

Abstract
Intracellular Ca2+ release through ryanodine receptor (RyR) and inositol trisphosphate receptor (IP3R) channels is supported by a complex network of additional proteins that are located in or near the Ca2+ release sites. In this review, we focus, not on RyR/IP3R, but on other ion-channels that are known to be present in the sarcoplasmic/endoplasmic reticulum (ER/SR) membranes. We review their putative physiological roles and the evidence suggesting that they may support the process of intracellular Ca2+ release, either indirectly by manipulating ionic fluxes across the ER/SR membrane or by directly interacting with a Ca2+-release channel. These channels rarely receive scientific attention because of the general lack of information regarding their biochemical and/or electrophysiological characteristics makes it difficult to predict their physiological roles and their impact on SR Ca2+ fluxes. We discuss the possible role of SR K+ channels and, in parallel, detail the known biochemical and biophysical properties of the trimeric intracellular cation (TRIC) proteins and their possible biological and pathophysiological roles in ER/SR Ca2+ release. We summarise what is known regarding Cl− channels in the ER/SR and the non-selective cation channels or putative ‘Ca2+ leak channels’, including mitsugumin23 (MG23), pannexins, presenilins and the transient receptor potential (TRP) channels that are distributed across ER/SR membranes but which have not yet been fully characterised functionally.
Introduction
The sarcoplasmic reticulum (SR) is a highly specialised intracellular Ca2+ store that controls the contractile cycle in striated muscle and provides a traditional model system for Ca2+ signalling (Fig. 1). In striated muscle, Ca2+ release from and uptake into the SR during the contraction cycle are mediated by the ryanodine receptor (RyR) and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), respectively. In contrast, the Ca2+ released via the inositol trisphosphate receptor (IP3R), which is activated by cytokine- and hormone-induced signalling pathways, primarily regulates cellular metabolic processes including gene expression. There are many unexplained aspects to the Ca2+ release events of muscle SR that occur via these two types of Ca2+ release channel. This should not be surprising since the SR contains a plethora of incompletely characterised proteins and these may regulate the activity of RyR or IP3R, directly or indirectly.
Figure 1.
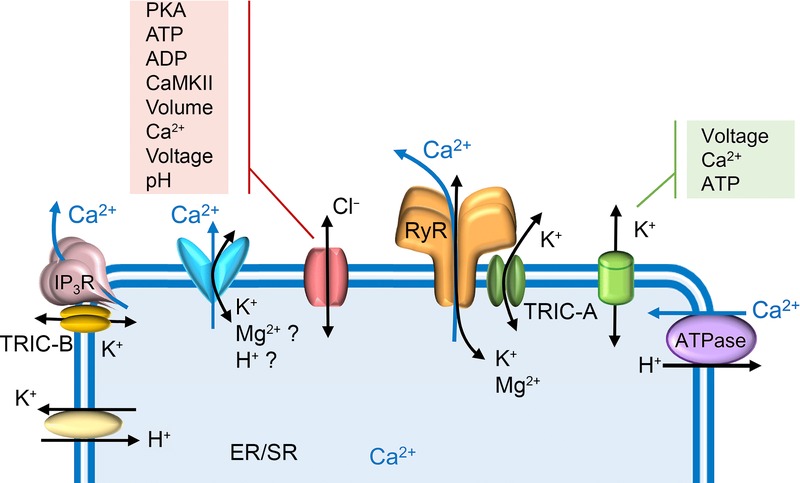
Ionic fluxes across the ER/SR compartment
RyR and IP3R are the acknowledged Ca2+ release channels of the ER/SR. The sarcoplasmic reticulum Ca2+ ATPase (SERCA) pumps Ca2+ into ER/SR with counter movement of H+ out. There are also numerous unidentified ionic pathways that are predicted from experimental work. These comprise the various SR anion and cation channels which appear to be triggered by many distinct activators. In addition, a putative ER/SR-residing K+–H+ exchanger may be responsible for pumping protons into the SR in exchange for K+ moving out. Many recognised ion channels are also located on ER/SR membranes although their physiological roles in this location have not been clearly assigned. These include the monovalent cation-conducting TRIC channels and several non-selective cation channels (for example, MG23, pannexins, presenilins and the TRP channels, TRPV1, TRPP2 and TRPM8), often described as Ca2+ leak channels.
An example of a debated aspect of SR function is how the membrane potential across the SR is maintained. Physiological SR Ca2+ release through RyR and IP3R channels can persist efficiently for several milliseconds and therefore counterion movements are required to balance the movement of positive charge that occurs as Ca2+ leaves the SR. Counter currents of ionic species such as K+, Mg2+ or Cl− are expected but there is controversy over which ion channels within the SR are involved (Yazawa et al. 2007; Gillespie & Fill 2008; Guo et al. 2013). It has, for decades, been assumed that the physiological role of SR K+ channels and/or SR Cl− channels is to enable rapid counter-ion fluxes across the ER/SR to compensate for the charge movements associated with Ca2+ release and re-uptake processes. More recently it has been suggested, however, that RyR can pass sufficient counter current to balance its own Ca2+ release and that other SR ion channels would not be required for this function (Gillespie & Fill 2008). Instead, it is argued that there are always some SR K+ channels opening irrespective of the membrane potential across the SR. These channels would allow the excess K+ (built up during Ca2+ release) to leave the SR following termination of a Ca2+-release event (hence equilibrating SR membrane potential). This would be important because RyR channels would be closed and not able to pass K+. This does not take into account the possible involvement of anion fluxes in controlling ER/SR membrane potential and leaves open the question as to the physiological role/s of the various types of SR Cl− channel. Also unknown, but perhaps related, is how the SR maintains luminal pH. SERCA catalyses the extrusion of protons (Fig. 1) as Ca2+ is pumped into the SR (Inesi & Tadini-Buoninsegni 2014) and therefore it is expected that a mechanism/s exists to balance the change in intraluminal pH. Acidification of isolated skeletal SR vesicles is observed during the release of Ca2+ from those vesicles and it has been suggested that proton movements could contribute a small proportion (<10%) towards the countercurrent required for charge compensation during SR Ca2+ release (Kamp et al. 1998). The pathway/s for rapid proton influx during the Ca2+ release process has not been identified but there is evidence for localisation of a K+–H+ exchanger in the ER/SR of some tissues which could be involved in pumping protons into the SR in exchange for K+ out (Kuum et al. 2012). A number of Ca2+-leak channels have been localised to the ER/SR and as these tend to show little specificity among cations, perhaps protons could be permeable in one or more of these channels. The identification and characterisation of SR ion channels and transporters remains a relatively unexplored area of muscle biology. This short review discusses recent progress in the field and highlights what we do and do not know about the ion channels, other than RyRs and IP3Rs, in ER/SR membranes.
TRIC and K+ channels in the SR; voltage is the main regulator
TRIC (trimeric intracellular cation) channel subtypes, namely TRIC-A and TRIC-B, are derived from distinct genes in the mammalian genome, and their counterparts are also detected in the fruit fly and nematode genomes (Yazawa et al. 2007). In the gene-expression profile among mouse tissues, TRIC-A is preferentially detected at high levels in excitable tissues, while relatively low levels of TRIC-B are ubiquitously observed in various tissues. In muscle cells, TRIC proteins are localised in the SR and nuclear membranes. TRIC proteins are composed of ∼300 amino acid residues and contain three putative membrane-spanning segments to form a bullet-shaped homo-trimeric structure. When either native or recombinant TRIC-A and TRIC-B proteins were incorporated into artificial membranes under voltage-clamp conditions, monovalent cation-selective channel events were observed, suggesting that both TRIC subtypes function as K+ channels in cells (Yazawa et al. 2007; Pitt et al. 2010). These single channel current fluctuations share certain characteristics with those of the previously reported SR K+ channels obtained from muscle SR from a variety of species (Miller 1978; Labarca & Miller 1981; Shen et al. 1993) and tissues (Gray & Williams 1985; Picher et al. 1996) thus indicating, since muscle SR contains abundant TRIC-A, that the TRIC subtypes form the SR K+ channel.
The physiological importance of the TRIC subtypes is gradually becoming realised through the study of TRIC-knockout (KO) mice and there are now several reports of TRIC mutations in human disease. In TRIC-KO mice, we have observed impaired ER/SR Ca2+ release in several cell types. The Tric-a-KO mice developed hypertension due to vascular hypertonicity (Yamazaki et al. 2011). In the mutant vascular smooth muscle cells, insufficient RyR-mediated Ca2+ sparks for inducing hyperpolarisation were detected (Yamazaki et al. 2011). The Tric-b-knockout mice exhibited neonatal respiratory failure, demonstrating the essential physiological role of this TRIC subtype in the lungs. In the mutant alveolar epithelial cells devoid of TRIC-B, the IP3R-mediated Ca2+ release which controls surfactant production and handling was markedly reduced (Yamazaki et al. 2009). The double-knockout mice lacking both TRIC subtypes developed embryonic heart failure, and SR Ca2+ handling was severely impaired in the mutant cardiac myocytes (Yazawa et al. 2007). These studies demonstrate the indispensable role of the TRIC proteins to the Ca2+ release process in a variety of tissues and show that the proteins are important both for RyR- and IP3R-mediated Ca2+ release.
Recent studies have also identified two distinct associations between TRIC channels and human diseases. In Japanese population-based studies, single nucleotide polymorphisms (SNPs) in the TRIC-A gene are associated with hypertension risk and sensitivity to common antihypertensive medications (Yamazaki et al. 2011). In the TRIC-A risk variant, gene expression may be restricted to vascular smooth muscle, thus elevating resting vascular tone. It has also been found that two deletion mutants in the TRIC-B gene are responsible for the autosomal recessive disease osteogenesis imperfecta, a condition which leads to bone fragility and increased susceptibility to fractures (Shaheen et al. 2012; Rubinato et al. 2014). Both mutations lead to severe truncation of the TRIC-B protein but the pathological mechanisms leading to this novel type of osteogenesis imperfecta are not yet understood.
To understand the specific physiological roles of the TRIC subtypes in various tissues and how mutations can lead to human disease, comprehensive biophysical characterisation of each TRIC channel subtype is required. This is complicated, however, since both isoforms are present in most cell types and since purification of recombinant proteins has proved difficult (Venturi et al. 2013). The Tric-b-KO mouse dies at birth and neonatal SR is not sufficiently developed for the study of SR ion channels. The Tric-a-KO mouse survives. Tissue from Tric-a-KO mice has enabled characterisation of the native K+ channels that remain when the SR is devoid of TRIC-A (Venturi et al. 2013). These channels are selective for monovalent cations and are sensitive to voltage, being activated at voltages where the SR luminal side of the channels is negative with respect to the cytosolic side of the channels. Single-channel studies reveal complex sub-conductance gating behaviour. The channels appear to gate to at least four sub-conductance open states which are approximately 80%, 60%, 46% and 30% of the full open single-channel conductance (200 pS). Multiple sub-conducting states were not previously reported in early SR K+ channel studies; instead a single ‘noisy’ sub-level at 60–70% of the full conductance was repeatedly reported (for example, Tomlins et al. 1984; Fox 1985; Tomlins & Williams 1986). This discrepancy could be attributed to the fact that fast gating between different conductance levels would not be resolved at the filtering (100–200 Hz) applied in their analysis. It is notable that different investigators described slightly different current amplitudes for the ‘noisy’ sub-conductance state (for example, Tomlins & Williams 1986; Hill et al. 1989, 1990; Rousseau et al. 1992) and that, occasionally, markedly different sub-conductance state amplitudes were reported (Labarca & Miller 1981; Picher et al. 1996).
As described above, the main functional modulator of SR K+ channels appears to be voltage. The majority of the single SR K+ channels (82%) observed after incorporating vesicles of skeletal muscle SR from Tric-a-KO mice into bilayers, display voltage-dependent gating behaviour with increased open probability (Po) at positive holding potentials (cytosol relative to SR lumen) compared to negative holding potentials(Venturi et al. 2013). Thus, if the SR were to become negatively charged with respect to the cytosol, this would lead to more opening of SR K+ channels. A proportion of the channels (12%) do not appear to be sensitive to voltage and exhibit higher than usual activity at positive and negative voltages and an increased propensity for sub-conductance state gating behaviour (Venturi et al. 2013). The heterogeneity in gating behaviour is likely to ensure that there will always be a proportion of SR K+ channels opening irrespective of the potential across the SR membrane. This gating behaviour together with the voltage sensitivity of SR K+ channels suggests that these channels would be extremely effective at dissipating any asymmetry in [K+] that could occur across the SR membrane and in preventing SR membrane potential moving far from 0 mV. Apart from voltage, there is no other obvious physiological regulator of SR K+ channels. SR K+ channels do exhibit sensitivity to cytosolic and luminal pH (Miller 1978; Labarca et al. 1980; Bell 1985) and to membrane lipid composition (Bell & Miller 1984) but perhaps these factors become more important in pathological conditions such as ischaemia. It has been reported that SR K+ channel gating is regulated by cytosolic and luminal [Ca2+] but this is not observed by all investigators (Rousseau et al. 1992; Uehara et al. 1994; Wang & Best 1994; Picher et al. 1996). Luminal Ca2+ could also cause some degree of block of outward K+ flux if the luminal [Ca2+] was raised high enough (Liu & Strauss 1991). It is possible that TRIC-A and TRIC-B may have additional or different physiological roles to that of maintaining [K+] equilibrium across the SR or of permitting charge-compensating ionic fluxes. For example, there is evidence that TRIC-A may bind to or indirectly interact with RyR2 to enhance RyR2 opening (Zhou et al. 2014). It will be important to investigate this and other putative regulatory functions of TRIC-A and TRIC-B.
There are isolated reports of other types of K+ channels in ER/SR membranes and these include small (Kuum et al. 2012) and large (Yamashita et al. 2006) conductance Ca2+-activated K+ channels and ATP-sensitive K+ channels (Zhou et al. 2005). Unfortunately there is sparse information regarding the physiological relevance of these channels for ER/SR Ca2+ stores, and again, speculation on the role of the small conductance Ca2+-activated K+ channels is that of providing countercurrent of K+ during the Ca2+-uptake process (Kuum et al. 2012).
Anion channels of the ER/SR and their multiple mechanisms of activation
SR Cl− channel current fluctuations were first observed after incorporation of rabbit skeletal SR vesicles into artificial membranes (Miller 1978). Subsequent investigations of the biophysical properties of the SR Cl− channels have shown that there are multiple types of Cl− channel in the SR and that they exhibit a wide range of conductance and gating properties (for example, Tanifuji et al. 1987; Rousseau et al. 1988; Townsend & Rosenberg 1995). Several channels show Ca2+ dependence, some are regulated by pH and many are voltage sensitive (Kawano & Hiraoka 1993; Townsend & Rosenberg 1995). Some SR Cl− channels are regulated by ATP (for example, Ahern & Laver 1998; Kawano et al. 1999), some by protein kinase A (PKA; Kawano et al. 1992, 1999) or by Ca2+-calmodulin-dependent protein kinase (CaMKII; Kawano & Hiraoka 1993). Anion selectivity and single-channel conductance of the various SR Cl− channels also varies widely and even adenine nucleotides are reported to be permeant in certain channels (Kawano et al. 1999). It seems clear that, unlike the SR K+ channels, where voltage is the main regulator and no specific or potent ligand has been identified, the SR Cl− channels are functionally diverse and are regulated by different conditions and specific regulators. Thus, the SR Cl− channels may be involved in multiple functions. Unfortunately, the majority of SR Cl− channels that can be observed following incorporation of SR vesicles into bilayers appear to gate to multiple sub-conductance levels and this makes it extremely difficult to distinguish between the different types of channel, especially since multiple channels usually incorporate into the membrane.
The observed functional SR Cl− channel behaviour has not yet been linked to any identified protein. Several classes of Cl− channel are known and these include the CLC family of chloride channels and transporters (Stauber et al. 2012). In the mammalian genome, there are nine CLC genes, four of which encode cell-surface Cl− channels and five encode intracellular Cl−–H+ exchangers. The Cl−–H+ exchangers (ClC3–7) mainly reside on components of the endosomal–lysosomal pathway and influence vesicular acidification. There is sparse information regarding the possible localisation of ClC transporters in the ER/SR although one report indicates that ClC4 resides on this compartment (Okkenhaug et al. 2006). In addition to CLC proteins, members of the CLCA (Ca2+-activated Cl− channel; Patel et al. 2009), CLIC (intracellular Cl− channel; Jiang et al. 2014) and VRAC/LRRC (volume-regulated anion channel/leucine-rich repeat-containing; Voss et al. 2014) families could potentially also form functional SR Cl− channels. The CLIC proteins show sequence homology with glutathione-S-transferase (GST) and can function both as Cl− channels or as soluble, globular proteins that interact with and regulate the function of other proteins (Dulhunty et al. 2001). In this regard, it is interesting that CLIC2 has been shown to bind directly to RyR1 (Meng et al. 2009) and inhibit channel opening (Dulhunty et al. 2001; Meng et al. 2009).
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated Cl− channel expressed on the apical surface of epithelial cells in lungs, pancreas and intestine. CFTR is also found in muscle cells and there is recent evidence for its localisation in human skeletal muscle SR (Divangahi et al. 2009). It has been suggested that the dysregulation of this Cl− channel, which is seen in cystic fibrosis sufferers, could explain certain abnormalities in muscle function (Lamhonwah et al. 2010). No obvious similarity in the biophysical properties of any of the reported SR anion channels and those of CFTR is apparent although experimental conditions were not specifically designed for activation of CFTR.
The physiological role/s of the different SR Cl− channels and their involvement in Ca2+ signalling remains a subject of speculation. It has been assumed that they provide charge compensation during Ca2+ release and uptake processes. But if this was their only role, then why does the SR contain so many different types of anion channel? An interesting report of a Cl− channel from liver rough ER that was activated by Mg2+-ADP and inhibited by Mg2+-ATP, suggested that this Cl− channel could be activated by metabolic stress (Ashrafpour et al. 2012). The need for further study in this area is obvious and it must begin with identification of the proteins that constitute the SR Cl− channels so that structure-function studies can begin.
ER/SR Ca2+ leak channels: a growing community
Several non-selective cation channels are also found in the ER/SR membranes of a variety of cell types and may function as Ca2+ leak channels. These include mitsugumin23 (MG23), pannexin channels (PanX1 (Vanden Abeele et al. 2006), PanX2 (Ambrosi et al. 2010), and PanX3 (Ishikawa et al. 2011)), presenilins (Tu et al. 2006; Zhang et al. 2010) and several ion-channels belonging to the transient receptor potential (TRP) family: TRPV1, TRPP2 and TRPM8 (Dong et al. 2010; Taylor & Dale 2012). Why so many different types of non-selective cation channel are expressed on ER/SR Ca2+ stores is not known but the possible physiological consequences of activating such channels are numerous. Passive Ca2+ leak from intracellular compartments is observed in many cell types (Lomax et al. 2002; Giunti et al. 2007) and the ionic pathway is usually expected to be that of RyR or IP3R. In cardiac cells, diastolic SR Ca2+ leak is facilitated in certain pathophysiological conditions including heart failure and catecholaminergic polymorphic ventricular tachycardia (CPVT) and may lead to fatal cardiac arrhythmias (Bers, 2014). There is evidence, however, for RyR-independent mechanisms of SR Ca2+ efflux, and that these ionic fluxes may be more strongly activated in disease states (Zima et al. 2010). The participation of one or more of the newly discovered Ca2+-permeable ion channels located on ER/SR membranes in generating the ER/SR Ca2+ leak from various cells is a subject that deserves detailed investigation. We describe those Ca2+ leak channels that have been identified and discuss what little is known regarding their biophysical properties. Figure 2 summarises some of the putative cellular roles of these channels.
Figure 2.
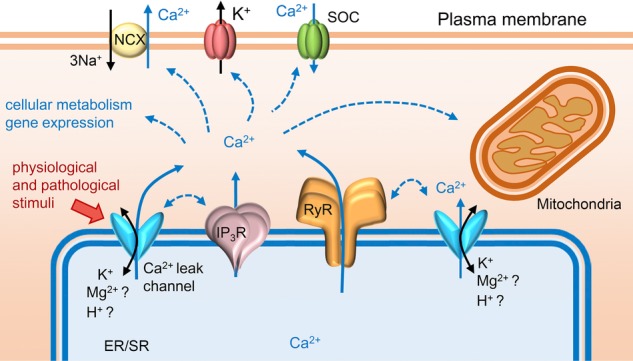
ER/SR Ca2+ leak
Ca2+ signals from the ER/SR of different cell types can originate through distinct mechanisms and can also be mediated/enhanced by Ca2+ leak channels located on the ER/SR network. Some of these non-selective cation channels, thought to be novel pathways for Ca2+ release, have also been shown to directly affect the function of the main Ca2+-release channels, RyR and IP3R. Certain pathological stimuli, such as accumulated reactive oxygen and nitrogen species, might be involved in upregulation of ER/SR Ca2+ leak, but usually the mechanisms underlying the activation of Ca2+ leak channels are unclear. Rises in cytosolic Ca2+ influence many cellular processes and can lead to altered cellular metabolism, deranged gene expression and mitochondrial-induced apoptosis.
Mitsugumin 23
MG23 is a 23 kDa transmembrane protein found in ER/SR and nuclear membranes of striated muscle and other tissues including epithelial cells, secretory organs and the brain (Nishi et al. 1998). Electron microscopy and 3D particle reconstruction revealed the unstable and peculiar morphology of MG23 (Venturi et al. 2011). Two types of particle were consistently observed; small asymmetric particles and large bowl-shaped particles of hexameric symmetry. It was hypothesised that the MG23 bowl configuration is constructed from six asymmetric particles and can be readily constructed and disassembled.
Recombinant purified MG23 proteins reconstituted into planar lipid bilayers behave as voltage-sensitive cation-conducting channels which are equally permeable to Ca2+ and K+ (Venturi et al. 2011). MG23 exhibits very unusual gating behaviour characterised by brief ‘flickery’ opening events and apparently co-ordinated gating of multiple channels; possibly the instability of the bowl-shaped assembly formed by MG23 molecules underlies the observed unusual ion-channel activity observed in bilayer experiments. The biophysical properties of MG23 may allow its involvement in charge compensation and/or in SR Ca2+ leak. The use of the MG23 KO mouse (Yamazaki et al. 2010) may help to expose the functional roles of MG23. It is interesting that two recent studies have reported MG23 to play a role in generating lethal signals from the ER after stressed-induced DNA damage (Yamazaki et al. 2010; Yamashita et al. 2013). It is known that changes in the Ca2+ content and leak from ER/SR compartments can affect the incidence of apoptotic cell death (Pinton et al. 2008) but the molecular identity of this pathway remains unknown. The cation permeability of MG23 suggests that it could be implicated in this phenomenon.
TRP channels
TRPV1 is highly expressed in the plasma membrane of a variety of cellular components of the central nervous system but is also present in non-neuronal tissues, for example in kidney, liver and vascular smooth muscle (Veronesi & Oortgiesen, 2006). TRPV1 has also been detected in the ER and the Golgi system (Veronesi & Oortgiesen, 2006; Dong et al. 2010). In particular, in skeletal muscle, it has been proposed to act as a functional SR Ca2+ leak channel (Xin et al. 2005; Lotteau et al. 2013), and that this particular mechanism for intracellular Ca2+ mobilisation may be directly involved in RyR1 activation (Lotteau et al. 2013).
TRPP2 (polycystin-2) is widely expressed in various cell types, with highest levels found in kidney. Loss-of-function mutations of the gene encoding TRPP2 are associated with autosomal dominant polycystic kidney disease (ADPKD; Koulen et al. 2002). This cation-selective channel is mainly located on the plasma membrane but it is also abundant in the ER of kidney epithelial and smooth muscle cells (Cai et al. 1999; Koulen et al. 2002; Geng et al. 2008). The specific physiological functions of TRPP2 in the distinct cellular compartments where it is located are still not fully understood. It has been suggested that, in the ER, TRPP2 functions as a Ca2+-release channel which can interact with IP3Rs and that it is triggered to open by local rises in Ca2+ concentration (Koulen et al. 2002; Geng et al. 2008; Sammels et al. 2010). It has also been proposed that TRPP2 could be involved in protecting cells from apoptosis by reducing the releasable ER Ca2+ pool in response to apoptotic stimuli (Wegierski et al. 2009).
TRPM8 is predominantly found in the sarcolemma of somatosensory neurons but it is also abundant in the ER membranes of human prostate epithelial cells, where has been identified as a novel pathway for the release of Ca2+ (Thebault et al. 2005). TRPM8 is activated by cold temperatures and menthol and is sensitive to pH (Mahieu et al. 2010). Again, the physiological relevance of the expression of this particular channel in the ER is not understood.
Pannexin channels
PanX1, a member of the ubiquitously expressed pannexin family of channels (Bruzzone et al. 2003), has been shown to form mechanosensitive, cation- and ATP-permeable channels which enable intercellular Ca2+ mobilisation (Bao et al. 2004; Vanden Abeele et al. 2006; Penuela et al. 2014). It has been reported that overexpression of PanX1 drastically increases ER Ca2+ permeability implicating a role in ER Ca2+ leak (Vanden Abeele et al. 2006; Penuela et al. 2014). PanX1 is not the only pannexin channel residing in intracellular membranes; the PanX2 (Ambrosi et al. 2010; Wicki-Stordeur et al. 2013) and PanX3 (Ishikawa et al. 2011) isoforms have also been detected in the ER network. In particular, PanX3 is suggested to contribute to the ER Ca2+ leak mechanism that is required to stimulate osteoblast differentiation (Ishikawa et al. 2011).
Presenilins
The Ca2+ permeability of the presenilins, gene mutations of which have been linked to familial Alzheimer diseases, has been extensively documented in several studies (Tu et al. 2006; Nelson et al. 2010; Zhang et al. 2010). In these reports, a speculative role for presenilins as participants in ER Ca2+ leak has been proposed although these conclusions are disputed (Shilling et al. 2012).
The subcellular distribution of these different types of Ca2+-permeable channels is, without doubt, fascinating, even though their physiological roles are primarily topics for speculation. Further investigation of their single-channel properties, their possible modulation by endogenous ligands and the development of specific pharmacological regulators are needed to clarify their contribution to physiological and pathophysiological ER/SR Ca2+ fluxes.
TMEM16E: a novel SR protein of unknown function but possible role in ion transport
As yet, TMEM16E cannot be listed as an anion channel, K+ channel, or Ca2+ leak channel and so must belong to its own unique class of putative SR ion-channel. Ten members of the TMEM16/anoctamin family were identified from the mammalian genome and share a homologous structure containing eight putative transmembrane segments with cytoplasmic amino and carboxyl-terminal tails (Pedemonte & Galietta, 2014). TMEM16 family members are expected to be involved in ion transport and phospholipid scrambling based on the functional properties of TMEM16A, TMEM16B and TMEM16F (Pedemonte & Galietta, 2014). TMEM16A and TMEM16B function as cell-surface Ca2+-activated Cl− channels, and contribute to apical Cl− secretion of epithelial cells and membrane potential regulation of smooth muscle. TMEM16F seems to function as both a Ca2+-dependent lipid scramblase and as a Ca2+-activated Cl− channel in blood cells. In Scott syndrome patients, TMEM16F mutations impair the phospholipid scrambling of the platelet cell membrane leading to a bleeding disorder (Suzuki et al. 2010). TMEM16E is an unusual family member as it is immunochemically detected in the ER/SR of skeletal and cardiac muscle cells, chondrocytes and osteoblasts (Mizuta et al. 2007). The expression profile suggests that TMEM16E plays multiple biological roles in the musculoskeletal system. TMEM16E was originally identified as the gene GDD1; missense mutations in this gene cause gnathodiaphyseal dysplasia (GDD), a rare bone disorder (Tsutsumi et al. 2004). TMEM16E nonsense mutations are associated with proximal limb-girdle muscular dystrophy (LGMD2L) and distal Miyoshi myopathy (MMD3), both of which are recessive types of muscular dystrophy (Bolduc et al. 2010). Linkage of TMEM16E mutations with muscular dystrophies, together with the subcellular distribution of this protein has led to suggestions that TMEM16E may mediate SR Cl− currents or phospholipid scrambling and may be important for muscle-specific cellular functions such as efficient SR Ca2+ handling and membrane repair after cell-membrane wounding. Aside from speculation, however, the function of TMEM16E in the ER/SR is still totally unknown.
Concluding remarks
We have described the biophysical properties of the various ion-channels of the SR where known, and tried to relate these properties to possible physiological roles. We have particularly been considering how the ionic fluxes would be important to the process of ER/SR Ca2+ release (Figs 1 and 2). However, the ER/SR is a multi-functional organelle and controls other cellular processes including secretory protein folding and modification, sterol and fatty acid biosynthesis, and ER stress signalling. Such divergent functions require specific ionic conditions within luminal ER/SR domains and changes to those conditions may cooperatively modulate more than one process. For example, SERCA inhibitors not only diminish Ca2+-release signalling, but also disrupt protein folding to induce the ER stress response because major ER chaperones are Ca2+ dependent. Moreover, the ER/SR functionally communicates with the cell membrane, mitochondria and nuclei. The use of SERCA inhibitors to reduce Ca2+ store content also triggers store-operated Ca2+ entry via the cell membrane and possibly reduces Ca2+-dependent mitochondrial metabolism and nuclear transcription. This diverse range of functions may explain the need for multiple types of ER/SR anion channel and the variety of the so-called Ca2+ leak channels.
Since the ER/SR controls a number of crucial cellular functions and since precise, spatiotemporal Ca2+ release events from ER/SR are also required to initiate or regulate many processes within cells, it is important to understand the molecular basis of ionic fluxes across the ER/SR. Functional investigations of ER/SR ion channels are difficult because of their inaccessibility for in situ voltage-clamp studies hence adding to the challenges of protein purification and reconstitution procedures. However, since there remain many fundamental, unanswered questions regarding intracellular Ca2+ signalling, it is essential to persist in the identification and characterisation of ER/SR channels.
Glossary
- ADPKD
autosomal dominant polycystic kidney disease
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CFTR
cystic fibrosis transmembrane conductance regulator
- CLC
Cl− channel family
- CLIC
intracellular Cl− channel family
- CLCA
Ca2+-activated Cl− channel
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- ER
endoplasmic reticulum
- GDD
gnathodiaphyseal dysplasia
- GST
glutathione-S-transferase
- IP3R
inositol trisphosphate receptor
- KO
knockout
- LGMD2L
limb girdle muscular dystrophy 2 L
- MG23
mitsugumin23
- MMD3
Distal Miyoshi-like myopathy 3
- PanX
pannexin
- PKA
protein kinase A
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SNPs
single nucleotide polymorphisms
- SR
sarcoplasmic reticulum
- TRIC
trimeric intracellular cation channel
- TRP
transient receptor potential
- VRAC/LRRC
volume-regulated anion channel/leucine-rich repeat-containing
Biographies
Since studying for his PhD at Kyoto University, Japan, Hiroshi Takeshima has been focusing on identifying new sarcoplasmic reticulum (SR) components including the ryanodine receptor (RyR), junctophilin and TRIC proteins. He has developed many knockout mice models to shed light on the physiological roles of the various SR proteins. After his appointment as Professor at Kurume University and Tohuko University, his group moved to the Graduate School of Pharmaceutical Sciences at Kyoto University where he is now based.
ElisaVenturi investigated the single-channel properties of RyR and other cation channels in the SR to earn her PhD in 2011 from the University of Bristol. She is now a post-doctoral research assistant in the Department of Pharmacology at the University of Oxford where she is purifying novel SR membrane proteins such as TRIC for subsequent structure-function studies.
Rebecca Sitsapesan obtained her PhD at the University of Strathclyde and following an appointment as British Heart Foundation Basic Science Lecturer at Imperial College, London, she moved to Bristol University and then to the University of Oxford where she is currently Professor of Pharmacology. Her group investigates the biophysical properties of RyR and other ion channels present on intracellular organelles and that are involved in the process of intracellular Ca2+ release, particularly in regard to cardiac physiology and pathophysiology.
Additional information
Competing interests
None of the authors has any conflicts of interests.
Funding
Our collaborative work was supported in part by the British Heart Foundation and JSPS Core-to-core program grants.
References
- Ahern GP. Laver DR. ATP inhibition and rectification of a Ca2+-activated anion channel in sarcoplasmic reticulum of skeletal muscle. Biophys J. 1998;74:2335–2351. doi: 10.1016/S0006-3495(98)77943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi C, Gassmann O, Pranskevich JN, Boassa D, Smock A, Wang J, Dahl G, Steinem C. Sosinsky GE. Pannexin1 and pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J Biol Chem. 2010;285:24420–24431. doi: 10.1074/jbc.M110.115444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafpour M, Babaei JF, Saghiri R, Sepehri H. Sharifi H. Modulation of the hepatocyte rough endoplasmic reticulum single chloride channel by nucleotide–Mg2+ interaction. Pflugers Arch. 2012;464:175–182. doi: 10.1007/s00424-012-1121-z. [DOI] [PubMed] [Google Scholar]
- Bao L, Locovei S. Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–68. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Bell J. Protons decrease the single channel conductance of the sarcoplasmic reticulum K+ channel in neutral and negatively charged bilayers. Biophys J. 1985;48:349–353. doi: 10.1016/S0006-3495(85)83790-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE. Miller C. Effects of phospholipid surface charge on ion conduction in the K+ channel of sarcoplasmic reticulum. Biophys J. 1984;45:279–287. doi: 10.1016/S0006-3495(84)84154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. 2014;76:107–127. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- Bolduc V, Marlow G, Boycott KM, Saleki K, Inoue H, Kroon J, Itakura M, Robitaille Y, Parent L, Baas F, Mizuta K, Kamata N, Richard I, Linssen WH, Mahjneh I, de Visser M, Bashir R. Brais B. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86:213–221. doi: 10.1016/j.ajhg.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Hormuzdi SG, Barbe MT, Herb A. Monyer H. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A. 2003;100:13644–13649. doi: 10.1073/pnas.2233464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R. Somlo S. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem. 1999;274:28557–28565. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- Divangahi M, Balghi H, Danialou G, Comtois AS, Demoule A, Ernest S, Haston C, Robert R, Hanrahan JW, Radzioch D. Petrof BJ. Lack of CFTR in skeletal muscle predisposes to muscle wasting and diaphragm muscle pump failure in cystic fibrosis mice. PLoS Genet. 2009;5:e1000586. doi: 10.1371/journal.pgen.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X-P, Wang X. Xu H. TRP channels of intracellular membranes. J Neurochem. 2010;113:313–328. doi: 10.1111/j.1471-4159.2010.06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty A, Gage P, Curtis S, Chelvanayagam G. Board P. The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J Biol Chem. 2001;276:3319–3323. doi: 10.1074/jbc.M007874200. [DOI] [PubMed] [Google Scholar]
- Fox JA. Conductance and selectivity properties of a substate of the rabbit sarcoplasmic reticulum channel. Biophys J. 1985;47:573–576. doi: 10.1016/S0006-3495(85)83953-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Boehmerle W, Maeda Y, Okuhara DY, Tian X, Yu Z, Choe C-u, Anyatonwu GI, Ehrlich BE. Somlo S. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci U S A. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie D. Fill M. Intracellular calcium release channels mediate their own countercurrent: the ryanodine receptor case study. Biophys J. 2008;95:3706–3714. doi: 10.1529/biophysj.108.131987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunti R, Gamberucci A, Fulceri R, Bánhegyi G. Benedetti A. Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: A mechanistic study on rat liver microsomes. Arch Biochem Biophys. 2007;462:115–121. doi: 10.1016/j.abb.2007.03.039. [DOI] [PubMed] [Google Scholar]
- Gray MA. Williams AJ. Multiple conducting states in the K+ channel of rabbit cardiac muscle SR; effect of block by a bis-quaternary ammonium compound. J Physiol. 1985;369:183P. [Google Scholar]
- Guo T, Nani A, Shonts S, Perryman M, Chen H, Shannon T, Gillespie D. Fill M. Sarcoplasmic reticulum K+ (TRIC) channel does not carry essential countercurrent during Ca2+ release. Biophys J. 2013;105:1151–1160. doi: 10.1016/j.bpj.2013.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Jr, Coronado R. Strauss HC. Potassium channel of cardiac sarcoplasmic reticulum is a multi-ion channel. Biophys J. 1989;55:35–45. doi: 10.1016/S0006-3495(89)82778-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Jr, Coronado R. Strauss HC. Open-channel subconductance state of K+ channel from cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 1990;258:H159–H164. doi: 10.1152/ajpheart.1990.258.1.H159. [DOI] [PubMed] [Google Scholar]
- Inesi G. Tadini-Buoninsegni F. Ca2+/H+ exchange, lumenal Ca2+ release and Ca2+/ATP coupling ratios in the sarcoplasmic reticulum ATPase. J Cell Commun Signal. 2014;8:5–11. doi: 10.1007/s12079-013-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Iwamoto T, Nakamura T, Doyle A, Fukumoto S. Yamada Y. Pannexin 3 functions as an ER Ca2+ channel, hemichannel, and gap junction to promote osteoblast differentiation. J Cell Biol. 2011;193:1257–1274. doi: 10.1083/jcb.201101050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Phang JM, Yu J, Harrop SJ, Sokolova AV, Duff AP, Wilk KE, Alkhamici H, Breit SN, Valenzuela SM, Brown LJ. Curmi PM. CLIC proteins, ezrin, radixin, moesin and the coupling of membranes to the actin cytoskeleton: A smoking gun. Biochim Biophys Acta. 2014;1838:643–657. doi: 10.1016/j.bbamem.2013.05.025. [DOI] [PubMed] [Google Scholar]
- Kamp F, Donoso P. Hidalgo C. Changes in luminal pH caused by calcium release in sarcoplasmic reticulum vesicles. Biophys J. 1998;74:290–296. doi: 10.1016/S0006-3495(98)77786-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano S. Hiraoka M. Protein kinase A-activated chloride channel is inhibited by the Ca2+–calmodulin complex in cardiac sarcoplasmic reticulum. Circ Res. 1993;73:751–757. doi: 10.1161/01.res.73.4.751. [DOI] [PubMed] [Google Scholar]
- Kawano S, Kuruma A, Hirayama Y. Hiraoka M. Anion permeability and conduction of adenine nucleotides through a chloride channel in cardiac sarcoplasmic reticulum. J Biol Chem. 1999;274:2085–2092. doi: 10.1074/jbc.274.4.2085. [DOI] [PubMed] [Google Scholar]
- Kawano S, Nakamura F, Tanaka T. Hiraoka M. Cardiac sarcoplasmic reticulum chloride channels regulated by protein kinase A. Circ Res. 1992;71:585–589. doi: 10.1161/01.res.71.3.585. [DOI] [PubMed] [Google Scholar]
- Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE. Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- Kuum M, Veksler V, Liiv J, Ventura-Clapier R. Kaasik A. Endoplasmic reticulum potassium–hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. J Cell Sci. 2012;125:625–633. doi: 10.1242/jcs.090126. [DOI] [PubMed] [Google Scholar]
- Labarca P, Coronado R. Miller C. Thermodynamic and kinetic studies of the gating behaviour of a K+-selective channel from the sarcoplasmic reticulum membrane. J Gen Physiol. 1980;76:397–424. doi: 10.1085/jgp.76.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca P. Miller C. A K+-selective, three-state channel from fragmented sarcoplasmic reticulum of frog leg muscle. J Membr Biol. 1981;61:31–38. doi: 10.1007/BF01870750. [DOI] [PubMed] [Google Scholar]
- Lamhonwah A-M, Bear CE, Huan LJ, Chiaw PK, Ackerley CA. Tein I. Cystic fibrosis transmembrane conductance regulator in human muscle: Dysfunction causes abnormal metabolic recovery in exercise. Ann Neurol. 2010;67:802–808. doi: 10.1002/ana.21982. [DOI] [PubMed] [Google Scholar]
- Liu QY. Strauss HC. Blockade of cardiac sarcoplasmic reticulum K+ channel by Ca2+: Two-binding-site model of blockade. Biophys J. 1991;60:198–203. doi: 10.1016/S0006-3495(91)82043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax RB, Camello C, Van Coppenolle F, Petersen OH. Tepikin AV. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J Biol Chem. 2002;277:26479–26485. doi: 10.1074/jbc.M201845200. [DOI] [PubMed] [Google Scholar]
- Lotteau S, Ducreux S, Romestaing C, Legrand C. Van Coppenolle F. Characterization of functional TRPV1 channels in the sarcoplasmic reticulum of mouse skeletal muscle. PLoS ONE. 2013;8:e58673. doi: 10.1371/journal.pone.0058673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu F, Janssens A, Gees M, Talavera K, Nilius B. Voets T. Modulation of the cold-activated cation channel TRPM8 by surface charge screening. J Physiol. 2010;588:315–324. doi: 10.1113/jphysiol.2009.183582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Wang G, Viero C, Wang Q, Mi W, Su X-D, Wagenknecht T, Williams AJ, Liu Z. Yin C-C. CLIC2-ryr1 interaction and structural characterization by cryo-electron microscopy. J Mol Biol. 2009;387:320–334. doi: 10.1016/j.jmb.2009.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C( Voltage-gated cation conductance channel from fragmented sarcoplasmic reticulum: Steady-state electrical properties. J Membr Biol. 1978;40:1–23. doi: 10.1007/BF01909736. [DOI] [PubMed] [Google Scholar]
- Mizuta K, Tsutsumi S, Inoue H, Sakamoto Y, Miyatake K, Miyawaki K, Noji S, Kamata N. Itakura M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem Biophys Res Commun. 2007;357:126–132. doi: 10.1016/j.bbrc.2007.03.108. [DOI] [PubMed] [Google Scholar]
- Nelson O, Supnet C, Liu H. Bezprozvanny I. Familial Alzheimer’s disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J Alzheimers Dis. 2010;21:781–793. doi: 10.3233/JAD-2010-100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Komazaki S, Iino M, Kangawa K. Takeshima H. Mitsugumin23, a novel transmembrane protein on endoplasmic reticulum and nuclear membranes. FEBS Lett. 1998;432:191–196. doi: 10.1016/s0014-5793(98)00864-3. [DOI] [PubMed] [Google Scholar]
- Okkenhaug H, Weylandt K-H, Carmena D, Wells DJ, Higgins CF. Sardini A. The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus. FASEB J. 2006;20:2390–2392. doi: 10.1096/fj.05-5588fje. [DOI] [PubMed] [Google Scholar]
- Patel AC, Brett TJ. Holtzman MJ. The role of CLCA proteins in inflammatory airway disease. Annu Rev Physiol. 2009;71:425–449. doi: 10.1146/annurev.physiol.010908.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N. Galietta LJV. Structure and Function of TMEM16 Proteins (Anoctamins) Physiol Rev. 2014;94:419–459. doi: 10.1152/physrev.00039.2011. [DOI] [PubMed] [Google Scholar]
- Penuela S, Harland L, Simek J. Laird DW. Pannexin channels and their links to human disease. Biochem J. 2014;461:371–381. doi: 10.1042/BJ20140447. [DOI] [PubMed] [Google Scholar]
- Picher M, Decrouy A. Rousseau E. Conducting and voltage-dependent behaviors of potassium ion channels reconstituted from diaphragm sarcoplasmic reticulum: Comparison with the cardiac isoform. Biochim Biophys Acta. 1996;1279:93–103. doi: 10.1016/0005-2736(95)00239-1. [DOI] [PubMed] [Google Scholar]
- Pinton P, Giorgi C, Siviero R, Zecchini E. Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Park KH, Nishi M, Urashima T, Aoki S, Yamazaki D, Ma J, Takeshima H. Sitsapesan R. Charade of the SR K+-channel: two ion-channels, TRIC-A and TRIC-B, masquerade as a single K+-channel. Biophys J. 2010;99:417–426. doi: 10.1016/j.bpj.2010.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, Chabot H, Beaudry C. Muller B. Reconstitution and regulation of cation-selective channels from cardiac sarcoplasmic reticulum. Mol Cell Biochem. 1992;114:109–117. doi: 10.1007/BF00240305. [DOI] [PubMed] [Google Scholar]
- Rousseau E, Roberson M. Meissner G. Properties of single chloride selective channel from sarcoplasmic reticulum. Biophys J. 1988;16:143–151. doi: 10.1007/BF00261900. [DOI] [PubMed] [Google Scholar]
- Rubinato E, Morgan A, D’Eustacchio A, Pecile V, Gortani G, Gasparini P. Faletra F. A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene. 2014;545:290–292. doi: 10.1016/j.gene.2014.05.028. [DOI] [PubMed] [Google Scholar]
- Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, Cai Y, Somlo S. De Smedt H. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285:18794–18805. doi: 10.1074/jbc.M109.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, Mousa N, Alsinani A, Ansari S, Alzahrani F, Al-Owain M, Alzayed ZS. Alkuraya FS. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet. 2012;49:630–635. doi: 10.1136/jmedgenet-2012-101142. [DOI] [PubMed] [Google Scholar]
- Shen WK, Rasmusson RL, Liu QY, Crews AL. Strauss HC. Voltage and temperature dependence of single K+ channels isolated from canine cardiac sarcoplasmic reticulum. Biophys J. 1993;65:747–754. doi: 10.1016/S0006-3495(93)81100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilling D, Mak D-OD, Kang DE. Foskett JK. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem. 2012;287:10933–10944. doi: 10.1074/jbc.M111.300491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber T, Weinert S. Jentsch TJ. Comprehensive Physiology. John Wiley & Sons, Inc; 2012. Cell biology and physiology of CLC chloride channels and transporters. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ. Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- Tanifuji M, Sokabe M. Kasai M. An anion channel of sarcoplasmic reticulum incorporated into planar lipid bilayers. Single channel behavior and conductance. J Membr Biol. 1987;99:103–111. doi: 10.1007/BF01871230. [DOI] [PubMed] [Google Scholar]
- Taylor CW. Dale P. Intracellular Ca2+ channels – A growing community. Mol Cell Endocrinol. 2012;353:21–28. doi: 10.1016/j.mce.2011.08.028. [DOI] [PubMed] [Google Scholar]
- Thebault S, Lemonnier L, Bidaux G, Flourakis M, Bavencoffe A, Gordienko D, Roudbaraki M, Delcourt P, Panchin Y, Shuba Y, Skryma R. Prevarskaya N. Novel role of cold/menthol-sensitive transient receptor potential melastatine family member 8 (TRPM8) in the activation of store-operated channels in LNCaP human prostate cancer epithelial cells. J Biol Chem. 2005;280:39423–39435. doi: 10.1074/jbc.M503544200. [DOI] [PubMed] [Google Scholar]
- Tomlins B. Williams AJ. Solubilisation and reconstitution of the rabbit skeletal muscle sarcoplasmic reticulum K+ channel into liposomes suitable for patch clamp studies. Pflugers Arch. 1986;407:341–347. doi: 10.1007/BF00585312. [DOI] [PubMed] [Google Scholar]
- Tomlins B, Williams AJ. Montgomery RAP. The characterization of a monovalent cation selective channel of mammalian cardiac muscle sarcoplasmic reticulum. J Membr Biol. 1984;80:191–199. doi: 10.1007/BF01868775. [DOI] [PubMed] [Google Scholar]
- Townsend C. Rosenberg RL. Characterization of a chloride channel reconstituted from cardiac sarcoplasmic reticulum. J Membr Biol. 1995;147:121–136. doi: 10.1007/BF00233541. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Kamata N, Vokes TJ, Maruoka Y, Nakakuki K, Enomoto S, Omura K, Amagasa T, Nagayama M, Saito-Ohara F, Inazawa J, Moritani M, Yamaoka T, Inoue H. Itakura M. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD) Am J Hum Genet. 2004;74:1255–1261. doi: 10.1086/421527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G. Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked Mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara A, Yasukochi M. Imanaga I. Calcium modulation of single SR potassium channel currents in heart muscle. J Mol Cell Cardiol. 1994;26:195–202. doi: 10.1006/jmcc.1994.1022. [DOI] [PubMed] [Google Scholar]
- Vanden Abeele F, Bidaux G, Gordienko D, Beck B, Panchin YV, Baranova AV, Ivanov DV, Skryma R. Prevarskaya N. Functional implications of calcium permeability of the channel formed by pannexin 1. J Cell Biol. 2006;174:535–546. doi: 10.1083/jcb.200601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi E, Matyjaszkiewicz A, Pitt S, Tsaneva-Atanasova K, Nishi M, Yamazaki D, Takeshima H. Sitsapesan R. TRIC-B channels display labile gating: evidence from the TRIC-A knockout mouse model. Pflugers Arch. 2013;465:1–14. doi: 10.1007/s00424-013-1251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi E, Mio K, Nishi M, Ogura T, Moriya T, Pitt SJ, Okuda K, Kakizawa S, Sitsapesan R, Sato C. Takeshima H. Mitsugumin 23 forms a massive bowl-shaped assembly and cation-conducting channel. Biochemistry. 2011;50:2623–2632. doi: 10.1021/bi1019447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronesi B. Oortgiesen M. The TRPV1 receptor: target of toxicants and therapeutics. Toxicol Sci. 2006;89:1–3. doi: 10.1093/toxsci/kfj034. [DOI] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T. Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344:634–638. doi: 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- Wang J. Best PM. Characterization of the potassium channel from frog skeletal muscle sarcoplasmic reticulum membrane. J Physiol. 1994;477:279–290. doi: 10.1113/jphysiol.1994.sp020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegierski T, Steffl D, Kopp C, Tauber R, Buchholz B, Nitschke R, Kuehn EW, Walz G. Köttgen M. TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J. 2009;28:490–499. doi: 10.1038/emboj.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki-Stordeur LE, Boyce AKJ. Swayne LA. Analysis of a pannexin 2-pannexin 1 chimeric protein supports divergent roles for pannexin C-termini in cellular localization. Cell Commun Adhes. 2013;20:73–79. doi: 10.3109/15419061.2013.791681. [DOI] [PubMed] [Google Scholar]
- Xin H, Tanaka H, Yamaguchi M, Takemori S, Nakamura A. Kohama K. Vanilloid receptor expressed in the sarcoplasmic reticulum of rat skeletal muscle. Biochem Biophys Res Commun. 2005;332:756–762. doi: 10.1016/j.bbrc.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Sugioka M. Ogawa Y. Voltage- and Ca2+-activated potassium channels in Ca2+ store control Ca2+ release. FEBS J. 2006;273:3585–3597. doi: 10.1111/j.1742-4658.2006.05365.x. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Taniwaki T, Kaikoi Y. Yamazaki T. Protective role of the endoplasmic reticulum protein mitsugumin23 against ultraviolet C-induced cell death. FEBS Lett. 2013;587:1299–1303. doi: 10.1016/j.febslet.2013.03.024. [DOI] [PubMed] [Google Scholar]
- Yamazaki D, Komazaki S, Nakanishi H, Mishima A, Nishi M, Yazawa M, Yamazaki T, Taguchi R. Takeshima H. Essential role of the TRIC-B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development. 2009;136:2355–2361. doi: 10.1242/dev.036798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Sasaki N, Nishi M. Takeshima H. Facilitation of DNA damage-induced apoptosis by endoplasmic reticulum protein mitsugumin23. Biochem Biophys Res Commun. 2010;392:196–200. doi: 10.1016/j.bbrc.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Yamazaki D, Tabara Y, Kita S, Hanada H, Komazaki S, Naitou D, Mishima A, Nishi M, Yamamura H, Yamamoto S, Kakizawa S, Miyachi H, Yamamoto S, Miyata T, Kawano Y, Kamide K, Ogihara T, Hata A, Umemura S, Soma M, Takahashi N, Imaizumi Y, Miki T, Iwamoto T. Takeshima H. TRIC-A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab. 2011;14:231–241. doi: 10.1016/j.cmet.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M, Lin PH, Pan Z, Komazaki S, Kato K, Nishi M, Zhao X, Weisleder N, Sato C, Ma J. Takeshima H. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature. 2007;448:78–82. doi: 10.1038/nature05928. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B. Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Tanaka O, Sekiguchi M, He H-j, Yasuoka Y, Itoh H, Kawahara K. Abe H. ATP-sensitive K+-channel subunits on the mitochondria and endoplasmic reticulum of rat cardiomyocytes. J Histochem Cytochem. 2005;53:1491–1500. doi: 10.1369/jhc.5A6736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lin P, Yamazaki D, Park KH, Komazaki S, Chen SRW, Takeshima H. Ma J. Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ Res. 2014;114:706–716. doi: 10.1161/CIRCRESAHA.114.301816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM. Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]