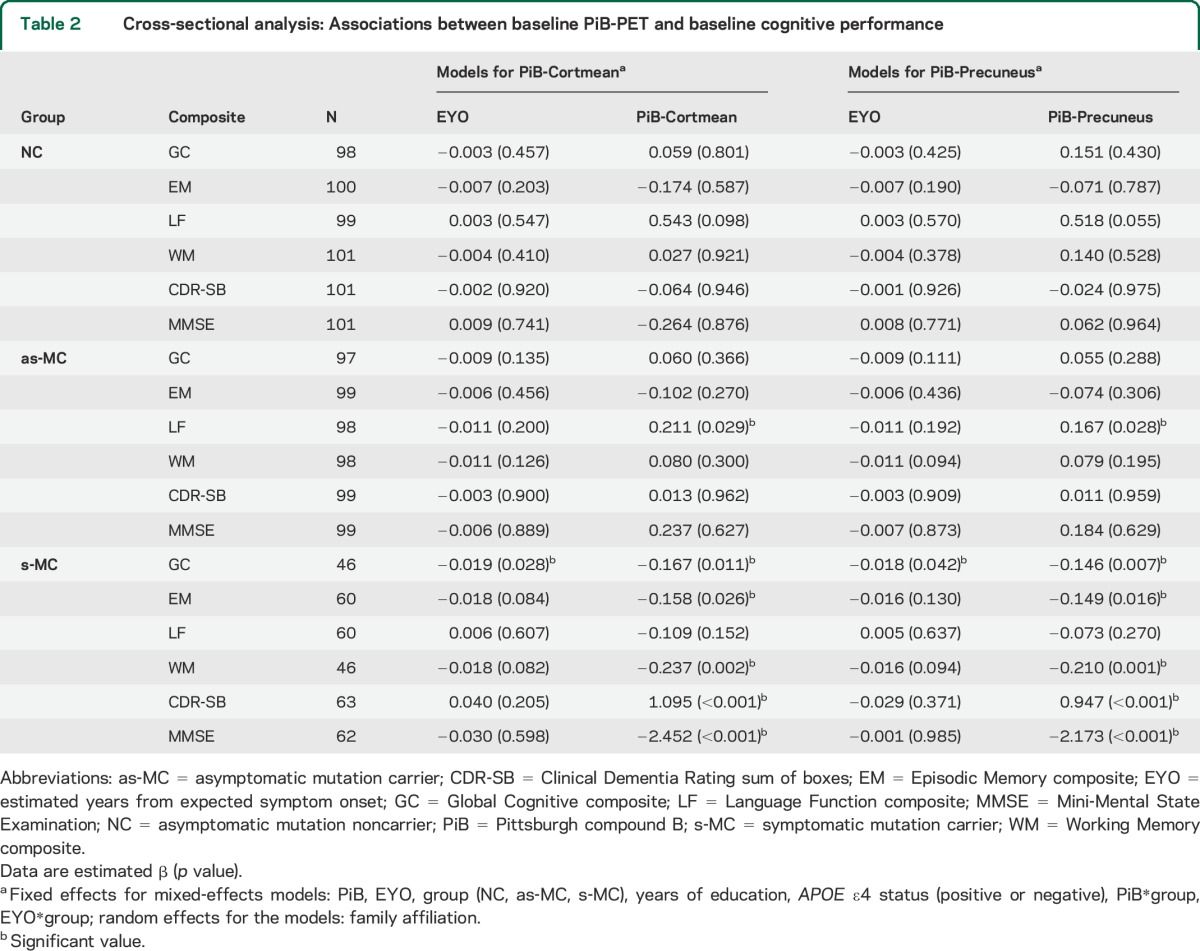
Figure 1. Estimated β values of cognition associated with PiB-PET in NC, as-MC, and s-MC groups.

Asterisk means that the estimated β is significant in this subgroup; *p < 0.05, **p < 0.01. as-MC = asymptomatic mutation carrier; EM = Episodic Memory composite; GC = Global Cognitive composite; LF = Language Function composite; NC = asymptomatic mutation noncarrier; PiB = Pittsburgh compound B; s-MC = symptomatic mutation carrier; WM = Working Memory composite.
Longitudinal analyses.
Baseline demographics and clinical features of participants in the longitudinal data set are shown in table 1. At baseline, as-MCs had similar age, EYO, and years of education compared with NCs. s-MCs were older and closer to EYO and had fewer years of education than NCs and as-MCs (p < 0.05). All other baseline features, including sex, APOE ε4 status, family mutation type, CDR scores, and PiB-PET values, were similar to those in the cross-sectional data set. The demographic features of those with and without longitudinal data are highly concordant (table e-3).
Relationships between baseline PiB-PET values and cognitive decline in each group are presented in table 3 and illustrated in figures 1 and 2. The β values presented in all tables represent the modulation of the longitudinal slope by amyloid. For these results, β refers to an effect on the annual change of a dependent variable. A β of −0.05 indicates that a 1-unit change in the predictor (e.g., PiB-Cortmean) leads to an additional 5% SD annual decline in the dependent variable (e.g., EM composite). In the as-MC group, greater baseline PiB values were only associated with lower EM scores (PiB-Cortmean: estimated β = −0.084, p = 0.043; PiB-Precuneus: estimated β = −0.073, p = 0.025).
Table 3.
Longitudinal analysis: Associations between baseline PiB-PET and future cognitive decline
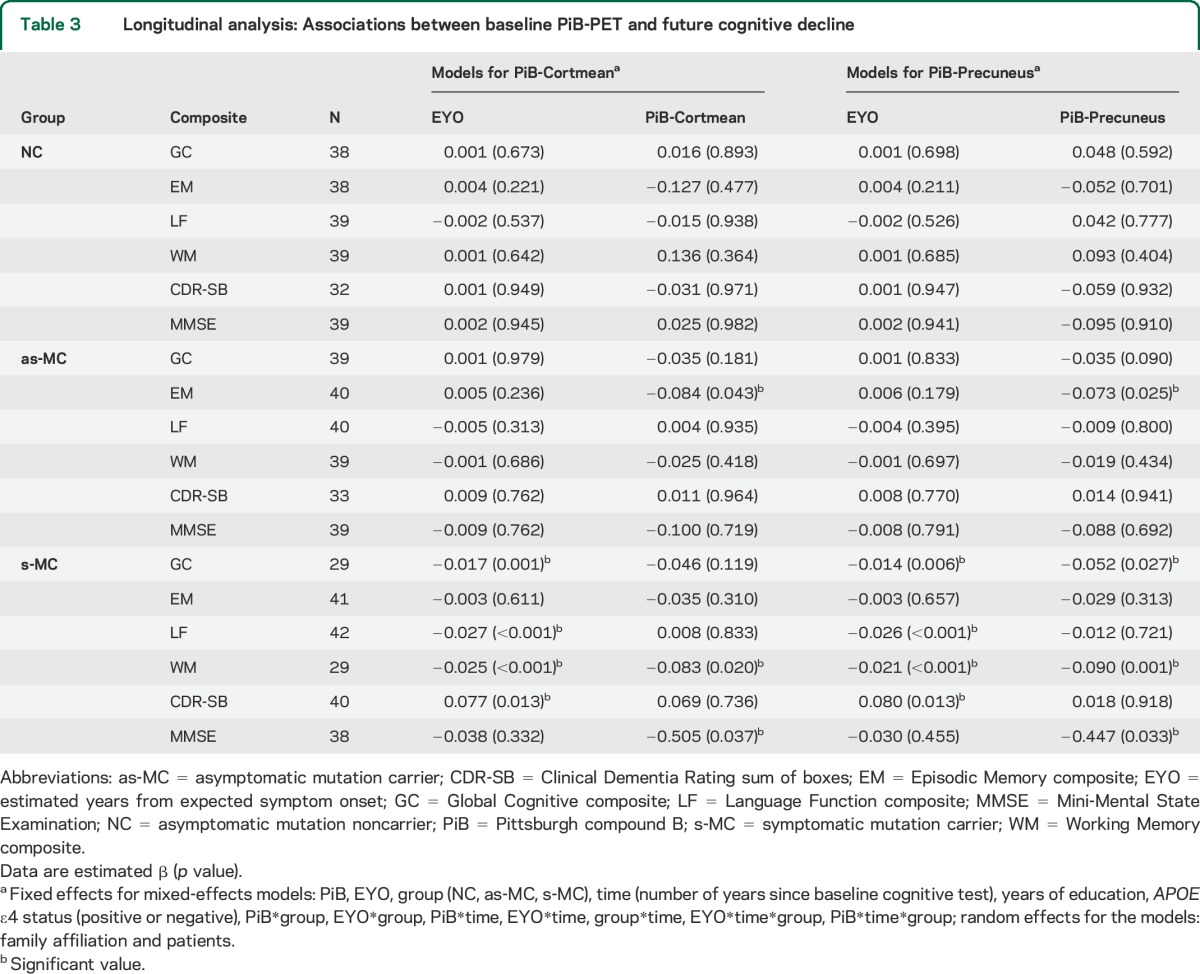
Figure 2. Cognitive decline over time as predicted by baseline PiB-PET values in NC, as-MC, and s-MC groups.

Baseline PiB values were centered. as-MC = asymptomatic mutation carrier; CDR-SB = Clinical Dementia Rating sum of boxes; EM = Episodic Memory composite; GC = Global Cognitive composite; LF = Language Function composite; MMSE = Mini-Mental State Examination; NC = asymptomatic mutation noncarrier; PiB = Pittsburgh compound B; s-MC = symptomatic mutation carrier; SUVR = standardized uptake value ratio; WM = Working Memory composite.
Higher baseline PiB-Cortmean values in s-MCs predicted greater decline in WM (estimated β = −0.083, p = 0.020) and MMSE (estimated β = −0.505, p = 0.037). Baseline PiB-Precuneus values in s-MCs predicted cognitive decline not only in WM (estimated β = −0.090, p = 0.001) and MMSE (estimated β = −0.447, p = 0.033) but also in GC (estimated β = −0.052, p = 0.027). Baseline EYO in s-MCs also had a significant effect on GC (estimated β = −0.017, p = 0.001), LF (estimated β = −0.027, p < 0.001), WM (estimated β = −0.025, p < 0.001), and CDR-SB (estimated β = 0.077, p = 0.013) (table 3).
The model significance and the p value for all the variables and interactions in the models are shown in tables e-4 and e-5. In addition, using the brainstem as an alternative reference region, similar results were found in both cross-sectional (table e-1) and longitudinal analyses (table e-2).
DISCUSSION
The results of the current study reveal that there is a significant association between fibrillar amyloid and cognitive impairment and decline in ADAD. Higher cerebral amyloidosis predicts greater longitudinal episodic memory decline in presymptomatic ADAD. Furthermore, in symptomatic ADAD, both multidomain cross-sectional impairment and longitudinal cognitive decline are associated with higher levels of amyloidosis. Our study was based on both cross-sectional and longitudinal observations of ADAD mutation carriers, who are destined to develop symptomatic AD and thus are an ideal population in which to assess the earliest cognitive changes associated with increasing amyloid burden.
Our results (figure 2) demonstrated that the effects of amyloid on longitudinal cognition occurred in a relatively continuous manner in as-MC and s-MC groups. This confirms the utility of analyzing continuous rather than dichotomized PiB-PET values, which increases the power of analyses to predict cognitive performance. The size of our effects was such that a 1-unit increase in amyloid deposition (PiB-Cortmean) led to between a 5% and 8% (SD) greater annual decline in cognitive scores.
Our prior reports from the DIAN study have revealed a highly significant relationship between EYO and actual age at onset and disease course,38 as well as EYO and cognition in ADAD.28,33 Our models accounted for EYO in estimating the additional effect of amyloidosis on cognitive impairment and decline. The unique role of EYO provides a biomarker-independent prospective estimate of each individual's relative stage in the disease process. Development of a multivariate model incorporating EYO and disease biomarkers, such as fibrillar amyloid levels, may enable cognitive decline to be predicted with greater precision.
Most prior studies of cognitively healthy older people demonstrated episodic memory decline associated with amyloid burden in the brain7,10–13,15,18,20,21,23 earlier than other cognitive domain declines.12,20,23 A meta-analysis including 16 independent cohorts (maximum of 1,278 participants) assessed the amyloid–cognition relationship in cognitively normal adults, and the results also showed that only episodic memory had a modest but significant negative relationship to amyloid burden detected by PiB-PET in presymptomatic AD.39 Similar to these results found in sporadic AD, our results in ADAD showed that cerebral amyloidosis predicted episodic memory decline over time in presymptomatic ADAD. The negative association between amyloidosis and episodic memory is relatively modest in the presymptomatic stage (β = −0.084, p = 0.043), likely due to the relatively subtle nature of such declines so early in disease progression. Larger sample sizes and longer follow-up time will further test this association in the future. The findings from both ADAD and sporadic AD imply that amyloidosis in the brain may be an indicator of early cognitive decline and thus provide more effective outcome measures for early-stage and prevention treatment trials.
In both our cross-sectional and longitudinal data sets, the strongest relationships between amyloid and cognition were found in the early stages of clinical ADAD. To date, only a few studies have reported the amyloid–cognition relationship in different stages of AD.7,16,17,19,23 Similar to our results, one longitudinal study19 found that baseline amyloid PET values significantly correlated with widespread cognitive decline across multiple cognitive domains in an MCI group but with only limited declines in cognitively normal and AD groups. Another longitudinal study23 also showed that cognitive decline associated with amyloidosis did not plateau after presymptomatic phases but extended into MCI and dementia. This reveals that the detrimental influences represented by measures of amyloid burden may not have plateaued after clinical onset.
Our findings have also confirmed the selective impairment of cognitive domains involved in the amyloid–cognition relationship. Episodic memory decline associated with amyloid burden appears earlier than other cognitive domain declines,12,20,23 and cognitive declines in global cognition and working memory occur in symptomatic stages.17,19,23 In our current findings examining s-MCs, amyloid PET was associated with cross-sectional deficits but not longitudinal declines in episodic memory, a finding at odds with some studies of late-onset symptomatic AD.19,23 One possible explanation for this is a floor effect on episodic memory tests observed in the s-MCs. It may be that at CDR 0.5 and 1 floor effects limited the ability to detect episodic memory decline in the tests used in our study. We did not find any negative association of amyloid burden with language. It is noteworthy that most s-MCs in our study had CDR scores of 0.5 and 1 (88.9% in cross-sectional and 95.2% in longitudinal data sets). Due to the lack of more advanced dementia cases (CDR 2–3), language was relatively preserved compared with other cognitive domains.
There are several limitations in this study. First, our study was based on ADAD, which accounts for less than 1% of all cases of AD,32,40 limiting the generalizability of our findings. However, increasing evidence supports that both ADAD and sporadic AD share a common pathophysiologic basis.28,32 Our results are similar to the prior findings for sporadic late-onset AD, suggesting that ADAD is a good model for the amyloid–cognitive relationships in sporadic AD. Second, this analysis lacked other AD biomarker measures, such as tau, atrophy, and hypometabolism. Future analyses incorporating these biomarkers may help inform the role of other biomarkers in cognitive decline. Furthermore, some individuals had incomplete psychometric measures at both baseline and longitudinal assessments. However, constructing psychometric composites using any available data (e.g., allowing as little as 1 test to generate a composite rather than requiring all scores) did not substantially change model results.
Last, although our results are important to further our understanding of the amyloid hypothesis and may generate more critical hypotheses to be tested in the future, they must be interpreted with caution because they are preliminary in nature and no rigorous multiplicity adjustment has been implemented.
Our findings support that cerebral amyloidosis is a useful marker of cognition in both presymptomatic and symptomatic ADAD, similar to findings in sporadic AD, supporting the definition of a presymptomatic stage of AD and providing outcome measures for early-stage treatment trials.
Supplementary Material
ACKNOWLEDGMENT
This manuscript has been reviewed by the DIAN study investigators for scientific content and consistency of data interpretation with previous DIAN study publications. The authors acknowledge the altruism of the participants and their families and the contributions of the DIAN research and support staff at each of the participating sites (http://www.dian-info.org/institutions_map.htm).
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAD
autosomal dominant AD
- as-MC
asymptomatic mutation carrier
- CDR
Clinical Dementia Rating
- CDR-SB
Clinical Dementia Rating sum of boxes
- DIAN
Dominantly Inherited Alzheimer Network
- EM
Episodic Memory composite
- EYO
estimated years from expected symptom onset
- GC
Global Cognitive composite
- LF
Language Function composite
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- NC
asymptomatic mutation noncarrier
- PiB
Pittsburgh compound B
- ROI
region of interest
- s-MC
symptomatic mutation carrier
- SUVR
standardized uptake value ratio
- WM
Working Memory composite
Footnotes
Supplemental data at Neurology.org
Editorial, page 750
Contributor Information
Collaborators: Fen Wang, Brian A. Gordon, Davis C. Ryman, Shengmei Ma, Chengjie Xiong, Jason Hassenstab, Alison Goate, Anne M. Fagan, Nigel J. Cairns, Daniel S. Marcus, Eric McDade, John M. Ringman, Neill R. Graff-Radford, Bernardino Ghetti, Martin R. Farlow, Reisa Sperling, Steve Salloway, Peter R. Schofield, Colin L. Masters, Ralph N. Martins, Martin N. Rossor, Mathias Jucker, Adrian Danek, Stefan Förster, Christopher A.S. Lane, John C. Morris, Tammie L.S. Benzinger, and Randall J. Bateman
AUTHOR CONTRIBUTIONS
Dr. Wang: writing the manuscript, study concept and design, analysis of data, statistical analysis. Dr. Gordon: revising the manuscript, study design, analysis of data. Dr. Ryman: revising the manuscript, study design, analysis of data. Ms. Ma: statistical analysis, analysis of data. Dr. Xiong: revising the manuscript, statistical analysis, analysis of data. Dr. Hassenstab: revising the manuscript, analysis of data. Dr. Goate: acquisition of data, analysis of data, revising the manuscript. Dr. Fagan: acquisition of data, analysis of data, revising the manuscript. Dr. Cairns: acquisition of data, analysis of data. Dr. Marcus: acquisition of data, analysis of data. Dr. McDade: acquisition of data, analysis of data, revising the manuscript. Dr. Ringman: acquisition of data, analysis of data, revising the manuscript. Dr. Graff-Radford: acquisition of data, analysis of data, revising the manuscript. Dr. Ghetti: acquisition of data, analysis of data, revising the manuscript. Dr. Farlow: acquisition of data, analysis of data, revising the manuscript. Dr. Sperling: acquisition of data, analysis of data, revising the manuscript. Dr. Salloway: acquisition of data, analysis of data, revising the manuscript. Dr. Schofield: acquisition of data, analysis of data, revising the manuscript. Dr. Masters: acquisition of data, analysis of data, revising the manuscript. Dr. Martins: acquisition of data, analysis of data, revising the manuscript. Dr. Rossor: acquisition of data, analysis of data, revising the manuscript. Dr. Jucker: acquisition of data, analysis of data, revising the manuscript. Dr. Danek: acquisition of data, revising the manuscript. Dr. Förster: acquisition of data, revising the manuscript. Dr. Lane: acquisition of data, revising the manuscript. Dr. Morris: revising the manuscript, study concept and design, analysis of data, study coordination, obtaining funding. Dr. Benzinger: revising the manuscript, study concept and design, overseeing imaging data collection, analysis of data. Dr. Bateman: revising the manuscript, study concept and design, analysis of data, study coordination, obtaining funding.
STUDY FUNDING
The DIAN is supported by NIA grant U19 AG032438 to J.C. Morris and by the generous support of F. Simmons and O. Mohan and an anonymous foundation, in addition to DIAN site support from the German Center for Neurodegenerative Diseases (DZNE), the NIHR Queen Square Dementia Biomedical Research Unit, and JO & JR Wicking Trust grants 13026 & 20821. The DIAN-TU (R.J. Bateman, PI) is supported by funding from the NIH U-01-AG042791, Alzheimer's Association, and the DIAN Pharma Consortium (Biogen, Eisai, Elan, Eli Lilly, Forum, Genentech, Hoffman La-Roche, Janssen, Mithridion, Novartis, Pfizer, and Sanofi-Aventis). The authors acknowledge Chinese Society of Neurology, the National Key Department of Neurology funded by Chinese Health and Family Planning Committee, and the National Natural Science Foundation of China (81100797) for their support for this study.
DISCLOSURE
F. Wang, B. Gordon, D. Ryman, S. Ma, C. Xiong, J. Hassenstab, and A. Goate report no disclosures relevant to the manuscript. A. Fagan reports consulting for Lilly, Roche, and IBL International. N. Cairns, D. Marcus, E. McDade, J. Ringman, N. Graff-Radford, B. Ghetti, M. Farlow, R. Sperling, and S. Salloway report no disclosures relevant to the manuscript. P. Schofield has received speaking fees from Janssen-Cilag. C. Masters, R. Martins, M. Rossor, M. Jucker, A. Danek, S. Förster, and C. Lane report no disclosures relevant to the manuscript. J. Morris has served as a consultant for Lilly USA, ISIS Pharmaceuticals, and Charles Dana Foundation. T. Benzinger reports research grants from Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly) and participation in clinical trials sponsored by Eli Lilly and LaRoche. R. Bateman reports research grants from Pharma Consortium (Biogen, Eisai, Elan, Eli Lilly, Forum, Genentech, Hoffman La-Roche, Janssen, Mithridion, Novartis, Pfizer, and Sanofi-Aventis). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184–185. [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42:631–639. [DOI] [PubMed] [Google Scholar]
- 4.Naslund J, Haroutunian V, Mohs R, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000;283:1571–1577. [DOI] [PubMed] [Google Scholar]
- 5.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004;62:925–931. [DOI] [PubMed] [Google Scholar]
- 6.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 2009;30:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain 2007;130:2837–2844. [DOI] [PubMed] [Google Scholar]
- 8.Tolboom N, van der Flier WM, Yaqub M, et al. Differential association of [11C]PIB and [18F]FDDNP binding with cognitive impairment. Neurology 2009;73:2079–2085. [DOI] [PubMed] [Google Scholar]
- 9.Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010;67:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pike KE, Ellis KA, Villemagne VL, et al. Cognition and beta-amyloid in preclinical Alzheimer's disease: data from the AIBL study. Neuropsychologia 2011;49:2384–2390. [DOI] [PubMed] [Google Scholar]
- 11.Kantarci K, Lowe V, Przybelski SA, et al. APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology 2012;78:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperling RA, Johnson KA, Doraiswamy PM, et al. ; AV45-A05 Study Group. Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging 2013;34:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol 2009;66:1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Resnick SM, Sojkova J, Zhou Y, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology 2010;74:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villemagne VL, Pike KE, Chételat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewers M, Insel P, Jagust WJ, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI). CSF biomarker and PIB-PET-derived beta-amyloid signature predicts metabolic, gray matter, and cognitive changes in nondemented subjects. Cereb Cortex 2012;22:1993–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim YY, Ellis KA, Pietrzak RH, et al. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology 2012;79:1645–1652. [DOI] [PubMed] [Google Scholar]
- 19.Doraiswamy PM, Sperling RA, Coleman RE, et al. : AV45-A11 Study Group. Amyloid-β assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology 2012;79:1636–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim YY, Pietrzak RH, Ellis KA, et al. Rapid decline in episodic memory in healthy older adults with high amyloid-β. J Alzheimers Dis 2013;33:675–679. [DOI] [PubMed] [Google Scholar]
- 21.Ellis KA, Lim YY, Harrington K, et al. Decline in cognitive function over 18 months in healthy older adults with high amyloid-β. J Alzheimers Dis 2013;34:861–871. [DOI] [PubMed] [Google Scholar]
- 22.Snitz BE, Weissfeld LA, Lopez OL, et al. Cognitive trajectories associated with β-amyloid deposition in the oldest-old without dementia. Neurology 2013;80:1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim YY, Maruff P, Pietrzak RH, et al. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer's disease. Brain 2014;137:221–231. [DOI] [PubMed] [Google Scholar]
- 24.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008;65:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchant NL, Reed BR, DeCarli CS, et al. Cerebrovascular disease, β-amyloid, and cognition in aging. Neurobiol Aging 2012;33:1006.e1025–e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Investig (Lond) 2012;2:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 31.Talbot C, Lendon C, Craddock N, Shears S, Morris JC, Goate A. Protection against Alzheimer's disease with apoE epsilon 2. Lancet 1994;343:1432–1433. [DOI] [PubMed] [Google Scholar]
- 32.Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS One 2012;7:e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storandt M, Balota DA, Aschenbrenner AJ, Morris JC. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology 2014;28:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benzinger TL, Blazey T, Jack CR, Jr, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci USA 2013;110:E4502–E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su Y, D'Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One 2013;8:e73377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage 2015;107:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452. [DOI] [PubMed] [Google Scholar]
- 38.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014;83:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 2013;80:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sassi C, Guerreiro R, Gibbs R, et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer's disease. Neurobiol Aging 2014;35:2881.e2881–e2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.