

Abstract
Promoter-proximal pausing by RNA polymerase II (Pol II) is a well-established mechanism to control the timing, rate, and possibly the magnitude of transcriptional responses. Recent studies have shown that cellular signaling pathways can regulate gene transcription and signaling outcomes by controlling Pol II pausing in a wide array of biological systems. Identification of the proteins and small molecules that affect the establishment and release of paused Pol II is shedding new light on the mechanisms and biology of Pol II pausing. This review will focus on the interplay between cellular signaling pathways and Pol II pausing during normal development and under disease conditions.
Keywords: GRO-seq, P-TEFb, RNA polymerase pausing, Signaling, Transcription
Pol II pausing as a rate-limiting step in transcription
Deciphering the mechanisms mediating the precise transcriptional control of gene expression by RNA polymerase II (Pol II) provides critical insights into the cellular response to developmental and environmental signals. The regulation of Pol II-mediated transcription occurs at each major stage of the transcription process, including initiation, elongation, and termination (Fig. 1). Historically, studies of transcription regulation have focused on the mechanisms by which gene-specific transcription factors recruit Pol II and basal transcription factors to gene promoters to form a transcription preinitiation complex (PIC). However, studies in recent years have revealed additional rate-limiting steps in transcription, including the release of promoter-proximal paused Pol II. Subsequent to recruitment and transcription initiation, Pol II often pauses after synthesis of a nascent RNA ~20–60 nt in length, and remains in a paused state until additional signals promote productive elongation. This promoter-proximal pausing serves as a rate-limiting step on more than 70% of metazoan genes and predominantly occurs at genes in stimulus-responsive pathways. After an introduction to Pol II pausing, we will highlight recent studies examining the interplay between cellular signaling pathways and Pol II pausing during normal development and under disease conditions (Table 1).
Figure 1. Pol II-mediated gene transcription is a multi-step process.
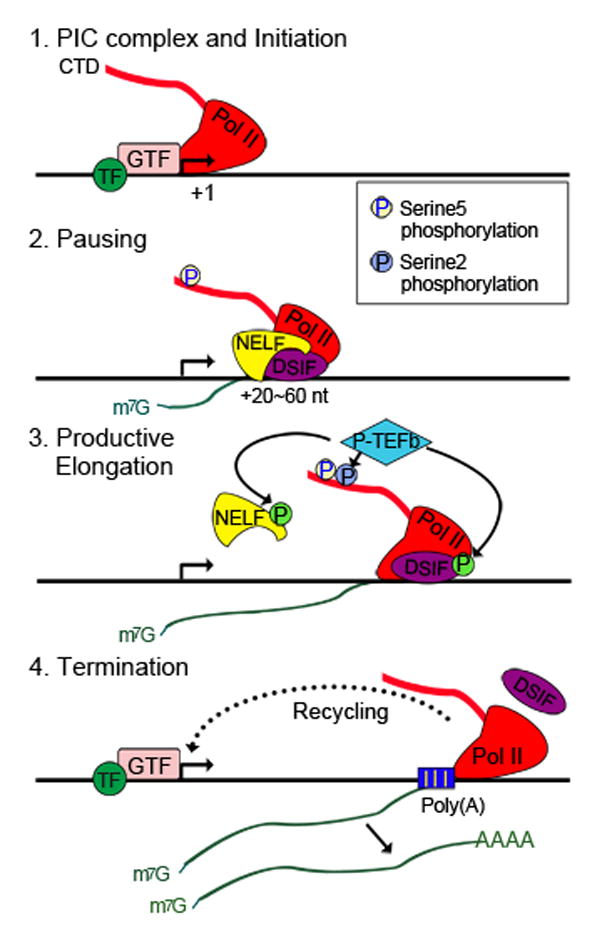
Step 1: Gene-specific transcription factors (TF) recruit general transcription factors (GTF) and polymerase II (Pol II) to form a preinitiation complex (PIC) at the promoter. Pol II is ready to initiate transcription.
Step 2: Pol II produces RNA of 20–60 nt long and pauses. The pausing factors DSIF (the DRB sensitivity-inducing factor) and NELF (the negative elongation factor ) are required for Pol II pausing. The Ser5 residues of the Pol II carboxy-terminal domain (CTD) repeats are phosphorylated. The short, nascent RNA remains associated with Pol II and is capped at the 5′end.
Step 3: P-TEFb recruitment releases Pol II from pausing. P-TEFb phosphorylates Ser2 of the Pol II CTD, DSIF and NELF, resulting in the dissociation of NELF. Phosphorylated DSIF becomes a positive elongation factor and travels with Pol II throughout the gene. Pol II enters the stage of productive elongation. Phosphorylation is indicated by a circled “P” on each factor.
Step 4: After transcribing through the gene body, Pol II passes the Poly(A) signal site and terminates transcription. Pol II is removed from the gene and the RNA is released. Free Pol II can be recycled back to promoter to reinitiate transcription.
Table 1.
Signals for Pol II pause release
| Signal | P-TEFb recruiting factors | References |
|---|---|---|
| Environmental signals | ||
| Heat shock | HSF | [58] |
| Hypoxia | HIF1α CDK8-containing mediator |
[53] |
| Serum stimulation | BRD4 CDK8-containing mediator |
[52], [60] |
| Proinflammation signals | ||
| LPS, TNFα | NF-κB BRD4 |
[62–63] |
| Hormones | ||
| Estrogen, testosterone, retinoic acid | ERα, AR, RAR | [65–67] |
| Lineage differentiation signals | ||
| Hematopoiesis | LDB1 complexes TIF1γ GATA-1 Ikaros |
[76–82] |
| ESC differentiation | c-Myc SEC KLF4 |
[10],[71],[85] |
| Disease signals | ||
| MLL-fusion leukemia | SECs BRD4 |
[89–90] |
| HIV infection | TAT SEC |
[91–96] |
| Cardiac hypertrophy | GATA4 BRD4 |
[97–102] |
Pol II Pausing: From Genes to Genomes
Chambon and colleagues proposed over 30 years ago that transcription elongation might be a rate-limiting step in metazoan gene expression [1]. Definitive evidence for the existence of transcriptionally engaged promoter-proximal paused Pol II came from Lis and colleagues [2]. Focusing on the Drosophila heat shock genes (hsp genes), they demonstrated that prior to heat shock induction, transcriptionally engaged Pol II accumulates ~20–60 bp downstream of the transcription start sites (TSSs) of hsp promoters and is associated with a short nascent RNA [2, 3]. Paused Pol II was also observed on other genes, including several mammalian immediate early genes (myc, fos, jun, etc.) [4–6], which respond rapidly to stimulation by extracellular growth factors. However, recent genome-wide mapping technologies have demonstrated that Pol II pausing is a widespread mechanism of transcription regulation in metazoans.
To detect paused Pol II, several genomic approaches have been developed (Table 2). The first approach uses chromatin immunoprecipitation with an antibody against Pol II coupled with deep sequencing (ChIP-seq) to identify sites of Pol II enrichment across the genome. Studies using this approach have revealed that the majority of gene promoters in metazoan cells have high levels of Pol II accumulation ~20–60 bp downstream of TSS, corresponding to the paused Pol II [7–10]. Subsequently, an RNA sequencing approach was developed to detect short, capped RNAs (scRNAs) enriched near the promoter, presumably produced by paused Pol II [11]. More recently, native elongating transcript sequencing (NET-seq) was developed to detect all transcripts associated with paused and elongating Pol II [12].
Table 2.
Comparison of sequencing-based methods for detecting genome-wide Pol II pausing.
| Methods | Description | Unique Aspects | References |
|---|---|---|---|
| Pol II ChIP-seq | Pol II-bound DNA is isolated by chromatin immunoprecipitation with a Pol II antibody and subjected to high-throughput sequencing. | Provides a genome-wide view of Pol II binding sites for all forms of Pol II, including Pol II in the PIC complex and transcriptionally engaged Pol II. | [7–10] |
| scRNA-seq | Short RNA species (<100 nt) that are 5′ capped and nuclear-localized are isolated and sequenced. | Uses the map of 3′ ends to define the location where Pol II pauses. | [11] |
| NET-seq | Transcripts associated with immunoprecipitated Pol II complexes are isolated and sequenced. | Detects all transcripts associated with Pol II in unperturbed cells. | [12] |
| GRO-seq | Nascent RNAs from transcriptionally engaged Pol II are isolated and sequenced. | Detects transcriptionally engaged Pol II that is either paused at promoters or actively elongating within gene bodies. Genome-wide application of classical transcription run-on assay. | [13–22] |
None of the approaches listed above, however, detect whether paused Pol II is elongation-competent, in contrast to global run-on sequencing (GRO-seq) [13]. GRO-seq is a deep sequencing-based method that can directly detect transcriptionally engaged Pol II through the incorporation of the nucleotide analog bromo-UTP (BrUTP) into the nascent RNA during a short (~100 nt), controlled run-on under conditions where new transcription initiation is blocked. Nascent RNAs from engaged (but paused) Pol II at promoters, or actively elongating Pol II in gene bodies, can be isolated using anti-BrUTP affinity chromatography and then processed for high throughput sequencing.
More recently, several new GRO-seq-based approaches, including precision run-on sequencing (PRO-seq) [14], GRO-cap [15, 16] and 5′GRO-seq [17], were developed to improve the mapping resolution, as well as to identify 5′ capped RNAs synthesized by paused Pol II. Studies using these approaches have yielded robust genome-wide maps of paused Pol II and have shown how Pol II pausing changes in response to activation of cellular signaling pathways [18–22] (Fig. 2).
Figure 2. GRO-Seq reveals the dynamics of Pol II elongation upon stimulation by signaling pathways.

(A) Genome browser views of GRO-seq data showing the Pol II elongation pattern of an estradiol (E2)-stimulated gene in MCF-7 human breast cancer cells. The green arrow indicates the leading edge of elongating Pol II after 40 min of treatment with E2. The elongation rate of Pol II can be calculated as: , for example. GRO-seq signals from positive and negative strand are represented by red and blue color, respectively.
(B) Metagene representations showing the average profile of GRO-seq reads ± 4 kb around the transcription start sites (TSS) of activated genes in E2-treated MCF-7 cells (top) and TNFα-treated AC16 cells (bottom). In MCF-7 cells, 40 min of treatment with E2 induces a more dramatic increase of Pol II density near the TSS compared to that in the gene body, suggesting that newly recruited Pol II is paused before entering productive elongation. By contrast, in AC16 cells, 30 min of treatment with TNFα results in a rapid increase and release of Pol II into the gene body, suggesting that TNFα signaling mainly regulates the pause release step of Pol II elongation. Adapted from [15,16].
Establishing Paused Pol II at Promoters
The status of paused versus elongating Pol II correlates with the phosphorylation pattern of the heptapeptide repeat (amino acid sequence: YSPTSPS) in the carboxy-terminal domain (CTD) of the largest Pol II subunit, Rpb1 [23]. Paused Pol II is enriched for phosphorylation at serine 5 of the CTD heptapeptide repeat, whereas elongating (i.e., pause-released) Pol II is phosphorylated at serine 2. Serine 5-phosphorylated Pol II (Pol II Ser5P) is held at promoters by two distinct pausing factors, the DRB sensitivity-inducing factor (DSIF) and the negative elongation factor (NELF) [24, 25] (Fig. 1 & 3). Although DSIF and NELF are sufficient to inhibit Pol II elongation in biochemical assays, other factors may also contribute to pausing in vivo. One such factor is Gdown1 (encoded by POLR2M) [26], which regulates the stability of paused Pol II by preventing premature termination of the elongating transcript. More recently, Trim28 (tripartite motif containing 28, also known as TIF1β) has been shown to facilitate pausing by stabilizing paused Pol II [27] (Fig. 3).
Figure 3. Pol II pausing and release are regulated by signaling pathways and multiple factors.
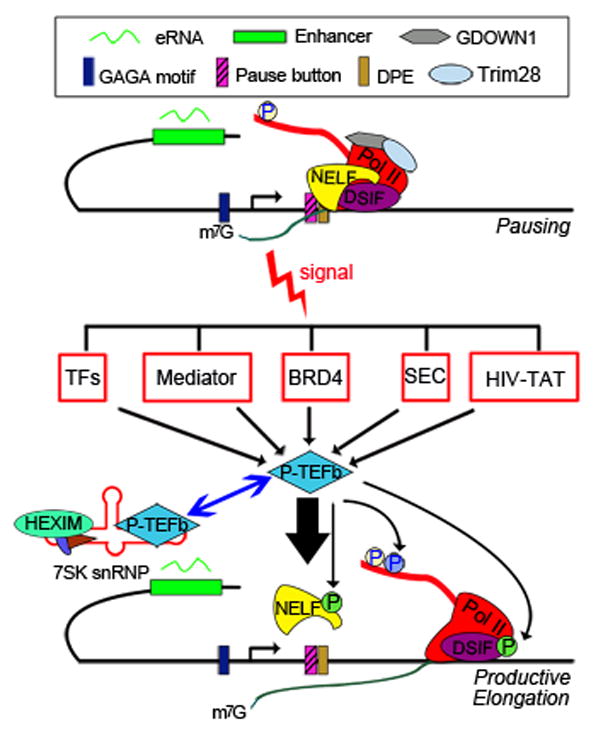
Pol II pausing requires DSIF and NELF, and is facilitated by GDOWN1 and Trim28. The promoter sequences may also contribute to the establishment of paused Pol II. DNA motifs such as the downstream promoter element (DPE), the “pause button” and GAGA factor binding motifs are enriched on paused promoters in Drosophila cells. In addition, enhancers and enhancer transcripts (eRNAs) are also involved in pausing establishment or release of Pol II. Environmental and developmental signals can trigger the release of paused Pol II by recruiting P-TEFb via the different types of activators listed, or by inducing P-TEFb release from the 7SK-HEXIM inhibitory complex.
Paused Pol II may be terminated prematurely and released from the gene or stably reside at the proximal promoter region, waiting for a signal to enter productive elongation. Using a variety of approaches, several studies have assessed the stability of paused Pol II in Drosophila or mammalian cells [28–30]. These studies revealed that paused Pol II is relatively stable, with an average half-life of more than six minutes. Such a stably paused Pol II would be beneficial for synchronous induction of expression across many genes [31, 32], and allow a temporal window to integrate signals and coordinate binding of signal-responsive transcription factors.
The mechanisms by which Pol II is paused remain elusive. Some nuclear receptors, such as the glucocorticoid receptor and estrogen receptor, have been found to recruit the pausing factor NELF to repress gene expression [33, 34], suggesting a direct role of signaling pathways in establishing Pol II pausing. In addition, studies in Drosophila and mammalian cells have suggested that the intrinsic property of promoters might contribute to Pol II pausing. Promoter elements such as the GAGA motif, the downstream promoter element (DPE), the “pause button”, and the TATA box have all been positively or negatively linked to Pol II pausing [11, 35–38], in some cases by recruiting transcription factors, including the GAGA factor (GAF) and the Motif 1 binding protein (M1BP) [39, 40].
Role of P-TEFb in the Release of Paused Pol II
The positive elongation factor b (P-TEFb) is a critical factor for promoting the release of paused Pol II [41–43]. P-TEFb is composed of Cyclin T and a kinase subunit CDK9 [44, 45], which phosphorylates serine 2 of the heptapeptide repeat in the Pol II CTD [46] (Fig. 1&3). It also phosphorylates NELF and DSIF, leading to the dissociation of NELF, while converting DSIF to an elongation-stimulating factor [47–49]. P-TEFb is necessary and sufficient for the release of paused Pol II. Inhibition of the kinase activity of P-TEFb with flavopiridol, a small molecule inhibitor, causes an increase in paused Pol II on most active genes in both Drosophila and mammalian cells [30, 50], indicating that P-TEFb kinase activity is required for pause release.
P-TEFb can be directly recruited to genes through association with DNA-binding transcription factors, or indirectly through association with transcription co-regulators and chromatin factors (Fig. 3). For example, the Mediator complex, which plays key roles in transcription initiation, has also been shown to function in the early stage of elongation by recruiting P-TEFb, overcoming the negative effect of pausing factors, or altering the phosphorylation status of the Pol II CTD [51–53]. Another important cofactor for P-TEFb recruitment is BRD4, a member of the bromodomain and extraterminal (BET) family of proteins, which bind to acetylated lysines in the amino-terminal tails of core histones through their bromodomains. BRD4 has been shown to recruit P-TEFb by binding to Cyclin T. Small molecule inhibitors of BET proteins can inhibit Pol II elongation on many genes in different cells types, supporting an important role for BRD proteins in the regulation of Pol II elongation [54].
Sequestration is another mechanism to regulate P-TEFb. In this regard, studies have shown that the majority of P-TEFb is sequestered in an inactive complex composed of 7SK RNA (an abundant small noncoding nuclear RNA), HEXIM (a double stranded RNA binding protein) and other subunits (Fig. 3). P-TEFb must be released from this nuclear ribonucleoprotein complex to become active [55, 56].
Taken together, these studies demonstrate that the recruitment of P-TEFb to gene promoters is a rate-limiting step for the regulated release of paused Pol II. Protein factors and cellular signals tightly regulate this key step, which we discuss in detail in the next section.
Pause Release Signals
The release of paused Pol II into active elongation is a key step in the transcription cycle. As such, it is tightly regulated, in many cases as an endpoint of cellular signaling pathways. In this section, we will discuss how environmental, developmental, and differentiation signals regulate Pol II pausing and release.
Environmental signals
Extracellular stress signals
Heatshock (hsp) genes and immediate early genes (IEGs) were the first types of genes found to be regulated by Pol II pausing. Studies from Lis and colleagues provided clear evidence that Pol II is preloaded on Drosophila hsp gene promoters under non-heat shock conditions, transcribing the initial 20–60 nt of RNA before pausing [3, 57]. Heat shock triggers Pol II release and productive elongation by recruiting P-TEFb. The recruitment of P-TEFb is dependent on heat shock factor (HSF), a signal-regulated transcription factor [58], implicating HSF in regulating the transition from Pol II pausing to elongation, rather than Pol II recruitment. Subsequent studies in mammalian cells have observed paused Pol II on many IEGs, such as c-myc, c-fos, c-jun and junB, under uninduced conditions [4–6]. Because these genes must respond quickly to environmental stimuli with a burst of transcription, a paused Pol II at their promoters may help to poise them for rapid induction. Disruption of pausing by depleting pausing factors compromises the rapid induction of IEGs, as shown in rat neurons [59].
Hypoxia, acting through the transcription factor HIF1α (hypoxia inducible factor1 alpha), is another stress signal that modulates gene expression programs controlled by Pol II pausing. Prior to induction, paused Pol II was found at the promoters of HIF1α target genes [53]. Hypoxia triggers the association of HIF1α with the CDK8-mediator complex, which in turn recruits P-TEFb to release the paused Pol II into productive elongation [53].
Histone modification may also act to trigger the release of paused Pol II, as shown for the serum-induced fosl1 gene [60]. Serum stimulation induces phosphorylation of serine 10 (Ser10) of histone H3 tail, creating a recognition site for the adaptor protein 14-3-3, which then recruits the MOF histone acetyltransferase to acetylate histone H4 at lysine 16. BRD4 binds to acetylated H4 and recruits P-TEFb to release Pol II [60]. Together, these studies illustrate how cellular signaling pathways can act through transcription factors and chromatin to promote a conversion from paused to elongating Pol II.
Proinflammatory signals
Early studies by Barboric et al. found that NF-κB, a key transcription factor regulating proinflammatory genes, recruits P-TEFb to stimulate Pol II elongation on the gene encoding IL-8 upon stimulation by TNFα (tumor necrosis factor alpha) [61]. Recent studies using Pol II-ChIP or GRO-seq have revealed that many proinflammatory genes in macrophages have paused Pol II on their promoters and are transcribed at basal level before stimulation [62, 63]. Exposure to bacterial lipopolysaccharide (LPS) triggers recruitment of P-TEFb to the promoters by Toll-like receptor (TLR) signaling pathways to induce productive elongation, rapidly amplifying gene expression [63].
Regulation of Pol II pausing is also involved in the repression of inflammatory genes [64]. For example, the glucocorticoid receptor (GR) has been shown to inhibit proinflammatory genes by acting at different stages of the transcription cycle. At genes controlled at the transcription initiation step, ligand-bound GR inhibits Pol II recruitment and transcription initiation upon LPS stimulation. In contrast, at genes controlled at the elongation step, GR promotes the accumulation of the pausing factor NELF. As expected, knockdown of NELF specifically abolishes GR-dependent repression of the elongation-controlled genes in macrophages [64].
Nuclear receptors (NR) and steroid hormone signaling pathways
The release of paused Pol II can be regulated by a number of nuclear receptor (NR)-dependent signaling pathways [65–67] (Table 1). Aiyar et al. found that by directly interacting with NELF-B, estrogen receptor α (ERα) recruits NELF to a number of target genes in response to estrogen stimulation, preventing Pol II release and thus repressing ERα-mediated gene activation in breast cancer cells [33]. Other studies, however, revealed that a large number of estrogen target genes have preloaded, paused Pol II in untreated MCF-7 breast cancer cells [68, 69]. Estrogen stimulation induces P-TEFb-dependent Pol II release and triggers gene activation. Therefore, both ERα-repressed and activated genes can be regulated by controlling Pol II release.
A genome-wide view of the immediate effects of estrogen on Pol II transcription was revealed recently by studies from the Kraus lab [18, 21]. Using GRO-seq, Hah et al. provided the first dynamic picture of Pol II transcription across the genome following stimulation by estrogen in MCF-7 cells [18] (Fig. 2A). In a follow-up study, Danko et al. developed a computational approach to mine GRO-seq data to determine the kinetics of transcription by following the progression of the Pol II wave after short treatments with estrogen in MCF-7 cells, or TNFα in AC16 cardiomyocytes [21] (Fig. 2B). They found that at TNFα-induced genes in AC16 cells, Pol II enters bodies of regulated genes almost immediately after treatment, suggesting a release of paused Pol II in response to activation of the TNFα signaling pathway. In contrast, at estrogen-induced genes in MCF-7 cells, the authors observed a genome-wide increase in Pol II accumulation at the promoters in response to estrogen signaling (Fig. 2B), suggesting that Pol II is recruited to the promoters of regulated genes in response to estrogen, either for preinitiation complex formation or the establishment of paused Pol II. Not surprisingly, the Pol II residence times at pausing sites positively correlates with the enrichment of NELF binding. This observation argues against pause release as the major rate-limiting step for estrogen/ERα-regulated genes. Instead, it supports a model that the primary effect of estrogen/ERα signaling is at a step prior to Pol II pausing, such as Pol II recruitment or initiation/reinitiation.
Together, these data emphasize the important point that having preloaded, paused Pol II at the promoters does not necessarily mean fast activation kinetics upon stimulation. Instead, Pol II pausing may be more important to allow synchronized gene activation in response to cellular signals, as has been observed in other biological systems [70, 71]. Together these studies provided strong evidence that different signaling pathways may induce gene activation by acting on distinct rate-limiting steps, with some affecting Pol II initiation (e.g. estrogen) and others affecting Pol II release (e.g. TNFα) (Fig. 4).
Figure 4. Signaling pathways regulate distinct steps in the transcription cycle.

The major steps in the transcription cycle are depicted. The two key rate-limiting steps, initiation and release, of paused Pol II, can be differentially regulated by signaling pathways. It is also possible that some signaling pathways regulate both steps to tightly control the transcription level.
Developmental and differentiation signals
Although Pol II pausing was initially discovered on genes responsive to environmental stimuli, increasing evidence suggests that the regulation of the paused-to elongating switch plays a fundamental role in gene expression during animal development, where cells have to integrate various developmental and differentiation signals to initiate organogenesis.
Embryonic development
Genetic and genomic studies in Drosophila, zebrafish, and mouse have all revealed a pivotal role for Pol II pausing factors in embryonic development, suggesting a link between developmental signals and the release of paused Pol II. In both Drosophila and zebrafish, disrupting the pausing function of DSIF causes tissue specific phenotypes in developing embryos [72, 73]. In mouse, deletion of the gene encoding the NELF subunit NELF-B leads to an inner cell mass deficiency and early embryonic lethality [74].
Genome-wide studies in early Drosophila embryos revealed an enrichment of paused Pol II on developmental control genes, such as Hox genes, and genes involved in cellular signaling pathways [8, 9]. In Drosophila, widespread Pol II pausing is first detected at the midblastula transition (MBT) stage [36]. GRO-seq assays have further revealed that ~55% of genes in early Drosophila embryos contain paused Pol II, and that pause release appears to be the dominant regulatory step for genes involved in anterior–posterior and dorsal-ventral axis patterning [75]. Using muscle development in Drosophila embryos as a model, Gaertner et al. have shown that at different embryonic stages, a large number of inactive genes gradually acquire paused Pol II during the developmental time course and are poised for future gene activation by tissue-specific signals [37]. Remarkably, in gastrulating Drosophila embryos, genes with paused Pol II at their promoters are more synchronously activated than non-paused genes [31, 32]. Importantly, replacement of a strongly paused promoter with moderately paused or non-paused promoters disrupts this synchrony, leading to a severe defect in cell movement during gastrulation [32].
These studies clearly indicate a critical role for Pol II pausing, and more specifically pause release, in organogenesis and animal development. The specific signals mediating these effects remain to be determined.
Hematopoietic differentiation
Hematopoietic lineage differentiation is tightly controlled by transcriptional regulation and signaling pathways. Multiple studies have revealed a physical interaction between P-TEFb and hematopoietic transcription factors, suggesting a vital role of Pol II elongation in hematopoietic gene regulation [76–82]. In erythroid differentiation in zebrafish embryos, loss of TIF1γ, a transcription co-factor essential for erythropoiesis, causes profound anemia [76]. Remarkably, this anemic phenotype can be largely reversed by genetic mutations that disrupt Pol II pausing. The physical interactions among TIF1γ, CDK9 and the SCL/GATA-1 hematopoietic transcription complexes further suggest a specific role of TIF1γ in recruiting P-TEFb to target genes of SCL/GATA-1 to release paused Pol II in response to signals during erythroid differentiation [76].
In the megakaryocyte lineage, recruitment of P-TEFb by the transcription factors GATA-1 and RUNX1 is required to promote megakaryocytic gene activation [79]. Moreover, calcium signaling triggers P-TEFb release from the 7SK-HEXIM complex through the calcium-dependent protease calpain 2. Intriguingly, calpain 2 is downregulated in Down syndrome patients with megakaryocytic neoplasia, and restoration of its expression corrects the megakaryocyte defects in a mouse model of Down syndrome, suggesting therapeutic possibilities [80]. Together, these studies illustrate how P-TEFb as a central component of signal-regulated pathways that drive hematopoietic lineage differentiation.
Differentiation of embryonic stem cells (ESCs)
In 2007, Guenther et al. reported that most transcriptionally inactive genes in human ESCs (hESCs) have promoter-associated Pol II that initiates transcription without productive elongation, providing some of the first evidence that Pol II pausing occurs genome-wide [7]. A later study in mouse ESCs confirmed widespread Pol II pausing on both active and inactive genes (mESCs), and further revealed the function of c-Myc in P-TEFb-mediated pause release in mESCs [10]. More recently, several studies carried out GRO-Seq in mESCs and revealed that pausing was prevalent at genes involved in metabolism, cell cycle, and signaling pathways, but not at genes controlling lineage differentiation and stem cell fate, or bivalent genes [19, 50, 83]. The role of pausing in cell cycle may explain the inner cell mass defect in NELF-B knockout mouse [74]. Accordingly, genetic depletion of NELF-B in mESCs leads to severe growth defects [83]. Interestingly, ESCs lacking NELF-B are more resistant to differentiation, owing to the markedly attenuated response to FGF/ERK signaling [83]. These data suggest that establishing Pol II pausing by NELF plays an important role in ESCs by maintaining appropriate cellular response to differentiation cues. Similarly, release of paused Pol II is also important during ESC differentiation. Lin et al. found that P-TEFb and its binding partner, the super elongation complex (SEC) (see details in the next section), are required for the induction of most genes during retinoid acid (RA)-induced mESC differentiation [71]. Moreover, ELL3, another elongation factor, binds to the enhancers at some RA-induced genes to promote the establishment of paused Pol II at promoters and recruit P-TEFb-containing SEC to release Pol II upon RA induction [84].
More recently, Pol II pausing was shown to be a rate-limiting step in the reprograming of induced pluripotent stem cells (iPSCs) from somatic cells. Pol II is engaged but paused at the promoters of several pluripotency genes during reprograming [85]. The release of paused Pol II requires BRD4-dependent release of P-TEFb from the 7SK-HEXIM complex, followed by P-TEFb recruitment to pluripotency promoters by the reprogramming factor KLF4. This study thus provides further evidence for the role of Pol II pause release in cell fate determination, although the signals promoting the effect remain to be determined.
In summary, numerous studies have demonstrated that Pol II pausing and release represent critical rate-limiting steps in metazoan gene expression. In response to environmental and physiological signals, various transcription factors and chromatin regulators work together with the pausing and elongation machinery to selectively fine-tune gene expression in a cell type- or tissue-specific manner. This added layer of regulation provides a different control point than Pol II recruitment to ensure precise patterns of spatial and temporal gene expression in response to cellular signaling.
Aberrant Release of Pol II Pausing by Cellular Signals in Disease
The critical role of the Pol II pausing-to-elongation transition in normal development and differentiation highlights its role as a key regulation checkpoint which, when disrupted, can lead to pathogenesis in many human diseases. In support of this view, aberrant expression of P-TEFb subunits CDK9 and Cyclin T1/T2 has been observed in various tumors [86]. Notably, c-Myc-dependent elongation through P-TEFb recruitment has been shown to cause transcriptional amplification in tumor cells, which may explain why c-Myc plays an oncogenic role in such a wide variety of human cancers [87]. In this section, we give some examples in which abnormal signal-dependent release of Pol II leads to different human diseases.
Super elongation complexes (SECs) and MLL-fusion leukemia
The mixed lineage leukemia (MLL) gene in human is involved in a large number of chromosomal translocations that produce chimeric oncogenic proteins [88]. Clues to the molecular mechanisms underlying MLL-fusion leukemia came with the identification of super elongation complexes (SECs) that contains P-TEFb, ELL1/2/3, AFF4 and several other MLL translocation partners [89]. These findings have led to a model in which the MLL-fused chimera proteins recruit P-TEFb-containing SEC to normal MLL target genes to cause premature activation of transcription elongation on genes that can be oncogenic when amplified, such as those within the HOX clusters. This dysregulation of gene expression is a driving force in MLL-associated leukemia. BRD4 may also contribute to SEC-dependent recruitment of P-TEFb via interaction with SEC, and BET inhibitors have shown significant therapeutic values in pre-clinical MLL leukemia models [90].
Human immunodeficiency virus (HIV) infection
Transcription of the HIV genome was one of the earliest model systems used to study transcription elongation [91]. At the early stages of HIV transcription, Pol II is paused after synthesis of a short RNA transcribed from the long terminal repeat (LTR) of the HIV-1 promoter. This short RNA forms a stem-loop structure termed the transactivation response element (TAR) that is recognized by the HIV-encoded transcriptional transactivator protein Tat. Tat, which is expressed early during active infection, recruits P-TEFb from the host cells to stimulate the production of full-length viral transcripts [43].
Recent studies using proteomic assays and structure analyses of Tat–associated proteins have provided more insights into Tat-P-TEFb dependent elongation [92–96]. These studies revealed that the Tat-P-TEFb complex also contains core subunits of SECs. The crystal structure of P-TEFb in complex with Tat and AFF4 has revealed a direct interaction between Tat and the SEC subunit AFF4 [96]. The presence of AFF4 in this complex enhances the ability of Tat-P-TEFb to recognize TAR. Thus, the association of Tat with SEC is critical for promoting P-TEFb-dependent elongation on the HIV promoter and ultimately the HIV life cycle.
Cardiac hypertrophy
Cardiac hypertrophy is characterized by enlarged myocyte size resulting from a cellular increase of mRNA and protein synthesis. The global increase of transcription in hypertrophic cardiomyocytes is mediated by ectopic activation of P-TEFb by disease signals [97], similar to what is observed in c-Myc amplified cancer cells. At least two mechanisms contribute to the increased P-TEFb activity. Studies in cultured cardiomyocytes have shown that hypertrophic signals trigger P-TEFb release from the 7SK-HEXIM complex [98]. In mice, genetic ablation of Clp-1, the gene encoding the mouse ortholog of human Hexim1, results in embryonic lethality [99]. Elevated expression of hypertrophy-related genes in these embryos is consistent with the role of 7SK-HEXIM in sequestering P-TEFb during cardiac growth. In addition, recruitment of P-TEFb to disease-promoting genes is increased during the development of hypertrophy, which may be caused by direct interactions between P-TEFb and transcription factors such as GATA4 [100] and NF-κB [101] that drive cardiac hypertrophy, or may be governed by the co-activator BRD4 [102]. Inhibition of BRD4 by BET inhibitors antagonizes the transcriptional output mediated by many transcription factors and suppresses Pol II release on disease-promoting genes during the development of hypertrophy in vivo [102]. These results suggest that the indispensable role of BRD4 occurs, at least in part, through its ability to recruit P-TEFb to promote pause release and transcriptional elongation.
The involvement of elevated P-TEFb activity, leading to enhanced release of paused Pol II, in disease pathogenesis emphasizes the necessity of keeping Pol II release under a tight control in physiological conditions. Understanding how abnormal Pol II pause release contributes to pathological gene expression will provide great insights into the biologic role of this key step in the transcription process. In addition, it highlights the importance of developing therapeutic approaches targeting Pol II pausing and release, as exemplified by the BET inhibitors and CDK9 inhibitors.
Pol II Pausing and Enhancers
Enhancers play critical roles in regulating signal-dependent gene transcription. Recent findings, including the discovery of enhancer-specific chromatin features and enhancer transcription that produces RNAs (eRNAs) [103], have provided new insights about enhancer function. New studies have connected paused Pol II to enhancer function.
Role of transcriptional elongation in signal-regulated enhancer function
Genome-wide deep sequencing approaches, including ChIP-seq, RNA-seq and GRO-seq, have revealed a global view of RNA transcription at active enhancers across many cell types in response to signal transduction events [103]. Using GRO-seq, several groups have detected a tight kinetic correlation between the transcription of enhancers and the transcription of cognate target genes in response to cellular signaling pathways [104–106], and have shown that enhancer transcription can be used to predict enhancer activity [104]. Similar to target gene promoters, the transcription of enhancers can be regulated at either transcription initiation or elongation, and is sensitive to elongation inhibitors, including flavopiridol and BET inhibitors [104, 105]. Inhibition of transcription elongation at enhancers by BET inhibitors and flavopiridol in TLR4-stimulated macrophages causes a reduction in H3K4 mono- and di-methylation at the enhancer, suggesting that enhancer transcription precedes enhancer H3K4 methylation [105]. In contrast, inhibition of transcription elongation at enhancers by flavopiridol in estrogen-stimulated breast cancer cells does not affect histone modification patterns and other molecular features of enhancers, supporting a model in which enhancer transcription occurs after the assembly of active enhancers or is not functionally linked to enhancer activity [104]. These differences may reflect context-dependent roles of enhancer transcription or eRNAs,
Release of Pol II pausing at promoters is facilitated by enhancers and eRNAs
Enhancers, enhancer transcription, and eRNAs have also been shown to contribute to Pol II elongation at target gene promoters. An early study of an androgen-regulated gene showed that Pol II is first recruited to a distal enhancer in response to androgen stimulation, and flavopiridol blocks the transfer of Pol II from the enhancer to the gene promoter [107]. This study suggests that enhancer-promoter looping may regulate Pol II elongation on androgen target genes. Indeed, high-resolution mapping of enhancer-promoter interactions in Drosophila embryos has revealed a frequent association of enhancer-promoter loops with paused Pol II [108].
A direct role of eRNA in the release of paused Pol II has been recently proposed [109]. eRNAs were found to be required for the induction of neuronal immediate early genes (IEGs) during neuronal activation. RNAi-mediated knockdown of eRNAs reduced gene expression of the target IEGs and the level of elongating Pol II along the gene bodies. Biochemical assays revealed a direct interaction between eRNAs and the RNA-binding domain of the NELF-E subunit, suggesting a model in which eRNAs may be acting as a decoy for nascent transcripts from IEG promoters to facilitate the release of paused Pol II [109].
These studies have revealed new facets of enhancer function and suggest that enhancers function to regulate Pol II pausing at target gene promoters. At present, the possible localization and function of paused Pol II at active enhancers is not clear.
Concluding remarks
The discovery of the prevalence of Pol II pausing on signal-responsive genes has provided important insights for understanding the underlying mechanism of signaling-regulated transcription programs. A growing number of transcription factors and chromatin regulators have been found to regulate the release of paused Pol II in response to cellular signaling pathways by interactions with pausing factors and/or P-TEFb. Different signaling pathways regulate transcription by selectively affecting Pol II recruitment, pause release or both. Although Pol II pausing was originally thought to be a mechanism set inducible genes in a poised state for rapid activation, growing evidence suggests that it may represent a transcriptional checkpoint to ensure precise, and perhaps synchronous, gene regulation in response to developmental cues, physiological states, cellular stresses, and various other signaling events. Still, many questions remain to be answered. These include the precise molecular and biochemical mechanisms that act to establish paused Pol II and influence of residence time of Pol II at pause sites. Understanding the full spectrum of the biological function of Pol II pausing will require more developmental models, as well as disease models of perturbed Pol II pausing. Finally, the dynamic interplay between Pol II pausing and chromatin structure is just beginning to be understood, but clearly much more remains to be uncovered. Addressing these exciting issues will be critical to uncovering therapeutic strategies to target pathological processes driven by signal-induced Pol II elongation.
Highlights.
Pol II pausing is a rate-limiting step in the transcription of many metazoan genes.
Pol II pausing can be regulated by cellular signaling pathways.
Pol II pausing and/or release can be regulated as an endpoint of signaling pathways.
Regulation of Pol II pausing and release allows fine-tuning of gene expression.
Acknowledgments
We thank Minho Chae for helping with figures. The research in the labs of X.B. and W.L.K are supported by grants from NIH/NIDDK (R00DK088963 and R01DK105287 to X.B., R01DK058110 and R01DK069710 to W.L.K.), and the Cancer Prevention Research Institute of Texas (CPRIT) (R1115 to X.B., RP130607 to W.L.K.). While we made every attempt to cite the most relevant literature, space limitation precluded us from citing all published works in this area.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gariglio P, et al. Clustering of RNA polymerase B molecules in the 5′ moiety of the adult beta-globin gene of hen erythrocytes. Nucleic Acids Res. 1981;9:2589–2598. doi: 10.1093/nar/9.11.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rougvie AE, Lis JT. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell. 1988;54:795–804. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- 3.Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strobl LJ, Eick D. Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 1992;11:3307–3314. doi: 10.1002/j.1460-2075.1992.tb05409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plet A, et al. Elongation and premature termination of transcripts initiated from c-fos and c-myc promoters show dissimilar patterns. Oncogene. 1995;10:319–328. [PubMed] [Google Scholar]
- 6.Law A, et al. Direct cloning of DNA that interacts in vivo with a specific protein: application to RNA polymerase II and sites of pausing in Drosophila. Nucleic Acids Res. 1998;26:919–924. doi: 10.1093/nar/26.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guenther MG, et al. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeitlinger J, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nechaev S, et al. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Core LJ, et al. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwak H, et al. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339:950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruesi WS, et al. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. Elife. 2013;2:e00808. doi: 10.7554/eLife.00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Core LJ, et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46:1311–1320. doi: 10.1038/ng.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam MT, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hah N, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min IM, et al. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011;25:742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Core LJ, et al. Defining the status of RNA polymerase at promoters. Cell Rep. 2012;2:1025–1035. doi: 10.1016/j.celrep.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danko CG, et al. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell. 2013;50:212–222. doi: 10.1016/j.molcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo X, et al. Dynamic reorganization of the AC16 cardiomyocyte transcriptome in response to TNFalpha signaling revealed by integrated genomic analyses. BMC Genomics. 2014;15:155. doi: 10.1186/1471-2164-15-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidemann M, et al. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta. 2013;1829:55–62. doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Hartzog GA, Fu J. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim Biophys Acta. 2013;1829:105–115. doi: 10.1016/j.bbagrm.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamaguchi Y, et al. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta. 2013;1829:98–104. doi: 10.1016/j.bbagrm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Cheng B, et al. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol Cell. 2012;45:38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunch H, et al. TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat Struct Mol Biol. 2014;21:876–883. doi: 10.1038/nsmb.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckley MS, et al. Kinetics of promoter Pol II on Hsp70 reveal stable pausing and key insights into its regulation. Genes Dev. 2014;28:14–19. doi: 10.1101/gad.231886.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, et al. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015;29:39–47. doi: 10.1101/gad.246173.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henriques T, et al. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell. 2013;52:517–528. doi: 10.1016/j.molcel.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boettiger AN, Levine M. Synchronous and stochastic patterns of gene activation in the Drosophila embryo. Science. 2009;325:471–473. doi: 10.1126/science.1173976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagha M, et al. Paused Pol II coordinates tissue morphogenesis in the Drosophila embryo. Cell. 2013;153:976–987. doi: 10.1016/j.cell.2013.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aiyar SE, et al. Attenuation of estrogen receptor alpha-mediated transcription through estrogen-stimulated recruitment of a negative elongation factor. Genes Dev. 2004;18:2134–2146. doi: 10.1101/gad.1214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. 2005;19:1116–1127. doi: 10.1101/gad.1297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amir-Zilberstein L, et al. Differential regulation of NF-kappaB by elongation factors is determined by core promoter type. Mol Cell Biol. 2007;27:5246–5259. doi: 10.1128/MCB.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen K, et al. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. Elife. 2013;2:e00861. doi: 10.7554/eLife.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaertner B, et al. Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep. 2012;2:1670–1683. doi: 10.1016/j.celrep.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendrix DA, et al. Promoter elements associated with RNA Pol II stalling in the Drosophila embryo. Proc Natl Acad Sci U S A. 2008;105:7762–7767. doi: 10.1073/pnas.0802406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuda NJ, et al. GAGA factor maintains nucleosome-free regions and has a role in RNA polymerase II recruitment to promoters. PLoS Genet. 2015;11:e1005108. doi: 10.1371/journal.pgen.1005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Gilmour DS. Distinct mechanisms of transcriptional pausing orchestrated by GAGA factor and M1BP, a novel transcription factor. EMBO J. 2013;32:1829–1841. doi: 10.1038/emboj.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall NF, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;12:2078–2090. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng J, et al. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998;273:13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- 45.Peng J, et al. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall NF, et al. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 47.Fujinaga K, et al. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wada T, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada T, et al. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 50.Jonkers I, et al. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conaway RC, Conaway JW. The Mediator complex and transcription elongation. Biochim Biophys Acta. 2013;1829:69–75. doi: 10.1016/j.bbagrm.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donner AJ, et al. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17:194–201. doi: 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galbraith MD, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153:1327–1339. doi: 10.1016/j.cell.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 55.Diribarne G, Bensaude O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol. 2009;6:122–128. doi: 10.4161/rna.6.2.8115. [DOI] [PubMed] [Google Scholar]
- 56.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 57.Lis J. Promoter-associated pausing in promoter architecture and postinitiation transcriptional regulation. Cold Spring Harb Symp Quant Biol. 1998;63:347–356. doi: 10.1101/sqb.1998.63.347. [DOI] [PubMed] [Google Scholar]
- 58.Lis JT, et al. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000;14:792–803. [PMC free article] [PubMed] [Google Scholar]
- 59.Saha RN, et al. Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat Neurosci. 2011;14:848–856. doi: 10.1038/nn.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zippo A, et al. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 61.Barboric M, et al. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 62.Adelman K, et al. Immediate mediators of the inflammatory response are poised for gene activation through RNA polymerase II stalling. Proc Natl Acad Sci U S A. 2009;106:18207–18212. doi: 10.1073/pnas.0910177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Escoubet-Lozach L, et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 2011;7:e1002401. doi: 10.1371/journal.pgen.1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupte R, et al. Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proc Natl Acad Sci U S A. 2013;110:14616–14621. doi: 10.1073/pnas.1309898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flajollet S, et al. The elongation complex components BRD4 and MLLT3/AF9 are transcriptional coactivators of nuclear retinoid receptors. PLoS One. 2013;8:e64880. doi: 10.1371/journal.pone.0064880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee DK, et al. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. J Biol Chem. 2001;276:9978–9984. doi: 10.1074/jbc.M002285200. [DOI] [PubMed] [Google Scholar]
- 67.Mitra P, et al. Estrogen receptor-alpha recruits P-TEFb to overcome transcriptional pausing in intron 1 of the MYB gene. Nucleic Acids Res. 2012;40:5988–6000. doi: 10.1093/nar/gks286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kininis M, et al. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090–5104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kininis M, et al. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol Cell Biol. 2009;29:1123–1133. doi: 10.1128/MCB.00841-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gilchrist DA, et al. Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev. 2012;26:933–944. doi: 10.1101/gad.187781.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin C, et al. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC) Genes Dev. 2011;25:1486–1498. doi: 10.1101/gad.2059211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo S, et al. A regulator of transcriptional elongation controls vertebrate neuronal development. Nature. 2000;408:366–369. doi: 10.1038/35042590. [DOI] [PubMed] [Google Scholar]
- 73.Jennings BH, et al. Locus-specific requirements for Spt5 in transcriptional activation and repression in Drosophila. Curr Biol. 2004;14:1680–1684. doi: 10.1016/j.cub.2004.08.066. [DOI] [PubMed] [Google Scholar]
- 74.Amleh A, et al. Mouse cofactor of BRCA1 (Cobra1) is required for early embryogenesis. PLoS One. 2009;4:e5034. doi: 10.1371/journal.pone.0005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saunders A, et al. Extensive polymerase pausing during Drosophila axis patterning enables high-level and pliable transcription. Genes Dev. 2013;27:1146–1158. doi: 10.1101/gad.215459.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bai X, et al. TIF1gamma controls erythroid cell fate by regulating transcription elongation. Cell. 2010;142:133–143. doi: 10.1016/j.cell.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bottardi S, et al. Direct protein interactions are responsible for Ikaros-GATA and Ikaros-Cdk9 cooperativeness in hematopoietic cells. Mol Cell Biol. 2013;33:3064–3076. doi: 10.1128/MCB.00296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bottardi S, et al. Ikaros interacts with P-TEFb and cooperates with GATA-1 to enhance transcription elongation. Nucleic Acids Res. 2011;39:3505–3519. doi: 10.1093/nar/gkq1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elagib KE, et al. Cross-talk of GATA-1 and P-TEFb in megakaryocyte differentiation. Blood. 2008;112:4884–4894. doi: 10.1182/blood-2008-03-145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elagib KE, et al. Calpain 2 activation of P-TEFb drives megakaryocyte morphogenesis and is disrupted by leukemogenic GATA1 mutation. Dev Cell. 2013;27:607–620. doi: 10.1016/j.devcel.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meier N, et al. Novel binding partners of Ldb1 are required for haematopoietic development. Development. 2006;133:4913–4923. doi: 10.1242/dev.02656. [DOI] [PubMed] [Google Scholar]
- 82.Song SH, et al. Multiple functions of Ldb1 required for beta-globin activation during erythroid differentiation. Blood. 2010;116:2356–2364. doi: 10.1182/blood-2010-03-272252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Williams LH, et al. Pausing of RNA Polymerase II Regulates Mammalian Developmental Potential through Control of Signaling Networks. Mol Cell. 2015;58:311–322. doi: 10.1016/j.molcel.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin C, et al. The RNA Pol II elongation factor Ell3 marks enhancers in ES cells and primes future gene activation. Cell. 2013;152:144–156. doi: 10.1016/j.cell.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu L, et al. Transcriptional pause release is a rate-limiting step for somatic cell reprogramming. Cell Stem Cell. 2014;15:574–588. doi: 10.1016/j.stem.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 86.Krystof V, et al. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr Pharm Des. 2012;18:2883–2890. doi: 10.2174/138161212800672750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin CY, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J Cell Biochem. 2005;95:234–242. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 89.Smith E, et al. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yankulov K, Bentley D. Transcriptional control: Tat cofactors and transcriptional elongation. Curr Biol. 1998;8:R447–449. doi: 10.1016/s0960-9822(98)70289-1. [DOI] [PubMed] [Google Scholar]
- 92.Gu J, et al. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle. 2014;13:1788–1797. doi: 10.4161/cc.28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.He N, et al. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sobhian B, et al. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tahirov TH, et al. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–751. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schulze-Gahmen U, et al. AFF4 binding to Tat-P-TEFb indirectly stimulates TAR recognition of super elongation complexes at the HIV promoter. Elife. 2014;3:e02375. doi: 10.7554/eLife.02375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kulkarni PA, et al. Phosphorylation of RNA polymerase II in cardiac hypertrophy: cell enlargement signals converge on cyclin T/Cdk9. Recent Prog Horm Res. 2004;59:125–139. doi: 10.1210/rp.59.1.125. [DOI] [PubMed] [Google Scholar]
- 98.Sano M, et al. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. 2002;8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 99.Huang F, et al. Ablation of the CLP-1 gene leads to down-regulation of the HAND1 gene and abnormality of the left ventricle of the heart and fetal death. Mech Dev. 2004;121:559–572. doi: 10.1016/j.mod.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 100.Sunagawa Y, et al. Cyclin-dependent kinase-9 is a component of the p300/GATA4 complex required for phenylephrine-induced hypertrophy in cardiomyocytes. J Biol Chem. 2010;285:9556–9568. doi: 10.1074/jbc.M109.070458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gutierrez SH, et al. Cardiac role of the transcription factor NF-kappaB. Cardiovasc Hematol Disord Drug Targets. 2008;8:153–160. doi: 10.2174/187152908784533702. [DOI] [PubMed] [Google Scholar]
- 102.Anand P, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lam MT, et al. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hah N, et al. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. doi: 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaikkonen MU, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Louie MC, et al. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci U S A. 2003;100:2226–2230. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghavi-Helm Y, et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;512:96–100. doi: 10.1038/nature13417. [DOI] [PubMed] [Google Scholar]
- 109.Schaukowitch K, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]