

Abstract
The calcium release complex is the major player in excitation–contraction coupling, both in cardiac and skeletal muscle. The core of the complex is the ryanodine receptor, and triadin is a regulating protein. Nevertheless, the precise function of triadin is only partially understood. Besides its function in the anchoring of calsequestrin at the triad/dyad, our recent results allow us to propose hypotheses on new triadin scaffolding functions, based on the studies performed using different models, from triadin knockout mice to human patients, and expression in non-muscle cells, taking into account the presence of multiple triadin isoforms.
Upon stimulation of the skeletal muscle at the neuromuscular junction, a membrane depolarization propagates along the plasma membrane up to the T-tubules, invaginations of the plasma membrane deep into the muscle fibre. This depolarization is then transformed into a large intracellular calcium release from the sarcoplasmic reticulum (SR), which in turn induces the muscle contraction. This step, called excitation–contraction (EC) coupling, is performed by the calcium release complex (CRC), localized exclusively in a specific region of skeletal muscle, the triad, composed of the close apposition of two terminal cisternae of sarcoplasmic reticulum on both sides of a T-tubule. The core of this complex is composed of two calcium channels, the dihydropyridine receptor (DHPR), a voltage-activated calcium channel in the T-tubule membrane, and the ryanodine receptor (RyR), the sarcoplasmic reticulum calcium channel (Lanner, 2012). A number of associated proteins are organized around these two channels (calsequestrin, triadin, junctin, …), and an efficient muscle contraction relies on the synchronous work of all these proteins. In skeletal muscle, both channels, even anchored in two different membranes, are physically associated (Marty et al. 1994): the membrane depolarization activates DHPR, which by a conformational change directly activates RyR and induces the calcium release from the SR (a mechanism called ‘voltage-induced calcium release’). The function of the calcium release complex is therefore based upon the precise organization of the triad and the implantation of the complex in this specific structure. In cardiac cells, a similar calcium release complex but with a slightly different activation, anchored in the cardiac dyad, is responsible for cardiac excitation–contraction coupling: membrane depolarization activates the cardiac DHPR, which induces an influx of the external calcium into the cell, and this calcium in turn activates RyR and induces a huge calcium release, a mechanism called ‘calcium-induced calcium release’ (CICR).
This extremely precise organization has to be maintained in the muscle fibre, a cell which undergoes large deformations during each contraction, and triadin could possibly be involved in this function. Triadin was first identified as a 95 kDa protein, specifically localized in the triad of the skeletal muscle (Brandt et al. 1990; Kim et al. 1990). Since its identification in 1990, the specific function of triadin has been the focus of numerous studies, in cardiac as well as in skeletal muscle, the two muscles in which triadin expression has been demonstrated. Because of its specific triad localization (at the origin of the name of the protein), and its association with RyR in the triads, involvement of triadin in excitation–contraction coupling has been presumed (Caswell et al. 1991; Knudson et al. 1993). Protein interaction studies have shown that molecular partners of triadin are RyR (Liu et al. 1994; Caswell et al. 1999; Groh et al. 1999), calsequestrin (CSQ), the protein which traps calcium inside the sarcoplasmic reticulum (Guo & Campbell, 1995; Kobayashi et al. 2000, Shin et al. 2000), and junctin (Zang et al. 1997). Functional studies have shown that triadin is able by itself to regulate the activity of the RyR calcium channel in vitro (Ohkura et al. 1998; Groh et al. 1999), or in vivo via its interaction with CSQ (Terentyev et al. 2007; Kucerova et al. 2012). The identification of different triadin isoforms raised the question of their ability to all perform the same functions.
Triadin, a multiprotein family with different localizations and partners
Triadin is in fact a multiple protein family, with different isoforms produced by the alternative splicing of a single TRDN gene (Thevenon et al. 2003). We have shown that at least three triadin isoforms are expressed in rat skeletal muscle (Marty et al. 2000; Vassilopoulos et al. 2005). We named these proteins Trisk 95, Trisk 51 and Trisk 32, according to their molecular weight (Fig.1). They have a common structure, and differ only by the length of their luminal segment and their unique C-terminal end. The smallest triadin, Trisk 32, is a minor skeletal muscle isoform, but it is the main, if not the only, cardiac triadin, also called CT1 (Kobayashi & Jones, 1999). In skeletal muscle it is localized not only in the triad but in the whole sarcoplasmic reticulum, and it is associated both with RyR1 in the triad and with the inositol trisphosphate (IP3) receptor in longitudinal SR, which it is able to regulate (Olah et al. 2011). In cardiac muscle Trisk 32 is colocalized with and associated to both RyR2 and the IP3 receptor. The two major skeletal muscle isoforms, Trisk 95 and Trisk 51, are exclusively expressed in skeletal muscle, in approximately equivalent amounts depending on the species (Marty et al. 2009). They are only localized in the triads of skeletal muscle, associated to RyR1 and CSQ1, and therefore are integral members of the calcium release complex (Marty et al. 2000).
Figure 1.
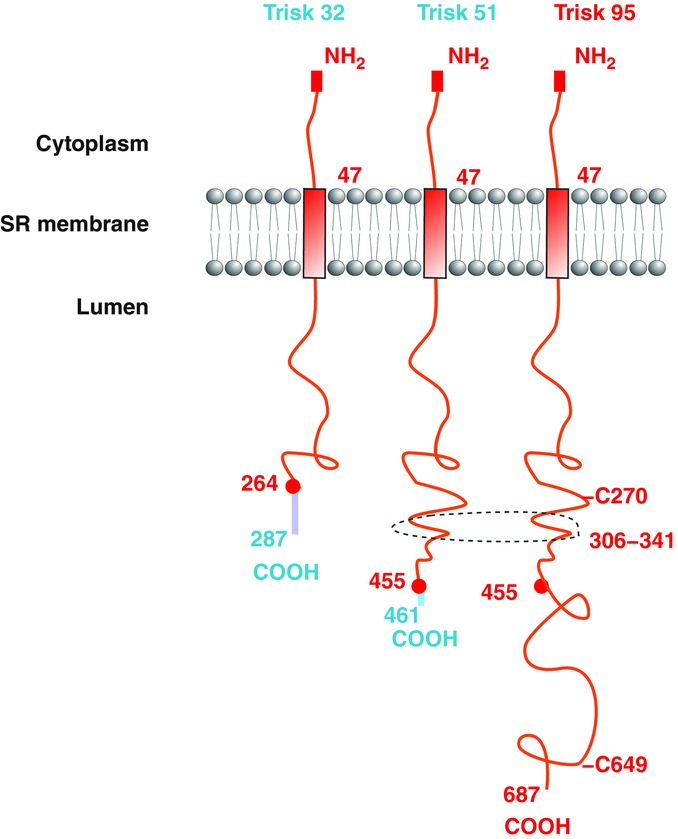
Structure of the main rat triadin isoforms The structures of the triadin isoforms in the sarcoplasmic reticulum are identical: a common N-terminal domain in the cytoplasm, a transmembrane helix, and a luminal part of variable length with a unique C-terminal end. The common parts are in red and the specific segments, starting at the red circles corresponding to the last common amino acids, are in a different colour. The domains involved in membrane deformation and indirect interaction with the microtubules are shown: the two cysteines C270 and C649, and the coiled-coil segment 306–341. The numbers refer to the rat sequence.
Function of triadin: development of knockout mouse models
In order to identify the function of triadin, two mouse models have been developed: one in the USA by the team of C. F. Perez and P. D. Allen (Shen et al. 2007), and one in France by our team (Oddoux et al. 2009). These mice have exactly the same phenotype: they are viable and fertile, and they have cardiac and skeletal muscle defects. From a structural point of view, the muscles of these mice are characterized by the presence of triads in an abnormal orientation, being longitudinal or oblique instead of the normal transversal orientation. Interestingly, in the two mouse models, the amount of triads in abnormal orientation is identical (30%). A moderate but clear muscle weakness, associated with reduction in the amplitude of calcium releases, was observed in the skeletal muscle of these mice (Shen et al. 2007; Oddoux et al. 2009). These mice also presented cardiac arrhythmia induced by isoproterenol (isoprenaline) injection, which mimics β-adrenergic stress (Chopra et al. 2009).
Triadin and human diseases
The cardiac phenotype of triadin knockout (KO) mice is quite similar to the phenotype of human patients affected by catecholaminergic polymorphic ventricular tachycardia (CPVT). CPVT is a rare genetic cardiac disease (Liu et al. 2008; Cerrone et al. 2009) characterized by stress-induced syncope or heart arrest, with an initial onset before the age of 10. Sixty to seventy per cent of affected patients have mutations in the cardiac ryanodine receptor gene RYR2 or in the cardiac calsequestrin gene CASQ2. Therefore CPVT has been proposed as a genetic disease of the cardiac calcium release complex. Based on the phenotype of triadin KO mice, and as triadin constitutes a link between RyR2 and CSQ2, we hypothesized that triadin mutation could be responsible for CPVT in humans. Therefore we searched for mutation in the triadin gene TRDN in a large cohort of 97 CPVT patients, in which no mutation had been identified in RYR2 or in CASQ2. We identified three triadin mutations in two families (Roux-Buisson et al. 2012). In the first family, the affected child was homozygous for a mutation (D18Afs*13) resulting in a STOP codon before the transmembrane segment. In the second family, the two affected brothers had two mutations, one on each allele: one amino acid substitution in position 59, in the transmembrane domain (T59R), and one STOP codon in position 205 (Q205*). The two premature STOP codons most probably resulted in the absence of protein, and we studied the effect of the third mutation, the T59R, in more detail. We induced the in vivo expression of the wild-type cardiac triadin Trisk 32, or the mutant Trisk 32-T59R, in the triadin KO mice, using systemic injection of recombinant adeno-associated virus (Roux-Buisson et al. 2012). One month after injection, the cardiomyocytes of these mice were isolated and we studied the expression level of the re-expressed triadin, its localization and its function. Whereas the wild-type Trisk 32 is perfectly re-expressed, colocalized with RyR2 in the cardiac dyad, the mutant Trisk 32-T59R can only be detected at the mRNA level, but not at the protein level. Expressing the wild-type Trisk 32 or the mutant Trisk 32-T59R in the model cell line COS-7 we observed that the mutant protein is degraded by the proteasome. Therefore, the three mutations identified in the patients resulted in the absence of protein, the patients being natural triadin KO, and CPVT in humans can be the result of the absence of triadin.
Triadin expression in non-muscle cells
Since the initial studies on triadin by the groups of K. P. Campbell and L. R. Jones (Guo & Campbell, 1995; Zang et al. 1997), triadin has been proposed to serve as an anchor for calsequestrin at the triad/dyad. But this cannot account for all the defects observed in triadin KO models. To get an insight into the additional functions of triadin, we studied the intrinsic properties of triadin in a system devoid of its usual muscle partners. We expressed triadin in a non-muscle cell line, the COS-7 cells, and analysed both the endoplasmic reticulum (ER) structure and the microtubule network, using specific antibodies (Fourest-Lieuvin et al. 2012). We observed that triadin induced huge modifications of the ER structure, forming rope-like ER structures, associated with a massive reorganization of the microtubule network, the microtubules being bundled at the periphery of the cell. Expression of triadin was also associated to constriction of the ER, which forms narrow tubes in the presence of triadin. These different experiments thus point to a new function of triadin. Triadin induces deformation of the reticulum, acts as a protein connecting the reticulum (or the calcium release complex) to the microtubule network, and allows the anchoring of triads to the microtubules network. We dissected the triadin’s sequences involved in this function, and we identified two domains: the two cysteines of Trisk 95, in positions 270 and 649, and a coiled-coil domain in position 306–341. These domains are partially present in Trisk 51, which has only one cysteine, and they are absent from Trisk 32 (Fig.1).
Conclusion: the possible functions of triadin
The studies performed on triadin allow us to propose multiple functions for triadin in skeletal and cardiac muscle. Trisk 95 is able to form large polymers because of its two cysteines (Froemming et al. 1999). These polymers induce deformation of reticulum membrane, improving the contact between RyR1 and DHPR and therefore the skeletal muscle excitation–contraction coupling. All the triadin isoforms are able to anchor CSQ, a soluble protein of the reticulum, at the cardiac dyad or skeletal muscle triad. Trisk 95 interacts indirectly with the microtubules via interaction with another protein of the reticulum membrane. This interaction allows the anchoring of the CRC and of the triad to the microtubules, and is involved in the formation or maintenance of triads during muscle development or contraction. Trisk 51 has only one cysteine, therefore its association with Trisk 95 induces a reduction in the size of triadin polymers, and probably a reduction in the membrane deformation. In the absence of triadin (KO mouse model), these functions are altered, as presented in Fig.2A and B for skeletal muscle defects, and Fig.2C and D for cardiac defects. In cardiac muscle, as the only triadin isoform, Trisk 32, is not able to induce membrane deformation, the main result of Trisk 32 deletion is the reduction in amount of calsequestrin in the dyads, which most probably explains the major part of the observed cardiac abnormalities. The interaction of Trisk 32 with microtubules has not been established yet. Some of these hypotheses are still speculative, and additional studies are necessary to confirm them. The schemes of Fig.2 should therefore be considered as a working hypothesis for the future studies, but some of the above defects can be observed in electron microscopy images of wild-type (WT) or triadin KO triads (Fig.3): the flattening of terminal cisternae resulting from CSQ reduction, and the increased distance between T-tubule and terminal cisternae.
Figure 2.
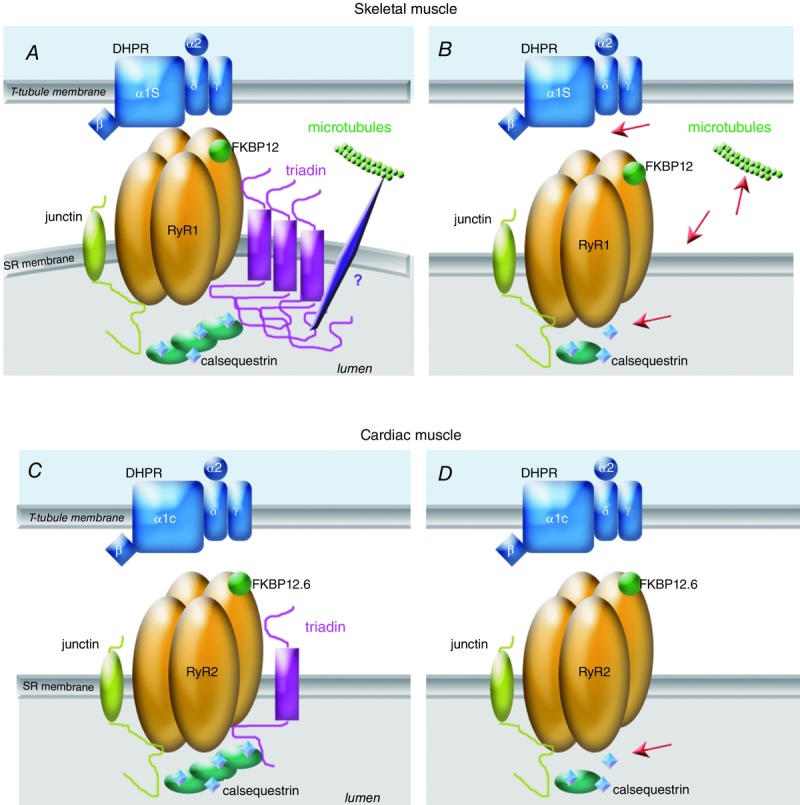
Function of triadin in skeletal and cardiac muscle, and alteration in triadin KO models A, in skeletal muscle of wild-type animals, the triadin isoform Trisk 95 forms polymers which induce membrane deformation, improving the DHPR–RyR contact and the EC coupling. Triadin anchors CSQ at the triad and via an indirect interaction allows the anchoring of the triad/CRC to the microtubules network. B, in the absence of triadin, the red arrows point to the major skeletal muscle defects: in the absence of membrane deformation, the EC coupling is altered, the amount of CSQ at the triad is reduced, the anchoring to the microtubules is altered, and some triads are not maintained in the normal transverse orientation. Association of the shorter skeletal muscle isoform Trisk 51 to Trisk 95 stops the elongation of these large Trisk 95 polymers. C, in cardiac muscle, the main function of the cardiac triadin Trisk 32 is the anchoring of CSQ in the vicinity of RyR2. D, this function is altered in triadin KO heart (red arrow). FKBP, FK506 binding protein.
Figure 3.
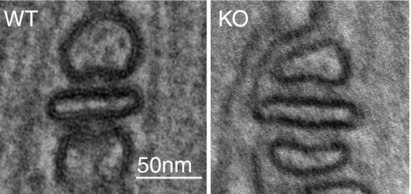
Triad alteration in skeletal muscle of triadin KO mouse In addition to the abnormal triad orientation observed in 30% of the triads, some alterations in the structure of all the triads can be visualized using electron microscopy on extensor digitorum longus muscle of triadin KO mice (Oddoux et al. 2009): a flattening of sarcoplasmic reticulum terminal cisternae resulting from the reduction in the amount of CSQ, and an increase in the distance between T-tubules and terminal cisternae.
Glossary
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CRC
calcium release complex
- CSQ
calsequestrin
- DHPR
dihydropyridine receptor
- EC
excitation–contraction
- ER
endoplasmic reticulum
- FKBP
FK506 binding protein
- KO
knockout
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
Biography
Isabelle Marty is a senior scientist (Research Director), working at INSERM, the French National Institute for Medical Research. She manages a research team ‘Muscle and Pathologies’, at the Grenoble Institute of Neurosciences, gathering basic scientists and geneticists, working on muscle pathologies related to defect in muscle calcium release. She develops two approaches: research on pathological human muscle, to identify the defects resulting of mutations, and research on cell and animal models, in order to determine the normal function of the muscle, and to develop therapies for these rare diseases.
Additional information
Competing interests
None declared.
Funding
This work was supported by grants from the Association Française contre les Myopathies (AFM), the Fondation Daniel Ducoin, the Institut National de la Santé et de la Recherche Médicale (INSERM), and the Vivier de la Recherche de la Faculté de Médecine de Grenoble.
References
- Brandt NR, Caswell AH, Wen SR. Talvenheimo JA. Molecular interactions of the junctional foot protein and dihydropyridine receptor in skeletal muscle triads. J Membr Biol. 1990;113:237–251. doi: 10.1007/BF01870075. [DOI] [PubMed] [Google Scholar]
- Caswell AH, Brandt NR, Brunschwig JP. Purkerson S. Localization and partial characterization of the oligomeric disulfide-linked molecular weight 95,000 protein (triadin) which binds the ryanodine and dihydropyridine receptors in skeletal muscle triadic vesicles. Biochemistry. 1991;30:7507–7513. doi: 10.1021/bi00244a020. [DOI] [PubMed] [Google Scholar]
- Caswell AH, Motoike HK, Fan H. Brandt NR. Location of ryanodine receptor binding site on skeletal muscle triadin. Biochemistry. 1999;38:90–97. doi: 10.1021/bi981306+. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Napolitano C. Priori SG. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm. 2009;6:1652–1659. doi: 10.1016/j.hrthm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C. Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci USA. 2009;106:7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourest-Lieuvin A, Rendu J, Osseni A, Pernet-Gallay K, Rossi D, Oddoux S, Brocard J, Sorrentino V, Marty I. Fauré J. Role of triadin in the organization of reticulum membrane at the muscle triad. J Cell Sci. 2012;125:3443–3453. doi: 10.1242/jcs.100958. [DOI] [PubMed] [Google Scholar]
- Froemming GR, Murray BE. Ohlendieck K. Self-aggregation of triadin in the sarcoplasmic reticulum of rabbit skeletal muscle. Biochim Biophys Acta. 1999;1418:197–205. doi: 10.1016/s0005-2736(99)00024-3. [DOI] [PubMed] [Google Scholar]
- Groh S, Marty I, Ottolia M, Prestipino G, Chapel A, Villaz M. Ronjat M. Functional interaction of the cytoplasmic domain of triadin with the skeletal ryanodine receptor. J Biol Chem. 1999;274:12278–12283. doi: 10.1074/jbc.274.18.12278. [DOI] [PubMed] [Google Scholar]
- Guo W. Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- Kim KC, Caswell AH, Talvenheimo JA. Brandt NR. Isolation of a terminal cisterna protein which may link the dihydropyridine receptor to the junctional foot protein in skeletal muscle. Biochemistry. 1990;29:9281–9289. doi: 10.1021/bi00491a025. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Stang KK, Moomaw CR, Slaughter CA. Campbell KP. Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268:12646–12654. [PubMed] [Google Scholar]
- Kobayashi YM, Alseikhan BA. Jones LR. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged β-strand in mediating the protein-protein interaction. J Biol Chem. 2000;275:17639–17646. doi: 10.1074/jbc.M002091200. [DOI] [PubMed] [Google Scholar]
- Kobayashi YM. Jones LR. Identification of triadin 1 as the predominant triadin isoform expressed in mammalian myocardium. J Biol Chem. 1999;274:28660–28668. doi: 10.1074/jbc.274.40.28660. [DOI] [PubMed] [Google Scholar]
- Kučerová D, Baba HA, Bokník P, Fabritz L, Heinick A, Mát’uš M, Müller FU, Neumann J, Schmitz W. Kirchhefer U. Modulation of SR Ca2+ release by the triadin-to-calsequestrin ratio in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2012;302:H2008–H2017. doi: 10.1152/ajpheart.00457.2011. [DOI] [PubMed] [Google Scholar]
- Lanner JT. Ryanodine receptor physiology and its role in disease. Adv Exp Med Biol. 2012;740:217–234. doi: 10.1007/978-94-007-2888-2_9. [DOI] [PubMed] [Google Scholar]
- Liu G. Pessah IN. Molecular interaction between ryanodine receptor and glycoprotein triadin involves redox cycling of functionally important hyperreactive sulfhydryls. J Biol Chem. 1994;269:33028–33034. [PubMed] [Google Scholar]
- Liu N, Ruan Y. Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51:23–30. doi: 10.1016/j.pcad.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Marty I, Fauré J, Fourest-Lieuvin A, Vassilopoulos S, Oddoux S. Brocard J. Triadin: What possible function 20 years later. J Physiol. 2009;587:3117–3121. doi: 10.1113/jphysiol.2009.171892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty I, Robert M, Villaz M, Lai Y, De Jongh KS, Catterall WA. Ronjat M. Biochemical evidence for a complex involving dihydropyridine receptor and ryanodine receptor in triad junctions of skeletal muscle. Proc Natl Acad Sci USA. 1994;91:2270–2274. doi: 10.1073/pnas.91.6.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty I, Thevenon D, Scotto C, Groh S, Sainnier S, Robert M, Grunwald D. Villaz M. Cloning and characterization of a new isoform of skeletal muscle triadin. J Biol Chem. 2000;275:8206–8212. doi: 10.1074/jbc.275.11.8206. [DOI] [PubMed] [Google Scholar]
- Oddoux S, Brocard J, Schweitzer A, Szentesi P, Giannesini B, Brocard J, Fauré J, Pernet-Gallay K, Bendahan D, Lunardi J, Csernoch L. Marty I. Triadin deletion induces impaired skeletal muscle function. J Biol Chem. 2009;284:34918–34929. doi: 10.1074/jbc.M109.022442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura M, Furukawa K, Fujimori H, Kuruma A, Kawano S, Hiraoka M, Kuniyasu A, Nakayama H. Ohizumi Y. Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry. 1998;37:12987–12993. doi: 10.1021/bi972803d. [DOI] [PubMed] [Google Scholar]
- Oláh T, Fodor J, Oddoux S, Ruzsnavszky O, Marty I. Csernoch L. Trisk 32 regulates IP3 receptors in rat skeletal myoblasts. Pflugers Arch. 2011;462:599–610. doi: 10.1007/s00424-011-1001-y. [DOI] [PubMed] [Google Scholar]
- Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Monnier N, Ray PF, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le Marec H, Lucet V, Mabo P, Probst V, Lacampagne A, Fauré J, Lunardi J. Marty I. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Franzini-Armstrong C, Lopez JR, Jones LR, Kobayashi YM, Wang Y, Kerrick WG, Caswell AH, Potter JD, Miller T, Allen PD. Perez CF. Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J Biol Chem. 2007;282:37864–37874. doi: 10.1074/jbc.M705702200. [DOI] [PubMed] [Google Scholar]
- Shin DW, Ma J. Kim DH. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 2000;486:178–182. doi: 10.1016/s0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Vedamoorthyrao S, Oduru S, Györke I, Williams SC. Györke S. Protein–protein interactions between triadin and calsequestrin are involved in modulation of sarcoplasmic reticulum calcium release in cardiac myocytes. J Physiol. 2007;583:71–80. doi: 10.1113/jphysiol.2007.136879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon D, Smida-Rezgui S, Chevessier F, Groh S, Henry-Berger J, Romero NB, Villaz V, DeWaard M. Marty I. Human skeletal muscle triadin: gene organization and cloning of the major isoform, Trisk 51. Biochem Biophys Res Comun. 2003;303:669–675. doi: 10.1016/s0006-291x(03)00406-6. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos S, Thevenon D, Smida Rezgui S, Urbani-Brocard J, Chapel A, Lacampagne A, Lunardi J, DeWaard M. Marty I. Triadins are not triad specific proteins: two new skeletal muscle triadins possibly involved in the architecture of sarcoplasmic reticulum. J Biol Chem. 2005;280:28601–28609. doi: 10.1074/jbc.M501484200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kelley J, Schmeisser G, Kobayashi YM. Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]