

Abstract
Prostate tumors develop resistance to androgen deprivation therapy (ADT) by multiple mechanisms, one of which is to express constitutively active androgen receptor (AR) splice variants lacking the ligand binding domain. AR splice variant 7 (AR-V7, also termed AR3) is the most abundantly expressed variant that drives prostate tumor progression under ADT conditions. However, the molecular mechanism by which AR-V7 is generated remains unclear. In this manuscript, we demonstrated that RNA splicing of AR-V7 in response to ADT was closely associated with AR gene transcription initiation and elongation rates. Enhanced AR gene transcription by ADT provides a pre-requisite condition that further increases the interactions between AR pre-mRNA and splicing factors. Under ADT conditions, recruitment of several RNA splicing factors to the 3’ splicing site for AR-V7 was increased. We identified two RNA splicing enhancers and their binding proteins (U2AF65 and ASF/SF2) that played critical roles in splicing AR pre-mRNA into AR-V7. These data indicate that ADT-induced AR gene transcription rate and splicing factor recruitment to AR pre-mRNA contribute to the enhanced AR-V7 levels in prostate cancer cells.
Keywords: Prostate Cancer, Androgen Deprivation Therapy, RNA Splice Variant, Alternative Splicing
Introduction
The primary treatment for metastatic prostate cancer (PCa) is androgen depletion therapy (ADT). Although initially effective, most tumors progress to castration-resistant PCa (CRPC) even under treatment with the most potent anti-androgens (e.g. MDV3100 (enzalutamide) and abiraterone). No curative therapy is available (1). It is commonly agreed that re-activation of the androgen receptor (AR) signaling contributes to CRPC (2, 3) through several proposed mechanisms including: AR gene amplification/mutation and AR protein overexpression (4, 5), intra-tumoral androgen synthesis (6, 7), aberrant expression of AR co-regulators (8) and alternative AR activation by cytokines and growth factors in the absence of androgens (9). In addition, recent findings indicate that the AR is also expressed as C-terminal truncated variants, called ARvs, through alternative RNA splicing (10-15). Lacking the ligand-binding domain of full length AR (AR), ARvs are constitutively active in driving AR-regulated transcription and promoting tumor progression, even under castrate conditions (11, 13, 15, 16). ARvs regulate a mitotic form of the AR-transcriptome rather than one associated with more differentiating functions (17, 18). Expression of ARvs occurs frequently in CRPC tumors (19). Although a number of ARvs have been described in PCa cell lines and xenografts, AR-V7 (also termed AR3) is the most commonly expressed ARv in human tissues (11, 13, 20). Its levels are correlated with increased risk of biochemical relapse (11, 13) and shorter survival time of CRPC patients (20). These results suggest a critical role of AR-V7 in supporting CRPC. However, the molecular mechanism by which AR-V7 mRNA is spliced remains unclear.
Pre-mRNA splicing involves stepwise assembly of RNA splicing factors to the regions containing 5’ and 3’ splicing sites, excision of the intron sequences and re-ligation of the adjacent exons (21). Alternative RNA splicing is the process whereby exons are selectively excised from the pre-mRNA, resulting in a different combination of exons in the final translated mRNA (22). AR-V7 mRNA is spliced at the alternative 3’ splice site (3’ss) next to a cryptic exon, exon 3B, rather than the 3’ss next to exon 4, resulting in translation of a C-terminal truncated form of the AR protein (11, 13). The decision as to which splicing site is excised is determined by both the regulatory RNA sequences (cis-elements) and their associated RNA splicing proteins (trans-elements). Depending upon the functional significance and location, some regulatory cis-elements are termed exonic splicing enhancers (ESE) or intronic splicing enhancers (ISE) (23, 24). In addition, RNA splicing is closely coupled with gene transcription (25). Both transcription initiation (26) and elongation rates (27, 28) have a significant impact on the outcome of splicing. This is achieved by the association of RNA splicing factors to the transcription machinery when transcription is initiated (29-31). These protein complexes move along the gene during transcription elongation, when transcribed pre-mRNA is screened by the RNA spliceosome to define and excise the splice sites, before transcription is terminated (32-34). Therefore, the abundance of a specific splice variant is controlled by both gene transcription rate and splicing factor recruitment to the pre-mRNA during the alternative splicing process.
The question that remains to be answered is whether ADT regulates the RNA splicing program that favors RNA synthesis of ARvs as a survival strategy for PCa in response to ADT. In this manuscript, we studied the molecular mechanisms by which AR-V7 was alternatively spliced in response to ADT.
RESULTS
AR and AR-V7 mRNA levels are increased in response to androgen deprivation
We first profiled AR and AR-V7 RNA levels in a panel of PCa cell lines: VCaP, LNCaP, LN(AI) and LN95. Both LN(AI) and LN95 cells are derived from LNCaP cells, but have been cultured under long-term ADT conditions, thus possessing an ADT-resistant phenotype. Using an absolute quantification method, real-time qPCR showed that VCaP cells expressed AR RNA that was 5-10 fold higher than LNCaP and LNCaP derived cells (Fig.1 and Suppl.1A), consistent with the report that VCaP cells have an increased AR gene copy number (35). AR RNA level was ~17.9 fold higher than AR-V7 in VCaP cells. LNCaP cells expressed extremely low levels of AR-V7 RNA. Both LN(AI) and LN95 cells expressed higher levels of AR-V7 than LNCaP cells. AR-V7 RNA levels in LN95 cells were 30-40% lower than that in VCaP cells. Western blotting assays showed consistently that VCaP cells expressed higher AR protein levels than other cell lines (Fig.1B and Suppl.1B), possibly due to the increase in AR gene copy number in VCaP cells. Both VCaP and LN95 cells expressed comparable levels of AR-V7 protein. LN(AI) cells expressed lower levels, while parental LNCaP cells had undetectable levels of AR-V7 protein.
Figure 1. AR and AR-V7 RNA levels in PCa cells.

(A) Total RNA was extracted from VCaP, LNCaP, LN(AI) and LN95 cells. AR and AR-V7 mRNA copy numbers within 20ng RNA were determined by real-time qPCR using absolute quantification as described in Materials and Methods. (B) AR and AR-V7 protein levels were detected by western blotting assays using AR(N-20) and AR-V7 antibodies. (C) VCaP, LNCaP, LN(AI) and LN95 cells were maintained in RPMI1640 medium containing 5% FBS or 5% charcoal stripped serum for 48 hours. Cells were treated with vehicle, 10nM DHT and/or 5uM MDV3100 for 24 hours. Relative RNA levels of AR and AR-V7 were determined by real-time qPCR using relative quantification to GAPDH. Results were obtained from three independent experiments with samples in triplicates and shown as means ± S.E.M. One-way ANOVA followed by student t-test was carried out using GraphPad Prism showing significance with P<0.01 as ** or P< 0.001 as ***. Primer sequences and standard curves for measuring RNA levels of AR and AR-V7 were presented in supplementary materials and figures.
ADT conditions were reported to increase both RNA and protein levels of AR in PCa cell lines and xenografts (17, 36). To determine whether ADT also regulated AR-V7 expression, we treated PCa cells with DHT and/or MDV3100 (Fig.1C). DHT reduced AR RNA levels significantly in VCaP and LNCaP cells, but only to a minor extent in LN(AI) cells. AR-V7 levels in LN(AI) cells were also repressed by DHT, following the changes of AR levels. In contrast, neither AR nor AR-V7 RNA levels were altered by DHT or MDV treatment in LN95 cells.
We found that AR-V7 expression was also reversibly regulated by DHT and MDV treatments. After VCaP cells were pre-treated with DHT for 24 hours, adding MDV to the culture medium dramatically up-regulated AR-V7 RNA and protein levels (Fig.2A-C). Vice versa, DHT significantly supressed AR-V7 expression after VCaP cells were pre-treated with MDV. Changes in AR-V7 RNA levels were correlated with AR levels. Using primary cultures from MDV-resistant VCaP tumor xenografts grown in mice (n=10), we further showed that both AR and AR-V7 RNA levels were maintained in relative high levels under maximum ADT conditions, but significantly decreased when DHT was added (Fig.2D). Since the doubling time of VCaP cells is 53 hours, changes in AR and AR-V7 RNA levels cannot be accounted by clonal selection. These results indicated that RNA splicing of AR-V7 was a dynamic and reversible process, which was regulated by AR signaling.
Figure 2. Alternative AR-V7 splicing is a reversible process regulated by AR signaling.
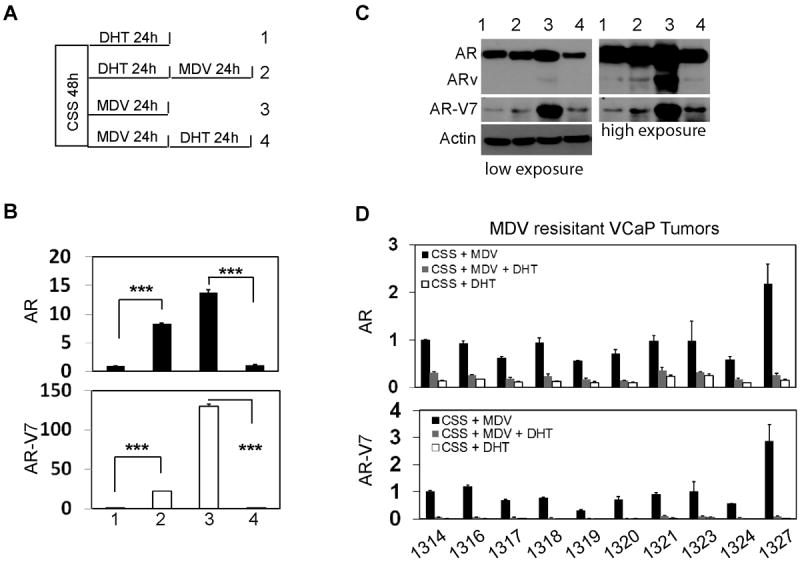
(A) Schematic diagram of experimental design shows the treatments to VCaP cells. VCaP cells were maintained in RPMI1640 medium containing 5% charcoal stripped serum for 48 hours. Cells were treated with 10nM DHT (sample 1 and 2) or 5uM MDV3100 (sample 3 and 4) for 24 hours. Cells were washed and replenished with medium containing 5uM MDV3100 (sample 2) or 10nM DHT (sample 4) for another 24 hours. RNA and protein samples were collected. (B) AR and AR-V7 mRNA level were measured by real-time qPCR using relative quantification to GAPDH. Results were obtained from three independent experiments with samples in triplicate and shown as means ± S.E.M. Student t-test showed statistical significances with P<0.001 as ***. (C) AR and AR-V7 protein levels were detected by Western blotting assays. (D) Primary cultured MDV-resistant VCaP cells were described in Material and Methods. Cells were treated with MDV3100, DHT or MDV3100 plus DHT for 24 hours. Total RNA was collected and sued to measure AR and AR-V7 RNA levels by real-time qPCR using relative quantitative method.
AR-V7 expression is important for VCaP cell proliferation under ADT conditions
The functional significance of AR-V7 expression was further tested in VCaP cells by comparing cell proliferation rates under conditions of AR versus AR-V7 knockdown by siRNA. SiRNA to exon 7 knocked down AR, siRNA to exon 3b knocked down AR-V7, and siRNA to exon 1 knocked down total AR (AR-V7 + AR). In the presence of androgen depletion and/or MDV treatments, AR-V7 or total AR, but not AR knockdown significantly reduced VCaP cell growth (Fig. 3A). However, cell growth relied on AR when DHT was present. Consistent with cell proliferation assays, expression levels of PSA and TMPSS2 were further dramatically decreased by siRNAs targeting AR-V7 or total AR, when compared with siRNA targeting AR alone under androgen depletion and/or MDV treatment conditions (Fig. 3B). In the presence of DHT, siRNA knocking down AR-V7 did not inhibit PSA and TMPSS2 expression. However, siRNA targeting total AR presented more potent suppressive effects to these genes when compared with siRNA targeting AR only. Interestingly, UGT2B17 and UBE2C were previously demonstrated to be AR-V7 regulated genes in prostate cancers (19). We further confirmed AR-V7 was required for UGT2B17 transcription in VCaP cells under ADT conditions. In contrast, DHT activated AR supressed UGT2B17 RNA levels, which effects were blocked by AR knockdown. Similar observation was also from UBE2C expression that was inhibited by AR but enhanced by AR-V7 (Suppl.2). These findings indicate that under ADT conditions AR-V7 can replace AR to sustain cell growth, and regulate a gene set distinct from AR. Our results support a critical role of AR-V7 that is responsible for prostate cancer phenotype shift from androgen sensitive to CRPC under castration stress. The efficiencies of siRNA knockdown of AR and or AR-V7 were shown by Western blotting in Fig. 3C. Together, our results indicate that AR-V7 maintains VCaP prostate cancer cell proliferation by an AR signaling mechanism under ADT conditions.
Figure 3.

VCaP cells were transfected with control siRNA or siRNA targeting AR Exon 1, Exon 7 or Exon 3b of the AR gene. (A) Cells were seeded in 96 well plates and treated with vehicle, 10nM DHT or 5uM MDV for 0-6 days. MTS assay was performed at each time point. Data were plotted as fold change over day 0. (B) After siRNA transfections, VCaP cells were treated with vehicle, DHT or MDV for another 24 hours. Relative RNA levels of PSA, TMPSS2 and UGT2B17 over GAPDH were measured by real-time qPCR. (C) Efficiencies of siRNA knockdown were confirmed by Western blotting assays with the indicated antibodies.
AR-V7 RNA splicing is coupled with AR gene transcription
Since the RNA splicing process was known to be coupled with gene transcription, and our data also indicated that AR-V7 RNA levels were correlated with AR RNA levels, we examined whether active AR-V7 splicing was controlled by the AR gene transcription rate. We applied three different approaches to inhibit AR gene transcription by treating cells with actinomycin D (ActD), benzimidazole (DRB) or trichostatin A (TSA). Cells were also co-treated with either 10nM DHT or 5uM MDV for 0, 1, 2, 4, 8 and 16 hours (Fig. 4). In the presence of vehicle, MDV maintained while DHT dramatically decreased both AR and AR-V7 RNA levels during 16 hours of treatment in VCaP cells, while AR and AR-V7 RNA levels in LN95 cells were maintained at relatively constant levels. However in both VCaP and LN95 cells, ActD and DRB eliminated, while TSA significantly reduced AR-V7 RNA levels correlating with changes of AR RNA levels. ActD forms complexes with double-strand DNA to prevent RNA pol II from forming the transcription initiation complex, while DRB is an inhibitor of C-terminal domain of pol II that inhibits gene transcription at the elongation step. TSA was confirmed to inhibit AR gene transcription in several different prostate cancer cell lines (37, 38). These results together indicated that AR-V7 splicing was dependent upon AR gene transcription initiation and elongation rates.
Figure 4. AR-V7 RNA splicing is coupled with AR gene transcription rate.

VCaP (A) and LN95 (B) cells were maintained in RPMI1640 medium containing 5% charcoal stripped serum and treated with 10nM DHT or 5uM MDV3100 in the presence of vehicle, 1uM actinomycin D (Act D), 5uM Benzimidazole (DRB) or 10nM Trichostatin A (TSA). RNA samples were collected at time points of 0, 1, 2, 4, 8, and 16 hours. AR and AR-V7 RNA levels were determined by relative quantification to 18s rRNA. Results were obtained from two independent experiments with samples in triplicates and shown as means ± S.E.M. Note: 18s rRNA levels were not altered by ActD, DRB or TSA treatment within 16 hours, therefore served as the internal control gene.
Recruitment, but not expression of RNA splicing factors contributes to AR-V7 splicing
Although RNA splicing is coupled with transcription, generation of a RNA splice variant requires splicing factors that recognize and excise the alternative splice sites. We chose a panel of splicing factors including U1A, U2AF65, AFS/SF2, hnRNP I, PSF and p54nrb that were demonstrated to play essential roles in RNA splicing (39) and measured their protein levels under DHT or MDV treatment. No changes in protein levels of these splicing factors were observed under different treatments and among different cell lines (Suppl.3).
We next determined whether recruitment of splicing factors to the AR gene was altered following ADT conditions. Chromatin immunoprecipitation (ChIP) assays were performed with primers amplifying the P1-P3 regions, corresponding to the 5’ and 3’ splice regions for AR and AR-V7 (Fig.5). The P4 region is located upstream of the 5’UTR of human GAPDH gene, a region of the gene where there are no active RNA splicing events. It therefore serves as a negative control. Consistent with ADT-induced AR gene transcription, the recruitment of pol II to P1, P2 and P3 regions were significantly higher in MDV treated VCaP cells. These changes were concurrent with increased recruitments of several RNA splicing factors (U1A, U2AF, ASF/SF2 and p54nrb) to P1, P2 and P3 regions. Exceptions were PSF (no change) and hnRNP I (decreased recruitment). These data suggested that MDV treatment increased spliceosome recruitment to the AR gene to process both AR and AR-V7 RNA splicing. In contrast, although MDV also enhanced pol II and U1A, U2AF and p54nrb onto P1 and P3 regions in LNCaP cells, their recruitments to the P2 region (containing AR-V7 3’ss) were not increased by MDV. These observations were consistent with low expression of AR-V7 in LNCaP cells. Together, these results suggested that spliceosome recruitment to the AR gene, rather than alterations in protein levels of splicing factors, contributed to AR-V7 splicing.
Figure 5. Recruitment of RNA splicing factors to the AR gene in PCa cells. (A).
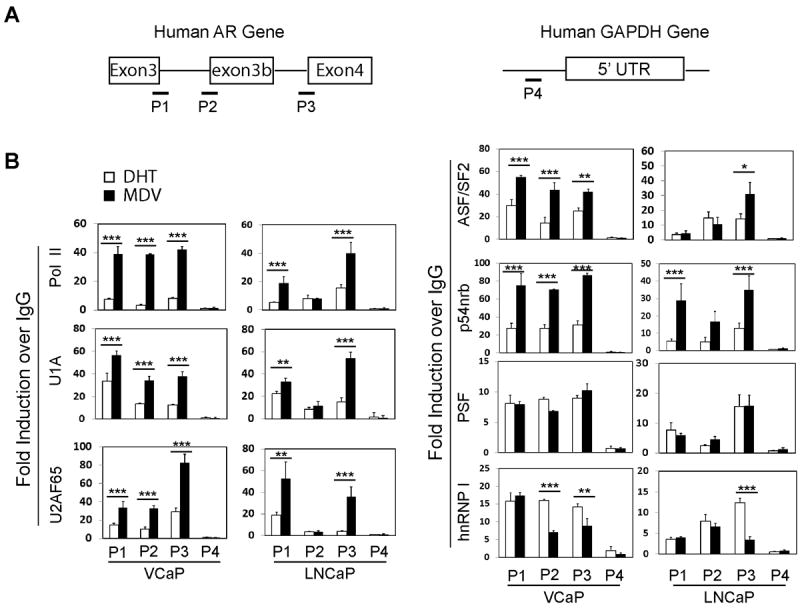
Schematic diagrams of the human AR gene and the GAPDH gene show the regions (P1-P4) that were amplified in ChIP assays. (B) VCaP and LNCaP cells were maintained in RPMI1640 medium containing 5% charcoal stripped serum for 48 hours. Cells were treated with either 10nM DHT or 5uM MDV3100 for another 24 hours. ChIP assays were performed using antibodies against pol II, U1A, U2AF65, ASF/SF2, p54nrb, PSF, hnRNP I and control IgG. Eluted DNA fragments were used as templates for real-time qPCR. Signals were calculated as percentage of input and blotted as fold changes over control IgG. ChIP data were derived from five independent experiments with triplicate samples per experiment. Student t-test showed statistical significance with P<0.05 as *, P<0.01 as ** and P<0.001 as ***.
Construction of AR-V7 minigene to identify RNA splicing enhancers
In order to identify any cis- and trans-element responsible for AR-V7 splicing, we constructed the AR-V7 minigene plasmid (Fig.6A), with exon 3B and its flanking ~400bp nucleotide sequence inserted in between exon 3 and exon 4 of the human AR gene. When transiently transfected, the minigene expressed 12-25 fold of AR and 8-300 fold of AR-V7 higher than the levels of endogenous AR transcripts in PCa cell lines (Fig.6B). Driven by the constitutively active CMV promoter, the levels of minigene transcribed AR-V7 were not affected by DHT or MDV treatment. These observations indicated that gene transcription rate, but not ADT condition per se, directly regulated AR-V7 RNA splicing.
Figure 6. Construction of AR-V7 minigene to identify RNA splicing enhancers.
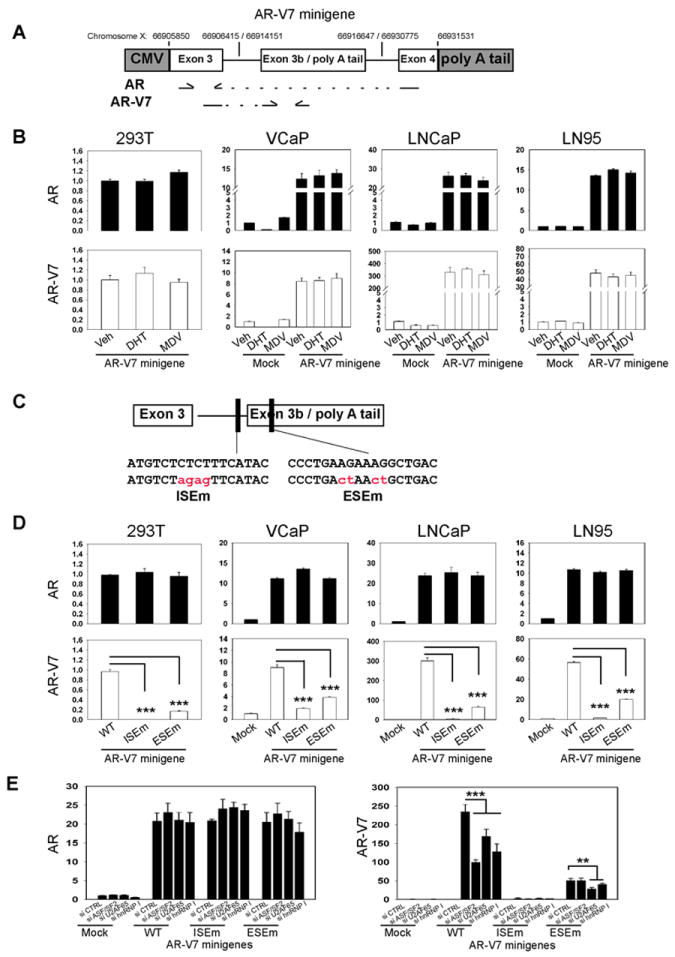
(A) Schematic diagram of AR-V7 minigene construct and locations of primers used in real-time qPCR. Three DNA fragments of human AR gene were cloned into pCMV2 vectors. Positions of each AR gene fragment in the chromosome X were marked. (B) 293T, VCaP, LNCaP and LN95 cells were transiently transfected with mock vector or AR-V7 minigene plasmid. Cells were treated with vehicle, 10nM DHT or 5uM MDV3100 for 24 hours. Relative mRNA levels of AR and AR-V7 mRNA levels to GAPDH were determined by real-time qPCR. (C) Using point mutagenesis, mutant AR-V7 mingenes were constructed with mutations at the ISE and ESE sites. (D) 293T, VCaP, LNCaP and LN95 cells were transiently transfected with mock, AR-V7 minigene (WT) or AR-V7 minigenes with mutations at ISE (ISEm) and ESE (ESEm). (E) LNCaP cells were transfected with indicated siRNAs followed with plasmids encoding mock or AR minigenes (WT, ISEm or ESEm). Relative RNA levels of AR and AR-V7 to GAPDH were determined by real-time qPCR. Results were obtained from three independent experiments and shown as means ± S.E.M. One-way ANOVA followed by student t-test was carried out using GraphPad Prism showing statistical significance with P< 0.001 as ***.
To locate potential RNA sequences responsible for AR-V7 splicing, we screened exon 3B and its flanking region using the Splicing Rainbow (40) and ESEfinder 3.0 (41) programs, two bioinformatic tools to predict potential splicing factor binding sites. One intronic splicing enhancer (ISE) and one exonic splicing enhancer (ESE) near the 3’ss of exon 3B were identified. The ISE was predicted to bind hnRNP I or U2AF65, while the ESE was a potential ASF/SF2 binding site. We applied mutagenesis and cloning techniques to further construct AR-V7 minigenes carrying point mutations at either ISE or ESE site (Fig.6C). When these mutant AR-V7 minigenes were introduced into PCa cells, only AR-V7 transcript levels, but not AR, were dramatically decreased (Fig.6D). To further confirm whether the functions of ISE and ESE were mediated through the predicted RNA splicing factors, we transfected AR-V7 minigenes into LNCaP cells in the presence of siRNAs against hnRNP I, U2AF65 and ASF/SF2. The levels of AR transcribed by AR-V7 minigenes were unchanged, regardless of siRNA knockdown or mutations at ESE or ISE with in the minigenes (Fig.6E). In contrast, levels of AR-V7 transcribed by the AR-V7(WT) minigene were dramatically decreased with hnRNP I, U2AF65 or ASF/SF2 knockdown. The AR-V7(ISEm) minigene expressed very low levels of AR-V7, which were insensitive to any knockdown of splicing factors. Interestingly, RNAi of U2AF65 and hnRNP I but not ASF/SF2 further decreased AR-V7 levels transcribed by the AR-V7(ESEm) minigene. These findings indicated that both the ISE and the ESE were specifically important for AR-V7 splicing through interactions with U2AF65, hnRNP I and ASF/SF2 respectively.
Identification of RNA splicing factors responsible for AR-V7 splicing
To further study binding proteins for the ISE and the ESE, we synthesized two 40bp RNA oligos to perform RNA pull-down assays (Fig.7A). Oligo 1 contains the ISE, while oligo 2 contains the ESE. Their sequences are listed in supplementary material. Both oligos were incubated with purified Flag-tagged U2AF65, hnRNP I and ASF/SF2. hnRNP I and U2AF65 bound oligo 1. These interactions were abolished when the ISE was replaced with the ISEm (Fig.7B). Interestingly, hnRNP I and U2AF65 can also be pulled down by oligo 2. However, those interactions were not affected by ESE point mutations. ASF/SF2 had a strong association with oligo 2, but not with oligo 1. This interaction was also dramatically decreased when ESE was replaced with the ESEm. In order to show specific U2AF65/hnRNP I-ISE and ASF/SF2-ESE interactions, we also used oligos 1 and 2 to pull down two other RNA splicing factors, U1A and Tra2β. U1A bound both oligos 1 and 2 even in the presence of ISEm and ESEm, whereas Tra2β had no association to either RNA oligo. These results indicate that ASF/SF2 specifically binds the ESE, while hnRNP I and U2AF65 bind oligo 1 at the specific ISE site. They also bind oligo 2 but possibly through sequences other than the ESE.
Figure 7. Characterization of RNA splicing factors that regulate AR-V7 splicing.

(A) Schematic diagram of the AR gene between Exon 3 and 4 and locations of AR pre-mRNA sequences used in RNA oligo pull-down assays (B) and primers used in RNA immunoprecipitation assays (C-D). (B) Flag tag purified RNA splicing factors U2AF65, hnRNP I, ASF/SF2, U1A and Tra2β were incubated with RNA oligos containing ISE, ESE and their point mutants. Oligo associated splicing factors were detected by western blotting assays. (C) VCaP cell lysates were immunoprecipitated with antibodies against ASF/SF2, U2AF65 and hnRNP I. Protein associated pre-mRNA were extracted and reverse transcribed and analyzed by regular PCR with primers amplifying P1-3 regions. (D) VCaP cells were treated with either 10nM DHT or 5uM MDV3100 for 2 hours. RNA immunoprecipitation assays were performed. Real-time qPCR quantified the enrichments of ASF/SF2, U2AF65 and hnRNP I on pre-mRNA at P1-3 regions. Signals were calculated as percentage of input, and blotted as fold changes over control IgG. (E) VCaP cells were transfected with siRNAs against control, ASF/SF2, U2AF65 and hnRNP I for 48 hours, after which cells were treated with either 10nM DHT or 5uM MDV3100 for another 24 hours. AR and AR-V7 RNA splicing products were measured by real-time qPCR using relative quantification against GAPDH. A primer set covering GAPDH exons 7 and 8 was used as a negative control to monitor GAPDH gene splicing in response to RNAi of ASF/SF2, U2AF65 and hnRNP I. (F-G) AR protein and mRNA were also detected by Western and Northern blotting assays. Densitometry analyses calibrate RNA and protein bands of the AR gene by 18S and Actin as the loading controls. In Fig 7G, both low and high exposures of Western blot with AR(N-20) antibody were shown. (H) LN95 cells were transfected with siRNAs against control, ASF/SF2, U2AF65 and hnRNP I for 48 hours. AR, AR-V7 and GAPDH splicing products were measured by real-time qPCR. Data were derived from three independent experiments and presented as means ± SEM. One-way ANOVA followed by student t-test was carried out using GraphPad Prism showing statistical significance with P<0.01 as** and P< 0.001 as ***.
RNA co-immnunoprecipitation (RIP) assays were further performed (Fig.7C). Antibodies against ASF/SF2, U2AF65 and hnRNP I, but not control IgG, precipitated AR pre-mRNA at the P2 region (containing both the ISE and the ESE). Interestingly, hnRNP I was absent in the P1 region (5’ss for both AR and AR-V7) and ASF/SF2 showed an extremely low level at the P3 region (3’ss for AR). These observations suggest that hnRNP I functions primarily at the 3’ss, while ASF/SF2 has a relatively higher specificity to the 3’ss for AR-V7, emphasizing its important role for AR-V7 splicing. We further used real-time qPCR to quantify the recruitments of ASF/SF2, U2AF65 and hnRNP I to the AR pre-mRNA at the AR-V7 3’ss under DHT or MDV treatment for 2 hours in VCaP cells (Fig.7D). MDV dramatically induced U2AF65 binding, but had no impact on ASF/SF2 and hnRNP I to the P1 region. However, all of these splicing factors had enhanced recruitment to the P2 region by MDV, while only U2AF65 and hnRNP I increased associations with the P3 region in response to MDV treatment. These results suggested that ASF/SF2-ESE and U2AF65/hnRNP I-ISE interactions were regulated by ADT condition in contributing to AR-V7 splicing.
To further confirm the functional significance of ASF/SF2, U2AF65 and hnRNP I for AR-V7 splicing, we knocked down each splicing factor in VCaP and LN95 cells and measured AR and AR-V7 levels by real-time PCR amplifying exon3/4 and exon3/3b (Fig.7E and 7H). RNA silencing of ASF/SF2, U2AF65 and hnRNP I significantly reduced AR-V7 RNA levels. In addition, hnRNP I knockdown also mildly reduced AR levels. To exclude the possibility that ASF/SF2 and U2AF65 silencing also affect other splicing events particularly that within the AR gene, we further showed that U2AF65 and ASF/SF2 knockdown did not affect AR levels by both Northern and Western blotting (Fig.7F-G). In addition, we also designed a pair of primers crossing exons 7/8 of the GAPDH gene to measure its RNA splicing as a control, and showed that RNA splicing of the GAPDH gene was not affected by RNA silencing of ASF/SF2 and U2AF65. The efficiency of RNA silencing was shown by Western blotting (Suppl.4). These results suggested that both ASF/SF2 and U2AF65, when compared with hnRNP I, were specifically important for AR-V7 splicing.
DISCUSSION
Although extensive studies show that constitutively transcriptional function of ARv is important for CRPC progression, the molecular mechanisms by which ARv is alternatively spliced remain unclear. Our studies demonstrate that AR gene transcription rates and splicing factor recruitment to AR pre-mRNA near the AR-V7 3’ss are two important factors contributing to AR-V7 splicing. The ADT condition does not directly regulate levels of AR-V7 splicing. Rather it enhances AR gene transcription rates and contributes indirectly to the generation of AR-V7 splice variant.
Our lines of evidence demonstrated that AR-V7 RNA synthesis depended upon active AR gene transcription. In various experimental conditions, changes in AR-V7 RNA levels were consistent with alterations of AR RNA levels in PCa cells. ADT induced the recruitment of RNA pol II to the AR gene, reflecting active AR gene transcription (Fig.4). Concurrently, higher AR and AR-V7 levels were observed under ADT conditions. Though ActD, DRB and TSA acted through different mechanisms to decrease AR gene transcription, they showed consistent repressive effects on AR-V7 RNA splicing (Fig.3). In addition, turning on or off of AR gene transcription by DHT and MDV reversibly controlled AR-V7 splicing (Fig.1-2). Previous publications showed that AR-V7 RNA was commonly expressed in PCa cells and even normal prostate epithelium cells (13, 15) and that the ratio of ARv:AR fluctuated between the 0.1-2.5% range depending upon active AR gene transcription by castration or DHT treatment (36). These results together confirmed that AR-V7 RNA splicing process was coupled with AR gene transcription rate.
Although AR gene transcription provides a favorable genetic environment for AR-V7 RNA splicing, the enzymatic reaction to excise the 3’ss for AR-V7 splicing requires recruitment of RNA splicing factor to AR pre-mRNA for AR-V7 RNA synthesis. We showed interactions of U2AF65-ISE and ASF/SF2-ESE were critical for this splicing event. Mutations at the ISE and the ESE abolished AR-V7 splicing, while RNA silencing of U2AF65 and ASF/SF2 decreased AR-V7 levels in both VCaP and LN95 cells. These results suggested that U2AF65 and ASF/SF2 by recognizing the ISE and the ESE acted as the pioneer factors to direct further recruitment of RNA spliceosome to the AR-V7 3’ss. Although U2AF65 and ASF/SF2 might participate in other RNA splicing events, such as splicing the 3’ss next to exon 4 for AR, RNA silencing of U2AF65 and ASF/SF2 did not affect AR RNA levels, nor do they affect the GAPDH (exon7/8) RNA splicing. Disruption of U2AF65-ISE or ASF/SF2-ESE interactions had no impact on AR RNA levels (Fig. 5B). It is likely that the recruitment of U2AF65 and ASF/SF2 to the AR-V7 3’ss is more efficient due to the presence of the ESE and the ISE near the AR-V7 3’ss. ASF/SF2 is a concentration-dependent regulator for alternative RNA splicing. Although ADT does not alter its expression level, more AR pre-mRNA is available as a substrate for ASF/SF2 to catalyse RNA splicing reactions. The existence of ESE within the exon 3B therefore is more attractive for ASF/SF2 to be recruited to the AR-V7 3’ss and synthesize AR-V7 mRNA. In addition, the involvement of U2AF65 and ASF/SF2 in excising 3’ss next to exon 4 might be compensated by other splicing factors. As excision of the AR-V7 3’ss is more sensitive to U2AF65 and ASF/SF2 protein levels, they may serve as rate limiting factors in controlling AR-V7 splicing efficiency. In contrast, although hnRNP I also binds the ISE, knocking down hnRNP I reduces both AR and AR-V7 levels, indicating hnRNP I serves as a general factor regulating both AR and AR-V7 splicing. Together, these results support that AR-V7 splicing is not generated by a random splicing error. Rather, it is executed by specific RNA splicing factors through recognizing specific RNA sequences near AR-V7 3’ss. This RNA splicing event is enhanced in response to ADT as AR transcription rates increase.
Our studies also demonstrated that neither AR gene amplification nor rearrangement were required for AR-V7 RNA splicing. Although VCaP cells have amplification of the AR gene, no such genetic aberrance is not reported in LN95 cells. However, LN95 cells express comparable AR-V7 RNA levels. Regardless of AR gene amplification, suppression of the AR gene transcription rate or RNA silencing of splicing factors reduced AR-V7 splicing in both LN95 and VCaP cells. In VCaP, LNCaP and LN(AI) cells, AR-V7 RNA levels were repressed by DHT treatment (Fig.1). Particularly, AR-V7 RNA levels in VCaP cells can be reversibly regulated by DHT or MDV treatment within 24 hours (Fig.2). These results indicate that AR gene amplification does not attribute to AR-V7 splicing. Rather AR gene amplification magnifies the levels of all transcripts by the AR gene, supporting VCaP cells as an ideal model to study alternative RNA splicing events of the AR gene. Interestingly, both castration resistant LN(AI) and LN95 cells express high levels of AR-V7 than their parent LNCaP cells, supporting the hypothesis that generation of AR-V7 contributes to CRPC progression.
An intragenic AR gene rearrangement was reported contributing to AR-V7 RNA synthesis in 22Rv1 cells (42). No such gene rearrangement was noted in VCaP or castration-resistant LN(AI) and LN95 cells (35). It could be possible that similar genomic disruption may exist in some other PCa cells. However, AR-V7 mRNA synthesis still requires the removal of intron sequences through the RNA splicing process. RNA splicing factors still need to recognize and excise the 3’ss, and ligate the exon 3 with exon 3B. This enzymatic reaction will depend on active AR gene transcription to create a permissible environment and recruitment of splicing factors to AR pre-mRNA. Our data showed that changes in recruitment of U2AF65 and ASF/SF2 occurred within 2 hours of MDV treatment (Fig.6D). Enhanced AR-V7 RNA levels by MDV can be lowered within hours by subsequent DHT treatment (Fig.2A-B), indicating AR-V7 RNA splicing is a dynamic and reversible process.
In summary, our data provide new insights to the complexity of AR gene splicing during CRPC progression of prostate cancers. It invites further investigation that may lead to therapeutic revenues to block AR-V7 expression in CRPC tumors and re-sensitize current anti-AR therapy.
MATERIALS AND METHODS
Cell Culture
The human PCa cell line VCaP (CRL-2876) was purchased from ATCC and LNCaP, C4-2B, LN(AI) and 293T cell lines were generously provided by Drs. Gleave, Rennie and Buttyan from the Vancouver Prostate Centre. LN95 cells were described previously and were a generous gift from Dr. Alan Meeker of Johns Hopkins University (18). VCaP and 293T cells were cultured in DMEM, while LNCaP and C4-2B cells in RPMI1640 medium. LN(AI) and LN95 cells were maintained in RPMI1640 medium with charcoal stripped serum. Charcoal stripped serum was used for steroid studies (Hyclone, Logan, Utah). Dihydrotestosterone (DHT), Actinomycin D (ActD), Benzimidazole (DRB) and Trichostatin A (TSA) were purchased from Cedarlane (Burlington, Canada). MDV3100 was from Haoyuan Chemexpress (Shanghai, China). Plasmids encoding Flag tagged RNA splicing factor are: U2AF65 (Dr. James Manley, Columbia University), ASF/SF2 (Dr. Gourisankar Ghosh, UC San Diego), hnRNP I (Dr. Allain Frédéric, Institute for Molecular Biology and Biophysics Eidgenössiche Technische Hochshule, Switzerland) and Tra2β (Dr. Stefan Stamm, University of Kentucky).
Reverse Transcriptase PCR and Real-time qPCR
Total RNA was extracted using TRIZOL reagent (Invitrogen) and treated with deoxyribonuclease at room temperature for 15 minutes to eliminate any DNA contamination. The reverse transcription reaction was performed using random hexamers and superscript II (Invitrogen), after which the product was used as a template for PCR. Real-time qPCR was performed on the ABI PRISM 7900 HT system (Applied Biosystems, Burlington, Canada) using the FastStart Universal SYBR Green Master mix (Roche, Laval, Canada) as we previously reported (43). Cycling was performed using default conditions of the 7900HT Software: 2 min at 50°C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Absolute quantification followed the previous report (44), AR and AR-V7 cDNAs were quantified by Nanodrop and their copy numbers were calculated by copy number (molecules/uL)=concentration(g/uL)/(bp size of double stranded product × 660) × 6.022 × 1023. A series of dilutions of the cDNAs were used as templates for real-time qPCR as standards for AR or AR-V7, respectively. A standard curve was drawn by plotting the CT value against the log of the copy number of molecules. The specific AR and AR-V7 copy numbers were calculated by the equation drawn from the graph. The relative quantification method has been described before using GAPDH or 18s rRNA as the internal control genes (43). All real-time qPCR assays were carried out using three technical replications as well as three independent cDNA syntheses. Primer information is listed in the supplementary materials.
Western Blot and Chromatin Immunoprecipitation (ChIP)
After treatments, cells were incubated with lysis buffer (50mM Tris pH8.0, 150mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) followed by a brief sonication to extract protein lysate. Lysates were immunoblotted with specific antibodies (detailed in supplementary materials). ChIP assays followed the protocol we previously reported (43, 45). DNA templates retrieved from ChIP were analyzed by real-time qPCR on the ABI PRISM 7900 HT system (Applied Biosystems, Burlington, Canada) using the FastStart Universal SYBR Green Master (Roche, Laval, Canada). Enrichments of immunoprecipitated DNA fragments were determined by the Ct value. Data were calculated as a percentage of input, and plotted as fold changes over control IgG. ChIP data were derived from five independent experiments with samples in triplicate. Data are presented as means ±SEM.
MDV resistant VCaP Xenografts
MDV resistant xenografts of VCaP cells were generated by injecting VCaP cells sc in castrate SCID mice (2×106 cells mixed 1:1 with Matrigel). When xenograft tumors reached 500-800 mm3, the animals were euthanized, tumors removed, collagenase digested and again injected sc into castrate SCID mice 1:1 with Matrigel. Mice were treated with MDV3100 (10mgm/kg) per gavage 5 days a week. When xenografts reach 800-1000 mm3 mice were euthanized, tumors final minced and 1/3 of each minced tumor was grown in a 100mm plastic culture dish with RPMI 1640 plus 5% CSS and 5uM gentamicin. Cells were treated with 10uM MDV3100, 1nM DHT or MDV3100 plus DHT for 24 hours. Total RNA was collected to measure AR and AR-V7 RNA levels by real-time qPCR. All animal studies were approved by the University of Washington IACUC.
Transfection and RNA Silencing
Transient transfection of plasmid DNA used the Lipofectamine 2000 (Invitrogen, Burlington, Canada). Transfection of siRNA oligos used the siLentFect Lipid Reagent (Bio-Rad, Mississauga, Canada) according to the provided protocols.
Cell Proliferation Assay
VCaP prostate cancer cells were first transfected with siRNA against the AR gene at indicated exons. Cells were seed in seeded in 96-well plates (5000 cells/well) with culture medium containing 10% charcoal stripped serum for another 24 hours. Cells were then treated with vehicle, DHT or MDV3100 for 0-6 days. The reagent of 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium (Promega, Madison, WI) was added at each time point. Cell proliferation rates were measured according to the manufacturer’s protocol.
AR-V7 Minigene Construction
The human genomic BAC clone (RP11-75E16) was provided by The Centre for Applied Genomics, The Hospital for Sick Children, University of Toronto. It was used as the template for PCR to amplify exon 3, exon 3B and exon 4 and their flanking intron regions (~300-400 base pairs) by Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Burlington, Canada). Three DNA fragments were cloned into the plasmid vector pCMV2 (Sigma, Oakville, Canada) between the EcoR I and Sal I sites. Full sequences of the vectors will be provided upon request. DNA sequencing confirmed the integrity of the final AR-V7 construct. DNA mutagenesis was further performed using the AR-V7 minigene as the template to construct AR-V7 (ESEm) and AR-V7 (ISEm).
RNA-protein interaction assay
RNA oligo pull-down was performed by first immobilizing 0.4nmol biotin-labeled RNA oligonucleotides (Invitrogen) onto 100μl of streptavidin beads (Pierce, Rockford, IL) in a final volume of 500μl of binding buffer (20mM Hepes-KOH, pH 7.9, 80mM potassium glutamate, 0.1mM EDTA, 1mM DTT, and 20% glycerol) at 4°C for 2 hours. RNA splicing factors, U2AF65, ASF/SF2, hnRNP I, U1A and Tra2β proteins were purified by transfecting plasmids encoding Flag tagged splicing factors into 293T cells, followed with purification using Anti-Flag M2 Affinity gel (Sigma) as reported (45). The immobilized RNA oligos were then incubated with 50ug purified splicing factors in binding buffer containing 30U/mL RNase OUT and 15ug/mL yeast tRNA in a final volume of 400μl at 4°C for 2 hours. The beads were washed three times with binding buffer, once with washing buffer (20mM HEPES-KOH, pH 7.9, 0.1mM EDTA, 1mM DTT, 75mM KCl, 20% glycerol), and suspended in 40μl of 2× sodium dodecyl sulfate sample buffer, and boiled for 5 min. Eluted proteins were analyzed by Western blot. RNA oligo sequences are listed in supplementary materials.
RNA co-immunoprecipitation (RIP)
RIPs were performed as previously reported (46, 47) with the following modifications. Cell nuclei were first extracted by isotonic buffer (10mM Tris/HCl, pH 7.4, 10mM NaCl, 2.5mM MgCl2, 1mM DTT, protease inhibitor cocktail, 30U/ml RNase OUT, 10mM β-glycerophosphate, and 0.5mM NaVO4). After incubation on ice for 7 minutes, nuclei were collected by centrifugation at 700 × g for 7 minutes, re-suspended in isotonic buffer supplemented with 90mM of NaCl and 0.5% Triton X-100, and briefly sonicated. Soluble nuclear extracts were first pre-cleared with protein A/G-Sepharose beads (Santa Cruz) and control IgGs, then immunoprecipitated with 2ug of ASF/SF2, hnRNP I or U2AF65 antibody. After extensive washes by isotonic buffer, the precipitated antibody-antigen complexes were first incubated with DNase I (RNase free, Ambion) for 15 minutes at 37°C, followed with 50 μg of proteinase K (Roche) treatment for 15 minutes at 37°C. Co-precipitated RNA was then extracted by TRIzol and used for cDNA synthesis and PCR analyses. Enrichment of precipitated RNA was determined by the threshold cycle (Ct) value. Data were calculated as percentage of input, and plotted as fold changes over control IgG. RIP data were derived from three independent experiments with samples in triplicate. Results were presented as means ± SEM.
Northern Blotting
VCaP cells were transfected with control siRNA or siRNA against U2AF65, ASF/SF2 and hnRNP I for 48 hours. Total RNA was extracted by using Trizol (Invitrogen). Twenty microgram of total RNA were used for Northern blotting analysis to detect AR and 18S RNA levels as described (48).
Statistics
Data were presented as means ± SEM that were calculated from three or more different experiments. Statistical significances were calculated by using one-way ANOVA and paired Student’s t-test. A p<0.05 was considered significant. * represents P<0.05, ** represents P<0.01 and *** represents P<0.001.
Supplementary Material
Suppl. 1. (A) Standard curves of AR and AR-V7 were calculated. AR and AR-V7 cDNA were amplified by PCR and sequenced to confirm their identities. DNA concentrations of the cDNAs were measured by Nanodrop (Thermo Scientific). After series of dilution, cDNAs were used as templates for real-time qPCR. The standard curve of AR and AR-V7 were created using Ct values and template cDNA quantities. The equation and R2 value were calculated and used to calculate AR and AR-V7 copy numbers in Fig. 1A. (B) VCaP, LNCaP, LN(AI) and LN95 cells were maintained in RPMI1640 medium containing 5% FBS or 5% charcoal stripped serum for 48 hours. Cells were treated with vehicle, 10nM DHT and/or 5uM MDV3100 for 24 hours. AR and AR-V7 protein levels were detected by western blotting assays.
Suppl. 2. AR-V7 regulate UBE2C mRNA levels under ADT condition. VCaP cells were transfected with control siRNA or siRNA targeting AR Exon 1, Exon 7 or Exon 3b of the AR gene. After siRNA transfections, VCaP cells were treated with vehicle, DHT or MDV for another 24 hours. Relative RNA levels of UBE2C over GAPDH were measured by real-time qPCR.
Suppl. 3. Expression levels of RNA splicing factors in PCa cells. VCaP, LNCaP, and C4-2B cells were maintained in medium containing 5% charcoal stripped serum for 48 hours. Cells were treated with 10nM DHT or 5uM MDV3100 for 24 and 48 hours. Western blot assays detected AR, AR-V7, U2AF65, ASF/SF2, hnRNP I, U1A, p54nrb, PSF protein levels. Vinculin and β-Actin were used as loading controls. All experiments were repeated in three independent experiments and one representative data was presented. Antibody information was listed in supplementary materials.
Suppl. 4. Efficiencies of RNA silencing in VCaP, LN95 and LNCaP cells. VCaP, LN95 and LNCaP cells were transfected with siRNA against control, ASF/SF2, U2AF65 and hnRNP I respectively. Efficiency of siRNA was measured by western blotting assays, with β-Actin as the loading control. All experiments were repeated in three independent experiments and one representative data was presented.
Acknowledgments
This study was supported by Pacific Northwest Prostate Cancer SPORE, National Cancer Institute (P50CA097186; X.D. and S.R.P.); Prostate Cancer Canada (RS2013-58; to X.D.) and Canadian Institute of Health Research (MOP-97934; X.D.); Department of Defence plus Veterans Administration Grants (S.R.P.).
Footnotes
Conflict of Interest: The authors declare no conflict of interest
References
- 1.Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007 Nov 30;28(4):665–76. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009 Oct;10(10):981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005 Nov 10;23(32):8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004 Jan;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004 Feb 15;91(3):483–90. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 6.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008 Aug 1;68(15):6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 7.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007 May 15;67(10):5033–41. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 8.Heemers HV, Schmidt LJ, Kidd E, Raclaw KA, Regan KM, Tindall DJ. Differential regulation of steroid nuclear receptor coregulator expression between normal and neoplastic prostate epithelial cells. Prostate. 2010 Jun 15;70(9):959–70. doi: 10.1002/pros.21130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999 Mar 19;274(12):7777–83. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 10.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008 Jul 1;68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009 Jan 1;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011 Nov;71(15):1656–67. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009 Mar 15;69(6):2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Guo Z, Sun F, Li W, Alfano A, Shimelis H, et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J Biol Chem. 2011 Oct 14;286(41):36152–60. doi: 10.1074/jbc.M111.265124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010 Aug;120(8):2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013 Jan 15;73(2):483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011 Oct 18;20(4):457–71. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012 Jul 15;72(14):3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6(11):e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6(4):e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006 Jul 1;119(Pt 13):2635–41. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 22.Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene. 2001 Oct 17;277(1-2):31–47. doi: 10.1016/s0378-1119(01)00695-3. [DOI] [PubMed] [Google Scholar]
- 23.Chew SL, Liu HX, Mayeda A, Krainer AR. Evidence for the function of an exonic splicing enhancer after the first catalytic step of pre-mRNA splicing. Proc Natl Acad Sci U S A. 1999 Sep 14;96(19):10655–60. doi: 10.1073/pnas.96.19.10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009 Nov;10(11):741–54. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornblihtt AR. Coupling transcription and alternative splicing. Adv Exp Med Biol. 2007;623:175–89. doi: 10.1007/978-0-387-77374-2_11. [DOI] [PubMed] [Google Scholar]
- 26.Auboeuf D, Honig A, Berget SM, O’Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002 Oct 11;298(5592):416–9. doi: 10.1126/science.1073734. [DOI] [PubMed] [Google Scholar]
- 27.Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. J Biol Chem. 2002 Nov 8;277(45):43110–4. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]
- 28.Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006 Jan;13(1):22–9. doi: 10.1038/nsmb1030. [DOI] [PubMed] [Google Scholar]
- 29.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, et al. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997 Jan 23;385(6614):357–61. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- 30.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002 Apr 4;416(6880):499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 31.Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009 Feb 20;136(4):688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011 Jan 7;144(1):16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornblihtt AR. Promoter usage and alternative splicing. Curr Opin Cell Biol. 2005 Jun;17(3):262–8. doi: 10.1016/j.ceb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Kadener S, Cramer P, Nogues G, Cazalla D, de la Mata M, Fededa JP, et al. Antagonistic effects of T-Ag and VP16 reveal a role for RNA pol II elongation on alternative splicing. EMBO J. 2001 Oct 15;20(20):5759–68. doi: 10.1093/emboj/20.20.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012 Jan 23;(2012):1–9. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010 Sep 28;107(39):16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009 Feb 1;69(3):958–66. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rokhlin OW, Glover RB, Guseva NV, Taghiyev AF, Kohlgraf KG, Cohen MB. Mechanisms of cell death induced by histone deacetylase inhibitors in androgen receptor-positive prostate cancer cells. Mol Cancer Res. 2006 Feb;4(2):113–23. doi: 10.1158/1541-7786.MCR-05-0085. [DOI] [PubMed] [Google Scholar]
- 39.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002 Apr;18(4):186–93. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 40.Stamm S, Riethoven JJ, Le Texier V, Gopalakrishnan C, Kumanduri V, Tang Y, et al. ASD: a bioinformatics resource on alternative splicing. Nucleic acids research. 2006 Jan 1;34(Database issue):D46–55. doi: 10.1093/nar/gkj031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic acids research. 2003 Jul 1;31(13):3568–71. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011 Mar 15;71(6):2108–17. doi: 10.1158/0008-5472.CAN-10-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie N, Liu L, Li Y, Yu C, Lam S, Shynlova O, et al. Expression and Function of Myometrial PSF Suggest a Role in Progesterone Withdrawal and the Initiation of Labor. Mol Endocrinol. 2012 Aug;26(8):1370–9. doi: 10.1210/me.2012-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010 Feb 19;327(5968):996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong X, Sweet J, Challis JR, Brown T, Lye SJ. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol Cell Biol. 2007 Jul;27(13):4863–75. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olshavsky NA, Comstock CE, Schiewer MJ, Augello MA, Hyslop T, Sette C, et al. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010 May 15;70(10):3975–84. doi: 10.1158/0008-5472.CAN-09-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007 Mar 26;176(7):929–39. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res. 2006 Nov 1;66(21):10613–20. doi: 10.1158/0008-5472.CAN-06-0028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. 1. (A) Standard curves of AR and AR-V7 were calculated. AR and AR-V7 cDNA were amplified by PCR and sequenced to confirm their identities. DNA concentrations of the cDNAs were measured by Nanodrop (Thermo Scientific). After series of dilution, cDNAs were used as templates for real-time qPCR. The standard curve of AR and AR-V7 were created using Ct values and template cDNA quantities. The equation and R2 value were calculated and used to calculate AR and AR-V7 copy numbers in Fig. 1A. (B) VCaP, LNCaP, LN(AI) and LN95 cells were maintained in RPMI1640 medium containing 5% FBS or 5% charcoal stripped serum for 48 hours. Cells were treated with vehicle, 10nM DHT and/or 5uM MDV3100 for 24 hours. AR and AR-V7 protein levels were detected by western blotting assays.
Suppl. 2. AR-V7 regulate UBE2C mRNA levels under ADT condition. VCaP cells were transfected with control siRNA or siRNA targeting AR Exon 1, Exon 7 or Exon 3b of the AR gene. After siRNA transfections, VCaP cells were treated with vehicle, DHT or MDV for another 24 hours. Relative RNA levels of UBE2C over GAPDH were measured by real-time qPCR.
Suppl. 3. Expression levels of RNA splicing factors in PCa cells. VCaP, LNCaP, and C4-2B cells were maintained in medium containing 5% charcoal stripped serum for 48 hours. Cells were treated with 10nM DHT or 5uM MDV3100 for 24 and 48 hours. Western blot assays detected AR, AR-V7, U2AF65, ASF/SF2, hnRNP I, U1A, p54nrb, PSF protein levels. Vinculin and β-Actin were used as loading controls. All experiments were repeated in three independent experiments and one representative data was presented. Antibody information was listed in supplementary materials.
Suppl. 4. Efficiencies of RNA silencing in VCaP, LN95 and LNCaP cells. VCaP, LN95 and LNCaP cells were transfected with siRNA against control, ASF/SF2, U2AF65 and hnRNP I respectively. Efficiency of siRNA was measured by western blotting assays, with β-Actin as the loading control. All experiments were repeated in three independent experiments and one representative data was presented.