

Abstract
Artificial oocyte activation is a critical step during somatic cell nuclear transfer (SCNT). Most of current activation protocols focus on inducing an increase in the intracellular free Ca2+ concentration of the oocyte. Here, we have utilized a zinc chelator, TPEN, to enhance the efficiency of oocyte activation during SCNT. TPEN treatment of matured pig oocytes resulted in the reduction of available Zn2+ in pig oocytes; however, the cytosolic Ca2+ concentration in the oocytes was not affected by the TPEN treatment. When various concentrations (100 – 250 μM) and incubation durations (45 min – 2.5 hours) of TPEN were used to activate oocytes, the efficiency of oocyte activation was not different from conventional activation methods. When oocytes that were activated by conventional activation methods were incubated with a lower concentration of TPEN (5 – 10 μM), a significant increase in embryos developing to the blastocyst stage was observed. In addition, when oocytes receiving a small Ca2+ stimulus were further activated by higher concentration of TPEN (100 – 200 μM), a significant increase in the frequency of blastocyst formation was observed, compared to a conventional activation method. This result indicated that TPEN can be a main reagent in oocyte activation. No increase in the cytosolic Ca2+ level was detected when oocytes were exposed to various concentrations of TPEN, indicating the ability of TPEN to induce oocyte activation is independent of an intracellular Ca2+ increase. We were able to produce clones through SCNT using the TPEN-assisted activation procedure, and the piglets produced through the process did not show any signs of abnormality. In this study, we have developed an efficient way to utilize TPEN to increase the developmental potential of cloned embryos.
1. Introduction
Somatic cell nuclear transfer (SCNT), i.e. cloning, can be used to generate genetically engineered animals, preserve endangered species, and produce animals with a select genetic background. For example, SCNT has been used to generate genetically engineered pigs that can be used for agriculture [1, 2] and biomedicine [3, 4]. Development of SCNT has dramatically expanded the application of pigs. However, current SCNT technology is inefficient as only one percent of SCNT generated pig embryos are able to develop to term. One of the factors that contribute to the poor efficiency is the ineffectiveness of current artificial oocyte activation procedures. Under physiological conditions, fusion of the sperm and oocyte plasma membranes leads to repetitive increases in the intracellular free calcium (Ca2+) concentration in the oocyte cytoplasm [5, 6]. These transient increases (termed oscillations) result from the release of phospholipase C-ζ (PLC-zeta) from the sperm head [7]. The Ca2+ oscillations activate Ca2+/calmodulin-dependent protein kinase II (CaMKII) [8] and the CaMKII phosphorylates early mitotic inhibitor 2 (EMI2, a.k.a. FBXO43) thus relieving the anaphase-promoting complex/cyclosome (APC/C) from FBXO43-mediated inhibition [9-11]. These events lead to the degradation of cyclin B [12, 13], a subunit of the M-phase promoting factor (MPF). These signaling pathways demonstrate that Ca2+ signaling is essential in the process of oocyte activation and in fact, disruption of Ca2+ signaling during oocyte activation can cause developmental defects [14].
Most artificial activation methods induce an increase in the intracellular free Ca2+ levels in the oocyte that mimic sperm-induced Ca2+ signaling. However, most methods are able to induce only a single Ca2+ rise in the ooplasm [15]. Compared to the repetitive Ca2+ increases observed after sperm-induced oocytes activation, a single Ca2+ spike is a relatively poor activator of oocytes. Different approaches have been attempted to increase the efficiency of oocyte activation. For example, it is possible to incubate activated oocytes with inhibitors in order to reduce the level of MPF [16, 17]; however, these inhibitors are not very specific toward MPF, and while they do degrade MPF, they also have a number of side effects.
Zinc (Zn2+) is important to maintain oocytes arrested at the MII stage. In mice there is a 50% increase in the intracellular amount of Zn2+ as oocytes develop from the germinal vesicle to MII stage [18]. Zn2+ is necessary in activating MPF as Zn2+ regulates the activity of CDC25 [19], a phosphatase that can dephosphorylate and thus activate cyclin-dependent kinase 1, a component of MPF. In addition, EMI2 (official gene symbol, FBXO43), a zinc-binding protein, is required to maintain high MPF activity during the MII arrest [12], and the increase in total intracellular Zn2+ during oocyte maturation directly controls FBXO43 activity [20]. A previous report demonstrated that removal of Zn2+ from MII stage oocytes can successfully induce oocyte activation, and thus permit oocytes to exit the MII stage [21]. This was the first report demonstrating that full-term development can occur without Ca2+ signaling. Although the efficiency of full term development was lower, the study demonstrated that the Ca2+ signal is not required for full-term development in mice.
Zn2+ is released from oocytes after fertilization indicating that removal of Zn2+ is a natural part of oocyte activation [22]. TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl) ethane-1,2-diamine), known to have a high specificity toward Zn2+, was used to lower the level of available Zn2+ to activate mouse oocytes [21]. In this study, we developed an efficient method to activate pig oocytes by reducing the intracellular level of Zn2+ using TPEN. We found that a combination of proper Ca2+ signal and TPEN treatment can increase the developmental potential of activated oocytes. We also demonstrated that TPEN can be used to efficiently activate oocytes during SCNT. This approach might be used to increase application of SCNT in pigs.
2. Materials and Methods
2.1 Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Missouri.
2.2 Chemicals
All chemicals in the experiments were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO) unless indicated otherwise.
2.3 Oocyte maturation
The oocytes used in the experiments were either obtained from pre-pubertal gilt ovaries collected at an abattoir (Farmland Foods Inc., Milan, MO) or were sow-derived oocytes purchased from A.R.T. (Monona, WI). Immature oocytes from gilt ovaries were aspirated from medium size (3–6 mm) follicles using an 18-gauge hypodermic needle attached to a 10-mL syringe. Oocytes with evenly dark cytoplasm and several layers of cumulus cells were then selected for maturation. Around 50 cumulus-oocyte complexes were placed in a well containing 500 mL of maturation medium, TCM 199 (Invitrogen, Grand Island, NY) with 3.05 mM glucose, 0.91 mM sodium pyruvate, 0.57 mM cysteine, 10 ng/mL epidermal growth factor (EGF), 0.5 mg/mL luteinizing hormone (LH), 0.5 mg/mL follicle stimulating hormone (FSH), 10 ng/mL gentamicin (APP Pharm, Schaumburg, IL), and 0.1% polyvinyl alcohol (PVA) for 42–44 h at 38.5°C, 5% CO2, in humidified air. The oocytes from sows were shipped overnight in maturation medium (TCM199 with 2.9 mM Hepes, 5 mg/mL insulin, 10 ng/mL EGF, 0.5 mg/mL p-FSH, 0.91 mM pyruvate, 0.5 mM cysteine, 10% porcine follicular fluid, 25 ng/mL gentamicin) and transferred into fresh medium at 24 hr. At the end of the maturation, the surrounding cumulus cells were removed from the oocytes by vortexing for 3 min in the presence of 0.03% hyaluronidase. Oocytes with a visible polar body were selected in manipulation medium (TCM199 with 0.6 mM NaHCO3, 2.9 mM Hepes, 30 mM NaCl, 10 ng/mL gentamicin, and 3 mg/mL bovine serum albumin [BSA]; osmolarity 305 mol/L) and then used for the experiments.
2.4 Oocyte activation
For parthenogenetic activation of pig oocytes, prepubertal gilt-derived oocytes were used. Different oocyte activation methods were used in the experiments. As conventional activation methods, either electrical or chemical activation was used. For electrical activation, mature oocytes were electroporated in activation medium (0.3 M mannitol, 1.0 mM CaCl2, 0.1 mM MgCl2, and 0.5 mM Hepes) by two direct-current (DC) pulses (1-sec interval) at 1.2 kV/cm for 30 μsec (using BTX Electro Cell Manipulator, Harvard Apparatus, Holliston, MA). For chemical activation, mature oocytes were incubated in Hepes-buffered Tyrode's lactate (TL-Hepes) medium in the presence of 200 μM thimerosal for 10 min in the dark followed by 8 mM dithiothreitol for 30 min. For TPEN-mediated activation, mature oocytes were incubated in TL-Hepes medium with various concentrations of TPEN (100 – 250 μM) for different durations. Two different approaches were used to activate oocytes with a combination of Ca2+ signaling and TPEN. First, mature oocytes were activated by conventional methods, either electrical or chemical, then incubated in TL-Hepes with 5 – 10 μM of TPEN. The other method was to provide a Ca2+ signal to mature oocytes by giving two direct-current (DC) pulses (1-sec interval) at 1.2 kV/cm for 30 μsec in a low Ca2+ media (0.3 M mannitol, 0.1 mM CaCl2, 0.1 mM MgCl2, and 0.5 mM Hepes), and then incubating the oocytes in TL-Hepes containing different concentrations of TPEN (100 – 200 μM) for various durations. Activated oocytes were washed three times in PZM3-MU1 [23, 24] then incubated at 38.5°C, 5% CO2 until the embryos are examined on either Day 6 or 7. At the end of the culture, where applicable, the frequency of cleaved embryos and blastocysts, and total cell number in the blastocysts were recorded. Hoechst 33342 (1.2 mg/mL) was used to stain nuclei and the embryos were then evaluated by epi-fluorescence microscopy.
2.5 Somatic cell nuclear transfer
Sow-derived mature oocytes were transferred into manipulation medium (TL-Hepes supplemented with 7.0 mg/mL cytochalasin B). By using micropipettes the polar body along with a portion of the adjacent cytoplasm was removed, and a donor cell was placed in the perivitelline space [25]. Genetically engineered cells, carrying a mutation on a gene, were used as donor cells for the in vitro study. For the in vivo study, either the same cell line used for the in vitro study or fetal fibroblast cells from Ossabaw miniature pigs served as donors. The reconstructed oocytes were then fused in a fusion medium (0.3 M mannitol, 0.1 mM CaCl2, 0.1 mM MgCl2, and 0.5 mM Hepes) by two DC pulses (1-sec interval) at 1.2 kV/cm for 30 μsec. After fusion, fused oocytes were fully activated using 200 μM thimerosal for 10 min in the dark followed by 8 mM dithiothreitol for 30 min [15]. Then a group of embryos were treated with either 5 μM TPEN for 30 min or 10 μM TPEN for 10 min. If necessary, embryos were then incubated in PZM3-MU1 with 0.5 mM Scriptaid, a histone deacetylase inhibitor, for 14–16 h. The embryos were then washed three times in PZM3-MU1 and incubated at 38.5°, under 5% CO2 in air until further evaluation At the end of the culture, SCNT-derived embryos were collected, and the frequency of blastocyst formation along with the total number of nuclei in the blastocysts was recorded.
2.6 Embryo transfer
For the in vivo study, at Day 1, the SCNT-derived embryos were surgically transferred into the ampullary-isthmic junction of surrogate gilts at 0 or 1 days after observed estrus. At the end of the gestation periods, the piglets were recovered through C-section.
2.7 Intracellular Ca2+ measurement
Matured oocytes were loaded with the Ca2+ indicator dye fura-2. For this purpose, they were incubated in the presence of 2 mM of the acetoxymethyl ester form of the dye and 0.02% pluronic F-127 (both from Invitrogen) for 40–50 min. The oocytes were transferred into a chamber with a cover-glass bottom; the chamber was then placed on the heated stage of an inverted microscope and changes in the intracellular free Ca2+ concentration in response to various TPEN treatments were recorded using InCyt Im2, a dual-wavelength fluorescence imaging system (Intracellular Imaging, Inc.; Cincinnati, OH). During measurements the emitted fluorescence was detected at 510 nm after exciting the dye alternately at 340 and 380 nm. The ratio of the two emitted fluorescence intensities was calculated and the data are presented as normalized fluorescence ratio values. In each treatment group, the measurements were repeated at least 5 times using different oocytes.
2.8 Intracellular Zn2+ measurement
After maturation, denuded oocytes were incubated in TL-Hepes medium in the presence of 2 μM FluoZin™-3, AM (Invitrogen) for 1 hour. Embryos were then washed in TL-Hepes medium and placed on an inverted fluorescent scope and viewed through a FITC filter (Nikon). Images of individual oocytes were taken at a constant exposure (3 seconds). Intensity of fluorescence from the images was quantified using the Image J software (NIH). The average level of Zn2+ in oocytes from each treatment was calculated from the measurement of eight oocytes.
2.9 Statistical analysis
Differences in the frequency of blastocyst formation were determined after analysis of variance (ANOVA) using the PROC MIX procedure of the Statistical Analysis System (SAS Institute, Cary, NC, USA) or Chi-square test. Percentage data were arcsin transformed prior to the ANOVA analysis. Average total numbers of nuclei in blastocysts and the level of Zn2+ in oocytes were compared by using the Student's T-test. Differences with P<0.05 were considered significant.
2.10 Experimental design
2.10.1 Effect of TPEN on intracellular Ca2+ and Zn2+ in oocytes
Since intracellular Ca2+ is a key element of oocyte activation and TPEN is known to lower the concentration of Zn2+, the level of intracellular Ca2+ and Zn2+ in mature oocytes after TPEN treatment was measured. These experiments will show if the target of TPEN is specific to Zn2+. In addition, in a separate experiment exogenous Zn2+ was added to the medium containing TPEN to identify whether the target of TPEN is Zn2+ specific. If the target of TPEN is specific to Zn2+, the effect of TPEN should be attenuated by the presence of exogenous Zn2+. Pronuclear formation of embryos after TPEN treatment with or without exogenous Zn2+ was monitored. Pronuclear formation was observed at 14 hours post TPEN treatment by staining the embryos with Hoechst 33342 (1.2 mg/mL) and viewing the epi-fluorescence.
2.10.2 Activation of oocytes with TPEN alone
To test if TPEN alone can successfully activate pig oocytes, various concentrations (100 – 250 μM) and incubation durations (45 min – 2.5 hours) of TPEN were used and subsequent development to blastocyst was recorded.
2.10.3 Treatment of activated oocytes with TPEN
To investigate whether TPEN treatment can enhance development of activated oocytes, various concentrations of TPEN (0.5 – 200 μM) were combined with conventional activation approaches used in SCNT. Optimum conditions identified from preliminary screening was used on activated oocytes and SCNT embryos, then subsequent development of the embryos was monitored.
2.10.4 TPEN as the main activating reagent
To investigate whether TPEN can be a main reagent of oocyte activation, oocytes which received the minimum amount of Ca2+ signaling through electroporation were incubated with different concentrations and durations of TPEN. Subsequent development was monitored to observe the effectiveness of the method.
2.10.5 In vivo development of SCNT embryos treated with TPEN
To test in vivo competency of SCNT embryos treated with TPEN, conventionally activated SCNT embryos were incubated with either 5 μM TPEN for 30 min or 10 μM TPEN for 10 min. A total of six embryo transfers were conducted; three groups were treated with Scriptaid, a histone deacetylase inhibitor.
3. Results
3.1 TPEN does not induce a Ca2+ increase in pig oocytes
To demonstrate that TPEN treatment stimulates signaling pathways independent of Ca2+ signaling, oocytes were exposed to various concentrations of TPEN and the changes in the intracellular Ca2+ levels were measured. As expected, no Ca2+ increase was detected when oocytes were exposed to 5, 10, or 200 μM TPEN. On the other hand, the oocytes incubated in the presence of thimerosal (which served as a positive control) showed massive Ca2+ increases (Figure 1). This indicates that the benefit of TPEN observed during oocyte activation is independent of Ca2+ signals.
Figure 1.
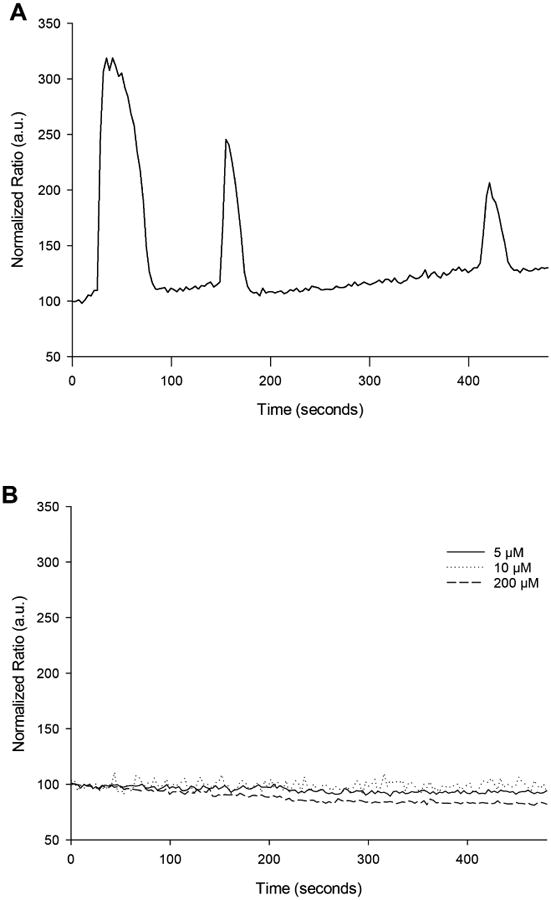
TPEN does not induce Ca2+ increase in MM oocytes. (A) Ca2+ increase after treating MII stage oocytes with thimerosal, a sulfhydryl-modifying agent. (B) Lack of Ca2+ increase after incubation in the presence of various concentrations of TPEN. The normalized fluorescence ratio on the Y-axis indicating Ca2+ changes is in arbitrary units.
3.2 Target of TPEN is Zn2+ specific
To demonstrate that TPEN can remove available Zn2+ in oocytes, mature pig oocytes were exposed to 200 μM of TPEN for 30 min then the level of intracellular Zn2+ was measured. As expected, TPEN treatment almost completely depleted available Zn2+ in the oocytes as we couldn't detect any fluorescent signal representing Zn2+ after the TPEN treatment (Figure 2A). When the level of fluorescence was quantified using Image J software, the difference between control and TPEN treated group was significant (p<0.05, Figure 2B). When the oocytes were treated with TPEN (200 μM, 30 min) with or without the presence of exogenous Zn2+, the presence of exogenous Zn2+ dramatically decreased the number of oocytes that formed a pronucleus at 14 hours post TPEN treatment; only one out of ten oocytes showed spontaneous pronuclear formation in the control group (Figure 2C).
Figure 2.
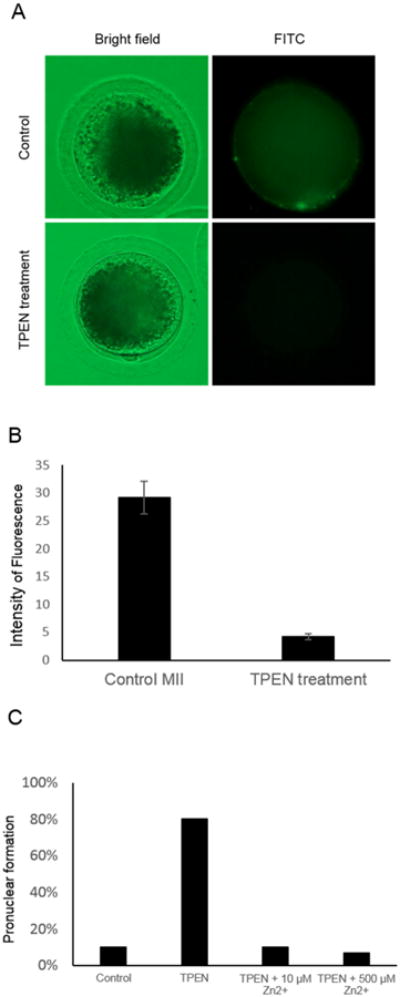
Target of TPEN is Zn2+ specific. (A) Mature oocytes incubated with TPEN have a lower concentration of intracellular Zn2+ compared to the control group. (B) Quantification of the level of Zn2+ in mature oocytes treated with TPEN and the control. Intensity of fluorescence was measured by Image J (n=8 in each group). The difference was significant (p<0.05) (C) Presence of exogenous Zn2+ inhibits oocyte activation induced by TPEN. Pronuclear formation after TPEN treatment was dramatically reduced with the addition of Zn2+.
3.3 Parthenogenetic activation of porcine oocytes by TPEN
We first tested if the known concentration/duration of TPEN (100 μM, 45 min) in the previous mouse study [21] had any effect on pig oocytes. The protocol could activate pig oocytes; however, the development was inferior to conventional artificial activation methods. The frequency of forming blastocysts on day 7 was comparable to the conventional oocyte activation method used (13% vs. 15%, respectively) but the rate of blastocyst formation was slower; no blastocyst formation was observed on day 5 with the TPEN treatment whereas 10% of activated embryos reached blastocysts in the control group. In addition, no expanded blastocysts were derived from the TPEN activation group on day 7, indicating lower developmental competency of the embryos derived from the TPEN activation method.
A wide range of concentrations (100 μM, 200 μM, and 250 μM) and durations (30 min to 2.5 hours) were tested to identify the optimum parameters for TPEN-induced oocyte activation (Table 1). Optimum embryo development was achieved by using 200 μM TPEN for 30 min and the frequency of blastocyst formation using the condition tended to be higher than a conventional activation method (p=0.1). However, a lower total number of nuclei in blastocysts was observed in the embryos derived from the optimal TPEN activation group (200 μM, 30 min), compared to the control (27.2 ± 1.92 vs. 32.6 ± 3.12, respectively). When the oocytes were incubated in 250 μM TPEN over 2.5 hours, a toxic effect of TPEN was observed. The 250 μM TPEN treatment over 2.5 hours had a negative effect on embryo development as there was no blastocyst formation. Here we identified an optimum concentration of TPEN which successfully induced development of activated oocytes, but the total number of nuclei in these embryos was lower compared to the conventional activation method.
Table 1.
Porcine oocytes activated with TPEN alone. When porcine oocytes were treated with TPEN, frequency of blastocyst formation was similar to conventional method; no beneficial effect was found. Longer exposure of oocytes to TPEN at high concentration was toxic. Frequency of blastocysts was recorded on Day 6.
| Treatment | Total number of oocytes | % blastocysts |
|---|---|---|
| Electroporation | 98 | 11.2% |
| 100 μM, 2 hr | 65 | 12.0% |
| 200 μM, 30 min | 25 | 23.0% |
| 200 μM, 1 hr | 79 | 20.0% |
| 250 μM, 2 hr | 30 | 10.0% |
| 250 μM, 2.5 hr | 25 | 0% |
3.4 Treatment of activated oocytes with TPEN can increase developmental potential in vitro
Next, we examined whether treating activated oocytes with TPEN could increase the efficiency of oocyte activation. When chemically activated oocytes were incubated with TPEN (100 μM, 45 min), no blastocyst formation was observed indicating the combination of the two methods is toxic to embryos. We hypothesized that the detrimental effect was due to excess stimuli from Ca2+ signaling and TPEN. Therefore, we introduced lower concentrations of TPEN after activating oocytes with a conventional method. Incubating activated oocytes with a lower concentration of TPEN increased the developmental potential of parthenogenetic embryos. Interestingly, when activated oocytes were incubated with a low concentration of TPEN (5-10 μM), the TPEN-treated group showed higher developmental potential compared to the control group. Specifically, the average percent of blastocyst formation in TPEN-treated oocytes (5 μM for 30 min) was 27.2 ± 1.7% but only 10.6 ± 2.5% developed to blastocyst in the control group (Table 2 and Figure 3). Moreover, the average number of nuclei in blastocysts was higher in TPEN-treated oocytes compared to the control group, which was activated by the thimerosal/DTT approach (Table 2). Next, the optimal concentration and duration of TPEN (5 μM, 30 min) was used to artificially activate oocytes reconstructed by SCNT. There was an increase in the frequency of blastocyst formation when activated SCNT oocytes were treated with TPEN (7.9% vs. 18.6%, respectively) (Table 3). This increase in development was comparable to the effect of Scriptaid, a histone deacetylase (HDAC) inhibitor, known to increase developmental potential of SCNT embryos. Thus, lower concentrations of TPEN (5–10 μM) can enhance embryo development when applied after conventional oocyte activation.
Table 2.
Incubation of TPEN can increase development potential of activated oocytes. Incubating activated porcine oocytes with lower concentrations of TPEN (5-10 μM) increased formation of blastocysts. Different letters indicate statistical difference (p<0.05). Frequency of blastocysts was recorded on Day 6.
| Treatment | Total number of embryos | % cleaved | % Blastocysts | Total cell number in blastocysts |
|---|---|---|---|---|
| THI/DTT | 113 | 59.3a | 10.6a | 28.2 ± 2.1a |
| THI/DTT + 5 μM TPEN, 30 min | 114 | 74.6b | 27.2b | 33.1 ± 2.6b |
| THI/DTT + 10 μM TPEN, 10 min | 83 | 81.9b | 21.7b | 31.2 ± 2.6b |
Figure 3.
Image of parthenogenetic embryos generated through the TPEN assisted activation method. Oocytes were activated by thimerosal/DTT, then half were incubated with 5 μM TPEN for 30 min. The pictures were taken on Day 6. (A) Control group. (B) Oocytes further activated with TPEN.
Table 3.
In vitro development of SCNT embryos treated with Scriptaid or TPEN alone. SCNT embryos were initially activated using thimerosal/DTT method. Treatment of TPEN can increase developmental potential of SCNT embryos compared to the control and compatible to Scriptaid treated embryos. Different letters indicate statistical difference (p<0.05). Frequency of blastocysts was recorded on Day 6.
| Treatment | Number of embryos | % Blastocysts |
|---|---|---|
| SCNT (control) | 76 | 7.9%a |
| SCNT + Scriptaid | 194 | 22.7%b |
| SCNT + 5 μM TPEN, 30 min | 102 | 18.6%b |
3.4 TPEN can serve as the main activating reagent
Because we hypothesized that a proper combination of Ca2+ signal and TPEN may be required to increase development of the activated oocytes, we tested if TPEN can be the main activating reagent. First, oocytes received an intracellular Ca2+ increase through electroporation; although the concentration of Ca2+ (0.1 mM) in the electroporation medium was not sufficient to induce oocyte activation. Then the oocytes were incubated with various concentrations of TPEN for various durations. The combination of a smaller Ca2+ increase and lower concentration of TPEN (10 μM) was not effective in inducing oocyte activation as no day 6 blastocysts were observed in the group. Surprisingly, a smaller Ca2+ increase followed by TPEN incubation at higher concentrations (100 μM or above) could successfully activate oocytes, and some combinations resulted in a superior in vitro development compared to the oocytes activated by a conventional method (Table 4). We have obtained the highest frequency of blastocysts, both day 6 and 7, when oocytes were activated by a smaller Ca2+ signal followed by incubating in 200 μM TPEN for 30 min.
Table 4.
Effect of TPEN as a main oocyte activating reagent. Different letters indicate statistical difference (p<0.05).
| Treatment | Number of embryos | Day 6 Blastocysts (%) | Day 7 Blastocysts (%) |
|---|---|---|---|
| Control - electroporation | 80 | 13.75a | 17.5a |
| 10 μM, 30 min | 40 | 0b | 5b |
| 10 μM, 1 hour | 40 | 15a | 22.5a |
| 10 μM, 2 hours | 40 | 15a | 25a |
| 100 μM, 10 min | 40 | 15a | 15a |
| 100 μM, 20 min | 40 | 25a | 30a |
| 200 μM, 30 min | 81 | 37.04c | 41.98c |
| 200 μM, 1 hour | 80 | 18.75a | 27.5a |
3.5 In vivo developmental competence of SCNT embryos following TPEN activation
To address in vivo competency of SCNT embryos whose development was stimulated by utilizing TPEN, a series of embryo transfers was performed. The embryos were transferred into six surrogates, two of which carried their pregnancy to term, each giving birth to two healthy piglets (Table 5). One of the term developments was from SCNT embryos that were not treated with a HDAC inhibitor, Scriptaid (Figure 4). Thus, TPEN treatment is compatible with term development after SCNT.
Table 5.
Result of embryo transfer.
| Treatment | Number of embryos transferred | Result |
|---|---|---|
| SCNT + 5 μM TPEN, 30 min + Scriptaid | 207 | 2 normal piglets were delivered |
| SCNT + 5 μM TPEN, 30 min + Scriptaid | 245 | Cycled |
| SCNT + 10 μM TPEN, 10 min + Scriptaid | 180 | Cycled |
| SCNT + 5 μM TPEN, 30 min | 280 | 2 normal piglets were delivered |
| SCNT + 5 μM TPEN, 30 min | 230 | Cycled |
| SCNT + 5 μM TPEN, 30 min | 280 | Cycled |
Figure 4.

Images of cloned pigs produced by this method. The animals were healthy and did not show any developmental abnormality.
4. Discussion
Artificial activation of oocytes is an essential process during SCNT. Unfortunately, current artificial activation methods used during SCNT are suboptimal as they cannot emulate the natural signaling pathway used by the fertilizing sperm. Most current artificial activation methods are able to induce only a single elevation in the intracellular free Ca2+ concentration of the oocyte [15, 26]; however, this single Ca2+ increase is thought to be less effective in degrading MPF compared to repetitive Ca2+ increases observed during fertilization [27]. This is believed to be a major reason for the low developmental potential of SCNT embryos [28]. Different strategies have been used to increase the efficiency of artificial oocyte activation; one of the most popular approaches being the reduction of MPF levels in the ooplasm using various inhibitors [16, 29]. However, due to potential toxicity of the chemicals applied, they are not widely used in cloned pig production.
Here we developed a system to increase the efficiency of oocyte activation by utilizing a membrane-permeable Zn2+ chelator, TPEN. The amount of Zn2+ in the oocyte increases during maturation and can regulate the timing of meiotic exit in mammalian oocytes [18]. This increase in the level of Zn2+ during oocyte maturation has also been reported in pigs [30]. It was suggested that the release of Zn2+ from FBXO43, a cytostatic factor with a Zn2+ binding domain, relieves APC/C from FBXO43-mediated inhibition, thus initiating the degradation of MPF [20]. Release of Zn2+ is observed during fertilization [22], suggesting that this is a natural process. By utilizing the Zn2+-specific chelator TPEN, we could stimulate the natural process of Zn2+ depletion seen during physiological oocyte activation, and thus increase the developmental potential of the activated oocytes.
The absence of cytosolic Ca2+ increase after a TPEN treatment indicates that the activation induced by the chelator does not utilize Ca2+ as a second messenger. Development of artificial oocyte activation procedures generally focused on inducing an increase in intracellular Ca2+ levels. Our strategy does not involve a Ca2+ increase but instead targets the signaling pathway downstream of the Ca2+ signal as the reduction in Zn2+ can directly affect the activity of APC/C thus by-passing the need of activating CaMKII. Treatment of oocytes with TPEN only reduced the level of Zn2+ but not Ca2+ indicating that the effect of TPEN is independent from Ca2+ signaling as previously described [21]. In addition, decrease in the number of embryos reaching the pronuclear stage when oocytes were treated with in the presence of exogenous Zn2+ indicates that the effect of TPEN is specific to Zn2+. This is similar to previous reports in pig oocytes [31].
Incubating mature porcine oocytes with TPEN could stimulate activation; however, the frequency of blastocyst formation was not superior to conventional Ca2+-mediated oocyte activation protocols. In fact, we observed fewer nuclei in blastocysts derived from the TPEN activation method. This is consistent with previous reports where mouse oocytes were activated by TPEN, and the efficiency was lower compared to natural sperm-induced activation [21] indicating that reducing the level of intercellular Zn2+ alone may not be enough to successfully induce oocyte activation. In the mouse study, a combination of Ca2+ increase and TPEN incubation had a deleterious effect on embryo development [21]. However, in those experiments SrCI2 was used to activate oocytes, a compound known to cause repetitive Ca2+ oscillations in mice [32]. TPEN was reported to disrupt Ca2+ signaling [18, 33]; therefore, we assumed that the detrimental effect of TPEN was due to its disruption of the oscillatory Ca2+ signal induced by SrCI2. We hypothesized that TPEN might be used in combination with an activation protocol which causes only a single Ca2+ increase. As we expected, proper concentration of TPEN (5 – 10 μM) could increase the developmental potential of the activated oocytes. Similarly, when we used TPEN as the main activating reagent, we also saw a significant increase in embryo development. This indicates that proper combinations of Ca2+ increase and Zn2+ depletion are effective in inducing meiotic exit of oocytes.
TPEN is known to be toxic and induces apoptosis in a concentration- and duration-dependent manner [34, 35]. Here, a similar effect was observed: long exposure to TPEN led to lower embryo development. This suggests that although a reduction in the intracellular Zn2+ concentration is important for oocyte activation, maintenance of a basal Zn2+ level is required for cellular survival. Our in vivo study indicates that we could produce viable clones using activation protocols that incorporate TPEN. The approach also allowed us to produce cloned piglets without treating SCNT embryos with Scriptaid, a HDAC inhibitor that is frequently used in SCNT protocols to increase developmental potential of SCNT embryos [36-39]. This full term development suggests that TPEN, at the concentration used in our experiments, is not toxic to the SCNT embryos. None of the animals displayed any health-related problems at birth as often seen in cloned animals. Moreover, they have already produced live progeny.
A recent study confirmed that TPEN can activate pig oocytes [31]. These authors demonstrated that events of oocyte activation such as cortical granule release and reduction in MPF occur when mature oocytes are incubated with TPEN. These events are a part of oocyte activation [40, 41] indicating that the depletion of Zn2+ by TPEN is sufficient to stimulate the machinery that is normally associated with meiotic resumption. However, the concentration of TPEN applied in the above-mentioned study was not effective in inducing embryo development as no blastocyst formation was observed. This is consistent with our findings as only higher concentrations of TPEN (higher than 100 μM) could activate pig oocytes and promote subsequent development.
In this study, the overall frequency of blastocysts from conventional activation or SCNT tended to be lower compared to our previous report [38]. This could be seasonal observation; however, we believe this is due to the difference in the source of oocytes. The majority of oocytes used in this study were collected from prepubertal gilts and it is known that such oocytes show poor development in pigs [3, 42]. Interestingly, the use of TPEN allowed us to observe a frequency of blastocysts in activated oocytes as high as 40%, indicating that TPEN activation can be particularly beneficial if oocytes with low developmental competency are used in the experiments.
In summary, our results demonstrate that TPEN, a Zn2+ chelator, can be utilized to increase the developmental potential of activated oocytes. When used in combination with electroporation after SCNT, the novel activation approach led to the production of live offspring and was also effective in enhancing overall proficiency of the technology.
Highlights.
A Zn2+ clelator, TPEN, can activate porcine oocytes
TPEN can enhance development of activated oocytes and SCNT embryos
TPEN can be a main oocyte activating reagent
TPEN does not induce a cytosolic Ca2+ increase in oocytes
TPEN-assisted oocyte activation method can be used to produce live clones
Acknowledgments
This study was partially funded by Food for the 21st Century, the National Institutes of Health by a grant to the National Swine Resource and Research Center (U42 OD011140) and by Agriculture and Food Research Initiative Competitive Grant no. 2011-67015-30006 from the USDA National Institute of Food and Agriculture (through ZM). We also acknowledge technical assistance provided by Mike L. Linville and Keith Giroux.
Footnotes
Conflict of Interest: The University of Missouri has filed a patent on using the above procedures to activate oocytes.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prather RS, Rowland RR, Ewen C, Trible B, Kerrigan M, Bawa B, et al. An intact sialoadhesin (Sn/SIGLEC1/CD169) is not required for attachment/internalization of the porcine reproductive and respiratory syndrome virus. Journal of virology. 2013;87:9538–46. doi: 10.1128/JVI.00177-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai L, Kang JX, Li R, Wang J, Witt WT, Yong HY, et al. Generation of cloned transgenic pigs rich in omega-3 fatty acids. Nature biotechnology. 2006;24:435–6. doi: 10.1038/nbt1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science (New York, NY) 2002;295:1089–92. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- 4.Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. The Journal of clinical investigation. 2008;118:1571–7. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline D, Kline JT. Repetitive calcium transients and the role of calcium in exocytosis and cell cycle activation in the mouse egg. Developmental biology. 1992;149:80–9. doi: 10.1016/0012-1606(92)90265-i. [DOI] [PubMed] [Google Scholar]
- 6.Swann K, Ozil JP. Dynamics of the calcium signal that triggers mammalian egg activation. International review of cytology. 1994;152:183–222. doi: 10.1016/s0074-7696(08)62557-7. [DOI] [PubMed] [Google Scholar]
- 7.Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, et al. PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development. 2002;129:3533–44. doi: 10.1242/dev.129.15.3533. [DOI] [PubMed] [Google Scholar]
- 8.Markoulaki S, Matson S, Ducibella T. Fertilization stimulates long-lasting oscillations of CaMKII activity in mouse eggs. Developmental biology. 2004;272:15–25. doi: 10.1016/j.ydbio.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Maller JL. Calcium elevation at fertilization coordinates phosphorylation of XErp1/Emi2 by Plx1 and CaMK II to release metaphase arrest by cytostatic factor. Current biology: CB. 2005;15:1458–68. doi: 10.1016/j.cub.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Rauh NR, Schmidt A, Bormann J, Nigg EA, Mayer TU. Calcium triggers exit from meiosis II by targeting the APC/C inhibitor XErp1 for degradation. Nature. 2005;437:1048–52. doi: 10.1038/nature04093. [DOI] [PubMed] [Google Scholar]
- 11.Hansen DV, Tung JJ, Jackson PK. CaMKII and polo-like kinase 1 sequentially phosphorylate the cytostatic factor Emi2/XErp1 to trigger its destruction and meiotic exit. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:608–13. doi: 10.1073/pnas.0509549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madgwick S, Hansen DV, Levasseur M, Jackson PK, Jones KT. Mouse Emi2 is required to enter meiosis II by reestablishing cyclin B1 during interkinesis. The Journal of cell biology. 2006;174:791–801. doi: 10.1083/jcb.200604140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones KT. Intracellular calcium in the fertilization and development of mammalian eggs. Clinical and experimental pharmacology & physiology. 2007;34:1084–9. doi: 10.1111/j.1440-1681.2007.04726.x. [DOI] [PubMed] [Google Scholar]
- 14.Vitullo AD, Ozil JP. Repetitive calcium stimuli drive meiotic resumption and pronuclear development during mouse oocyte activation. Developmental biology. 1992;151:128–36. doi: 10.1016/0012-1606(92)90220-b. [DOI] [PubMed] [Google Scholar]
- 15.Machaty Z, Wang WH, Day BN, Prather RS. Complete activation of porcine oocytes induced by the sulfhydryl reagent, thimerosal. Biology of reproduction. 1997;57:1123–7. doi: 10.1095/biolreprod57.5.1123. [DOI] [PubMed] [Google Scholar]
- 16.Nanassy L, Lee K, Javor A, Machaty Z. Effects of activation methods and culture conditions on development of parthenogenetic porcine embryos. Animal reproduction science. 2008;104:264–74. doi: 10.1016/j.anireprosci.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Nanassy L, Lee K, Javor A, Machaty Z. Changes in MPF and MAPK activities in porcine oocytes activated by different methods. Theriogenology. 2007;68:146–52. doi: 10.1016/j.theriogenology.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 18.Kim AM, Vogt S, O'Halloran TV, Woodruff TK. Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nature chemical biology. 2010;6:674–81. doi: 10.1038/nchembio.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Chai Y, Hannigan R, Bhogaraju VK, Machaca K. Zinc regulates the ability of Cdc25C to activate MPF/cdk1. Journal of cellular physiology. 2007;213:98–104. doi: 10.1002/jcp.21090. [DOI] [PubMed] [Google Scholar]
- 20.Bernhardt ML, Kong BY, Kim AM, O'Halloran TV, Woodruff TK. A zinc-dependent mechanism regulates meiotic progression in mammalian oocytes. Biology of reproduction. 2012;86:114. doi: 10.1095/biolreprod.111.097253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki T, Yoshida N, Suzuki E, Okuda E, Perry AC. Full-term mouse development by abolishing Zn2+-dependent metaphase II arrest without Ca2+ release. Development. 2010;137:2659–69. doi: 10.1242/dev.049791. [DOI] [PubMed] [Google Scholar]
- 22.Kim AM, Bernhardt ML, Kong BY, Ahn RW, Vogt S, Woodruff TK, et al. Zinc sparks are triggered by fertilization and facilitate cell cycle resumption in mammalian eggs. ACS chemical biology. 2011;6:716–23. doi: 10.1021/cb200084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshioka K, Suzuki C, Tanaka A, Anas IM, Iwamura S. Birth of piglets derived from porcine zygotes cultured in a chemically defined medium. Biology of reproduction. 2002;66:112–9. doi: 10.1095/biolreprod66.1.112. [DOI] [PubMed] [Google Scholar]
- 24.Bauer BK, Spate LD, Murphy CN, Prather RS. 1 Arginine Supplementation in vitro Increases Porcine Embryo Development and Affects mRNA Transcript Expression. Reproduction, Fertility and Development. 2010;23:107. [Google Scholar]
- 25.Lai L, Prather RS. Production of cloned pigs by using somatic cells as donors. Cloning and stem cells. 2003;5:233–41. doi: 10.1089/153623003772032754. [DOI] [PubMed] [Google Scholar]
- 26.Sun FZ, Hoyland J, Huang X, Mason W, Moor RM. A comparison of intracellular changes in porcine eggs after fertilization and electroactivation. Development. 1992;115:947–56. doi: 10.1242/dev.115.4.947. [DOI] [PubMed] [Google Scholar]
- 27.Kikuchi K, Naito K, Noguchi J, Shimada A, Kaneko H, Yamashita M, et al. Maturation/M-phase promoting factor: a regulator of aging in porcine oocytes. Biology of reproduction. 2000;63:715–22. doi: 10.1095/biolreprod63.3.715. [DOI] [PubMed] [Google Scholar]
- 28.Ducibella T, Huneau D, Angelichio E, Xu Z, Schultz RM, Kopf GS, et al. Egg-to-embryo transition is driven by differential responses to Ca(2+) oscillation number. Developmental biology. 2002;250:280–91. [PubMed] [Google Scholar]
- 29.Bing Y, Che L, Hirao Y, Takenouchi N, Rodriguez-Martinez H, Nagai T. Parthenogenetic activation and subsequent development of porcine oocytes activated by a combined electric pulse and butyrolactone I treatment. The Journal of reproduction and development. 2003;49:159–66. doi: 10.1262/jrd.49.159. [DOI] [PubMed] [Google Scholar]
- 30.Zhao MH, Kwon JW, Liang S, Kim SH, Li YH, Oh JS, et al. Zinc regulates meiotic resumption in porcine oocytes via a protein kinase C-related pathway. PloS one. 2014;9:e102097. doi: 10.1371/journal.pone.0102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao MH, Kim NH, Cui XS. Zinc depletion activates porcine metaphase II oocytes independently of the protein kinase C pathway. In vitro cellular & developmental biology Animal. 2014 doi: 10.1007/s11626-014-9784-8. [DOI] [PubMed] [Google Scholar]
- 32.Bos-Mikich A, Whittingham DG, Jones KT. Meiotic and mitotic Ca2+ oscillations affect cell composition in resulting blastocysts. Developmental biology. 1997;182:172–9. doi: 10.1006/dbio.1996.8468. [DOI] [PubMed] [Google Scholar]
- 33.Lawrence Y, Ozil JP, Swann K. The effects of a Ca2+ chelator and heavy-metal-ion chelators upon Ca2+ oscillations and activation at fertilization in mouse eggs suggest a role for repetitive Ca2+ increases. The Biochemical journal. 1998;335(Pt 2):335–42. doi: 10.1042/bj3350335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. TPEN induces apoptosis independently of zinc chelator activity in a model of acute lymphoblastic leukemia and ex vivo acute leukemia cells through oxidative stress and mitochondria caspase-3- and AIF-dependent pathways. Oxidative medicine and cellular longevity. 2012;2012:313275. doi: 10.1155/2012/313275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donadelli M, Dalla Pozza E, Costanzo C, Scupoli MT, Scarpa A, Palmieri M. Zinc depletion efficiently inhibits pancreatic cancer cell growth by increasing the ratio of antiproliferative/proliferative genes. Journal of cellular biochemistry. 2008;104:202–12. doi: 10.1002/jcb.21613. [DOI] [PubMed] [Google Scholar]
- 36.Lee K, Kwon DN, Ezashi T, Choi YJ, Park C, Ericsson AC, et al. Engraftment of human iPS cells and allogeneic porcine cells into pigs with inactivated RAG2 and accompanying severe combined immunodeficiency. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7260–5. doi: 10.1073/pnas.1406376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon DN, Lee K, Kang MJ, Choi YJ, Park C, Whyte JJ, et al. Production of biallelic CMP-Neu5Ac hydroxylase knock-out pigs. Scientific reports. 2013;3:1981. doi: 10.1038/srep01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee K, Redel BK, Spate L, Teson J, Brown AN, Park KW, et al. Piglets produced from cloned blastocysts cultured in vitro with GM-CSF. Molecular reproduction and development. 2013;80:145–54. doi: 10.1002/mrd.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao J, Ross JW, Hao Y, Spate LD, Walters EM, Samuel MS, et al. Significant improvement in cloning efficiency of an inbred miniature pig by histone deacetylase inhibitor treatment after somatic cell nuclear transfer. Biology of reproduction. 2009;81:525–30. doi: 10.1095/biolreprod.109.077016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ducibella T. The cortical reaction and development of activation competence in mammalian oocytes. Human reproduction update. 1996;2:29–42. doi: 10.1093/humupd/2.1.29. [DOI] [PubMed] [Google Scholar]
- 41.Ducibella T, Fissore R. The roles of Ca2+, downstream protein kinases, and oscillatory signaling in regulating fertilization and the activation of development. Developmental biology. 2008;315:257–79. doi: 10.1016/j.ydbio.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchal R, Feugang JM, Perreau C, Venturi E, Terqui M, Mermillod P. Meiotic and developmental competence of prepubertal and adult swine oocytes. Theriogenology. 2001;56:17–29. doi: 10.1016/s0093-691x(01)00539-8. [DOI] [PubMed] [Google Scholar]