

Abstract
Optic nerve head astrocytes (ONHAs) are the major glia cell type in the non-myelinated optic nerve head where they contribute critically to extracellular matrix synthesis during development and throughout life. In glaucoma, and in related disorders affecting the optic nerve and the optic nerve head, pathological changes include altered astrocyte gene and protein expression resulting in their activation and extracellular matrix remodeling. ONHAs are highly sensitive to mechanical and oxidative stress resulting in the initiation of axon damage early during pathogenesis. Furthermore, ONHAs are crucial for the maintenance of retinal ganglion cell physiology and function. Therefore, glioprotective strategies with the goal to preserve and/or restore the structural and functional viability of ONHA in order to slow glaucoma and related pathologies are of high clinical relevance. Herein, we describe the development of standardized methods that will allow for the systematic advancement of such glioprotective strategies. These include isolation, purification and culture of primary adult rat ONHAs, optimized immunocytochemical protocols for cell type validation, as well as plate reader-based assays determining cellular viability, proliferation and the intracellular redox state. We validated and standardized our protocols by performing a glioprotection study using primary ONHAs. Specifically, we measured protection against exogenously-applied oxidative stress using tert-butylhydroperoxide (tBHP) as a model of disease-mediated oxidative stress in the retina and optic nerve head by the prototypic antioxidant, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox). Levels of oxidative stress were increased in the response to exogenously applied tBHP and were assessed by 6-carboxy-2′, 7′ dichlorodihydrofluorescein diacetate (DCFDA) fluorescence. Normalized DCFDA fluorescence showed a maximal 5.1-fold increase; the half-maximal effect (EC50) for tBHP was 212 ± 25 μM. This was paralleled very effectively in the assays measuring cell death and cell viability with half-maximal effects of 241 ± 20 μM and 194 ± 5 μM for tBHP in the lactate dehydrogenase (LDH) release and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) conversion assays, respectively. Pre-treatment with 100 μM Trolox decreased the sensitivity of ONHAs to tBHP. Half-maximal effects increased to 396 ± 12 μM tBHP in the LDH release assay and to 383 ± 3 μM tBHP in the MTT assay. Vehicle treatment (0.1% v/v ethanol) did not significantly affect cellular responses to tBHP. Antioxidant treatment increases ONHA viability and reduces the deleterious effects of oxidative stress. Our experiments provide important feasibility data for utilizing primary rat ONHAs in plate reader-based assays assessing novel therapeutics for glioprotection of the optic nerve and the optic nerve head in glaucoma and related disorders. Furthermore, our novel, standardized protocols have the potential to be readily adapted to high-throughput and high-content testing strategies.
1. Introduction
Glaucoma is a multifactorial progressive ocular pathology, clinically presenting with damage to the retina and optic nerve (ON), ultimately leading to blindness (Casson et al., 2012). As the second leading cause of vision loss, glaucoma is estimated to affect more than 4 million individuals in the US alone (Klein and Klein, 2013) and affect an increasing number of patients worldwide (Foster et al., 2002; Quigley and Broman, 2006). However, the exact pathophysiological mechanisms underlying glaucoma still remain unknown (Bettin and Di Matteo, 2013; Chang and Goldberg, 2012). Elevated intraocular pressure (IOP) is the most clinically relevant biomarker for glaucoma and controlling IOP is currently the sole target of pharmaceutical and surgical intervention (Chang and Goldberg, 2012). While these therapies can lower IOP to slow ON damage, death of retinal ganglion cells (RGCs) and optic nerve head astrocytes (ONHAs) continues, resulting in progressive vision loss (Heijl et al., 2002; Quigley, 2011). In addition, IOP-lowering therapies often fail over time, have issues with patient compliance or have significant side effects, all of which are problematic in a chronic disease requiring long-term care. For some forms of glaucoma IOP-lowering therapies are ineffective altogether (Lee and Higginbotham, 2005). An increasing number of clinical and experimental studies suggest an IOP-independent mechanism of vision loss and highlight the need for complementary treatment approaches including neuroprotection (Chang and Goldberg, 2012; Farkas and Grosskreutz, 2001) and glioprotection (Noh et al., 2013; Qu and Jakobs, 2013). This is supported by a significant body of clinical evidence on normotensive glaucoma (Mozaffarieh and Flammer, 2013), and on glaucoma models where disease progression occurs without ocular hypertension (Mi et al., 2012). Indeed, it has been shown that neuroprotection can acieve preservation of visual function in the presence of continually elevated IOP as a disease-causing mechanism (Burroughs et al., 2011; Prokai-Tatrai et al., 2013).
Optic nerve head astrocytes (ONHAs) are the major glia cell type in the non-myelinated optic nerve head where they contribute critically to extracellular matrix synthesis (Hernandez, 2000). ONHAs exhibit complex intracellular signaling pathways (Kaja et al., 2015a), which are putative targets for glioprotection against oxidative stress. In glaucomatous retinopathy, changes in ONHAs include activation, migration, extracellular matrix remodeling, and alteration of gene and protein expression (Morrison et al., 2011).
Therefore, ONHAs are an important cellular target for drug discovery focused on glaucoma and related disorders affecting the ON and ON head. Specifically, rat primary ONHAs bear several advantages over ONHA cultures of human origin, foremost their lower genetic variability and their capacity for cryostorage.
We herein describe a complete experimental framework of optimized and validated protocols for plate reader-based assays for glioprotection using primary adult rat ONHAs.
These protocols include the ex vivo recovery and primary culture conditions of adult rat ONHAs, a detailed immunocytochemical characterization using a panel of relevant markers of glia cells, and protocols for plate reader-based assays determining cellular viability, proliferation, and intracellular redox state. The feasibility for testing drug candidates for glioprotection was established by testing the prototypic antioxidant 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) against exogenously-applied tert-butylhydroperoxide (tBHP)-induced oxidative stress, which is an established model for pathologic oxidative stress.
The purpose of the present study was to provide a framework and model system facilitating the discovery of novel therapeutic interventions for glaucoma and other pathologies of the ON and optic nerve head.
2. Materials and Supplies
For primary ONHA culture all buffers and media were obtained from Lonza (Walkersville, MD) except trypsin-EDTA (MediaTech Inc., Manassas, VA). Serum products were purchased from Gibco® (Life Technologies, Carlsbad, CA). Tissue culture plastics were from TPP® (Techno Plastic Products AG, Trasadingen, Switzerland; sourced from Midwest Scientific, St. Louis, MO), with the exception of tissue-culture coated black/clear bottom 96-well plates for fluorescent DCFDA assays (Nunc™; Thermo Fisher Scientific, Rockford, IL). Non-tissue culture-coated 96-well plates with either optical glass bottoms (for immunocytochemistry) or LDH assays were also from Nunc™ (Thermo Fisher Scientific, Rockford, IL).
Chemicals for coating of optical glass bottom 96-well plates were obtained from Sigma Aldrich Corp. (St. Louis, MO). Poly-L-lysine-coated 12 mm glass coverslips were purchased from BD Biosciences (San Jose, CA). All antibodies and dyes used for immunocytochemistry are commercially available and listed in Table 1.
Table 1.
Antibodies used for immunocytochemistry
| Catalog number | Manufacturer/Supplier | Target | Lot number | Dilution |
|---|---|---|---|---|
| ab416 | AbCam, Cambridge, MA | Rabbit polyclonal to EAAT1 | GR182524 | 1:200 |
| ab4674 | AbCam, Cambridge, MA | Chicken polyclonal to GFAP | GR1473 | 1:1,000 |
| ab11178 | AbCam, Cambridge, MA | Mouse monoclonal to S100β, clone SH-B1 | GR173178 | 1:1,000 |
| A-21429 | Life Technologies, Carlsbad, CA | Alexa Fluor® 555 goat anti-rabbit |
1107470 | 1:2,000 |
| A-21449 | Life Technologies, Carlsbad, CA | Alexa Fluor® 647 goat anti-chicken |
1081817 | 1:2,000 |
| A-10667 | Life Technologies, Carlsbad, CA | Alexa Fluor® 488 goat anti-mouse |
1304746 | 1:2,000 |
| D1306 | Life Technologies, Carlsbad, CA | 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | 1633083 | 1:50,000 |
| A12379 | Life Technologies, Carlsbad, CA | Alexa Fluor® 488 phalloidin | 1558760 | 1:50 |
tBHP (Sigma Aldrich Corp., St. Louis, MO) was applied exogenously to the cell culture medium and was used to chemically induce oxidative stress, in a defined an reproducible manner. DCFDA, calcein-AM, and MTT were purchased from Life Technologies (Carlsbad, CA).
3. Detailed Methods
3.1. Primary culture of rat optic nerve head astrocytes
The protocol for the studies presented herein was approved by the Institutional Animal Care and Use Committee at the University of Missouri – Kansas City, and was executed in accordance with the ARVO Statement on the Use of Animals in Ophthalmic and Visual Research.
Three month old male Brown Norway rats were euthanized and the optic nerve was carefully dissected and washed in a 35 mm dish containing ice-cold 0.1 M phosphate buffered saline pH 7.4 (PBS; Lonza, Walkersville, MD). Tissue was transferred into a second 35 mm dish containing 1 ml growth media, comprised of Dulbecco’s Modified Eagle’s Medium (Lonza, Walkersville, MD) supplemented with 20% fetal bovine serum (FBS; Gibco® Qualified; Life Technologies, Carlsbad, CA), 100 U/mL penicillin, and 100 μg/mL streptomycin (both from Lonza, Walkersville, MD). Optic nerve head tissue from 6 eyes was combined and subsequently cut into small pieces using a new disposable razor blade. The tissue suspension was transferred into a 5 ml microcentrifuge tube (Eppendorf North America, Hauppauge, NY) using a P1000 micropipette (Eppendorf North America, Hauppauge, NY) and digested for 20 min in a 37 °C water bath in growth media supplemented with 0.1% trypsin/EDTA in HBSS. Tissue was gently triturated every 5 min. with a P1000 micropipettor. Following digestion, the suspension was centrifuged in a swinging bucket centrifuge at 500 × g for 3 min. The supernatant was aspirated and the pellet resuspended in 1 mL growth media and triturated 15 × using a P1000 micropipettor. The cell suspension was then seeded into a 6 well plate (tissue equivalent from one animal, i.e. two optic nerve heads per well). Cells were monitored using a tissue culture microscope (DM IL, Leica Microsystems, Buffalo Grove, IL). At 7 days in vitro (DIV7; Fig. 1A), remaining tissue and media were aspirated by vacuum suction and fresh media was added. Media was subsequently refreshed every 72 hr. At DIV 14, when cells had reached approximately 90% confluency (Fig. 1A) ONHAs were sub-cultured into a T25 tissue culture flask (TPP®, Midwest Scientific, St. Louis, MO) by trypsinization (0.25% Trypsin, 2.21 mM EDTA in Hank’s Balanced Salt Solution; MediaTech Inc., Manassas, VA).
Figure 1.

A. Representative examples of ONHA culture after 6 days in vitro (DIV) prior to the first media change. Note the attached ONHAs and remaining tissue debris from the dissociation. The same culture at DIV14 after two media changes. ONHA passage 2 imaged 24 hr after seeding into a new tissue culture flask at a 1:5 dilution and the same culture, 5 days after seeding. B. ONHAs showed strong positive immunoreactivity for the astrocyte markers S100β, EAAT1, and GFAP. Representative, single confocal section is shown. Cells were from passage 5. Scale bar: 100 μm.
ONHA cultures were successfully maintained for more than 10 passages at a subculturing ratio of 1:5 every 72–96 hr (Fig. 1A). Passages 4 – 10 were used for the experiments described herein. ONHA cultures can be cryo-stored in complete media with 20% dimethylsulfoxide (DMSO) in the vapor phase of liquid nitrogen. We have successfully cryo-stored ONHA cultures for 2 years and have not been able to detect a functional difference between ONHA cultures before or after cyro-storage.
3.2. Immunocytochemistry and image acquisition
While astrocytes grow on non-coated glass coverslips, molecular substrates determine the morphological and functional properties of cells (Brewer, 1997; Brewer et al., 1993; He and Baas, 2003; Nam et al., 2007). In order to establish the potential for future co-culture experiments of ONHAs with neuronal cells, such as retinal ganglion cell which require substrate-coated glass for in vitro growth and differentiation, we tested various coverslip coatings to identify the optimal growth conditions for primary ONHAs.
Cells were seeded at 2,500 cells per well in proprietary substrate (CC2) coated optical glass bottom 96-well plates (Nunc™; Thermo Fisher Scientific, Rockford, IL) or optical glass bottom 96-well plates (Nunc™; Thermo Fisher Scientific, Rockford, IL) coated with either laminin, poly-L-lysine, poly-D-lysine, or poly-D-lysine/laminin (all from Sigma Aldrich Corporation, St. Louis, MO). After 48 hr in culture, cells were fixed for 15 min. in 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS) and stained with AlexaFluor® 488 phalloidin (1:50 dilution; Life Technologies, Carlsbad, CA) to label actin filaments and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Life Technologies, Carlsbad, CA) to label cell nuclei according to the manufacturer’s recommendations. Plates were imaged using a Cytation 3 imaging plate reader (Biotek, Winooski, VT) with a 63x objective and GFP and DAPI filter cubes. Images were acquired using the well-scan mosaic function in Gen5 software (Biotek, Winooski, VT). Qualitative analysis did not reveal any gross differences between the different tissue culture coatings (data not shown). Thus, primary ONHA culture is amenable to co-culture experiments with cell types requiring substrate-coated growth surfaces.
Immunocytochemistry was performed to positively identify cells as astrocytes using established astrocyte markers. The procedure was carried out essentially as described previously (Kaja et al., 2011; Kaja et al., 2012; Kaja et al., 2015a; Kaja et al., 2015b). Briefly, ONHAs were seeded onto 12 mm round poly-L-lysine coated glass coverslips (BD Biosciences, San Jose, CA) in a 24-well plate at a density of 7,500 cells. After two days, cells were rinsed in 500 μl PBS and subsequently fixed in 4% paraformaldehyde in PBS for 15 min. Coverslips were washed 3× in PBS and transferred into a humidified chamber using Dupont #5 forceps. Cells were incubated in blocking solution (1% bovine serum albumin [BSA], 10% donkey serum, 0.5% Triton-X100 in PBS) for 1 hr at room temperature. Blocking solution was aspirated by vacuum and the cells were incubated with primary antibody diluted in antibody diluent (1% BSA, 3% donkey serum, 0.5% Triton-X100, 0.005% NaN3 in PBS) overnight (16 hr) at 4 °C. All antibodies have previously been validated extensively for use in immunocytochemistry and are listed in Table 1. Coverslips were washed 3 × with PBS as above, and cells incubated with AlexaFluor®-labeled secondary antibodies (Table 1) in antibody diluent without NaN3 for 1 hr at room temperature in the dark. Cells were co-labeled with DAPI to label cell nuclei. Coverslips were subsequently washed 3 × with PBS, dipped in distilled, ultrapure (18 MΩ) H2O to remove residual salt and mounted on plain, frosted microscope slides (Mercedes Medical, Sarasota, FL) and mounted with Aqua-Poly/Mount (Polysciences Inc., Warrington, PA). Images were acquired using a Leica SP5X WLL microscope (Leica Microsystems Inc., Buffalo Grove, IL). Single optical sections are shown (Fig. 1B). All cells were immunoreactive for the calcium binding peptide S100β, the glutamate aspartate transporter (GLAST; a.k.a. excitatory amino acid transporter 1; EAAT1), and glial fibrillary acidic protein (GFAP; Fig. 1B), identifying them positively as astrocytes. Cells showed no immunoreactivity for myelin basic protein (an oligodendrocyte marker) and vimentin (a marker for fibroblasts and activated astrocytes; data not shown).
3.3. Induction of oxidative stress and drug treatment
Given the role of acute and chronically elevated cellular levels of oxidative stress in a number of neurodegenerative diseases, including glaucoma, we tested the feasibility of using primary ONHAs as a platform for screening drug candidates for glioprotection. We used chemically-induced, exogenously-applied oxidative stress as an in vitro insult to test the efficacy of the prototypic antioxidant, Trolox. We have previously validated the use of a standard oxidant, tBHP (Sigma Aldrich Corp.), for plate reader-based glioprotection studies using C6 astroglioma cells (Kaja et al., 2015b).
ONHAs were seeded in 96 well plates (TPP, Midwest Scientific, St. Louis, MO) at a density of 7,500 cells per well. 48 hr after seeding cells were exposed to increasing concentrations of tBHP (Sigma Aldrich Corp.) for 5 hr. For glioprotection studies, cells were pre-treated (1 hr) with the prototypic antioxidant Trolox (100 μM in 0.1% v/v ethanol; Sigma Aldrich Corp., St. Louis, MO) or ethanol vehicle (0.1%; Sigma Aldrich Corp., St. Louis, MO). Subsequently, plate reader-based assays quantifying ROS levels, cell viability, and cell proliferation were performed.
3.4. Assay measuring cellular ROS levels using DCFDA
ROS levels due to tBHP treatment were quantified using the membrane-permeable, fluorescent ROS indicator DCFDA essentially as previously described by us for neurons (Burroughs et al., 2012). ONHAs were seeded in 96-well plates were loaded with 10 μM DCFDA in complete media for 45 min and oxidative stress was chemically induced as described above. Cells were subsequently washed 2 × with 300 μl Hank’s Balanced Salt Solution (HBSS) supplemented with 2 mM CaCl2. DCFDA fluorescence was quantified using a multimodal plate reader (Synergy H1; Biotek, Winooski, VT) using the following settings: Excitation wavelength: λ = 490 nm, Emission wavelength: λ = 525 nm, 15 measurements per well, automatic gain. Fluorescence was blanked and normalized to the vehicle condition (no oxidative stress treatment; 0 μM tBHP). ROS levels reached a maximum of 5.8 ± 0.3 fold increase over the control condition (Fig. 2A; n = 5 plates per condition), when fitted using an asymmetric four-parameter non-linear curve fitting algorithm. Statistically significant increases of ROS levels over baseline were detected at tBHP concentrations of 150 μM tBHP or higher as determined by a One-Way ANVOVA with Bonferroni post-hoc test (Fig. 2A). The half-maximal effect (EC50) of tBHP on ROS levels was determined by fitting data points for each n individually and then calculating the group mean ± s.e.m. (224 ± 25 μM tBHP; n = 5; Fig. 2A).
Figure 2.
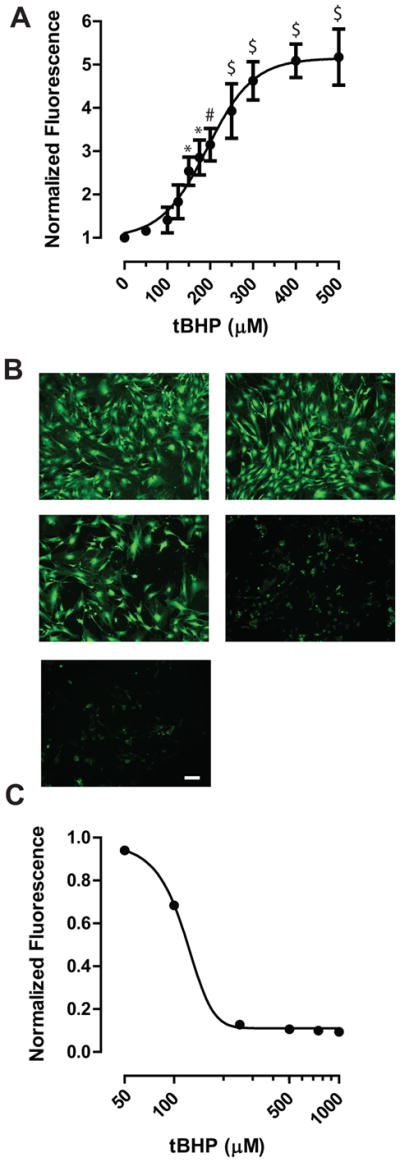
A. Generation of ROS was quantified using the DCFDA assay. tBHP treatment generated a maximal 5.8-fold increase compared to the control condition (0 μM tBHP). * P < 0.05, # P < 0.01, $ P < 0.001 compared to control (0 μM tBHP). Data is presented as mean ± s.e.m. (n = 5). Cells of passage 4 were used. B. Representative examples of calcein-labeled ONHAs. Increasing tBHP concentrations dose-dependently reduce calcein fluorescence. Cells of passage 5 were used. Scale bar: 25 μm. C. Calcein fluorescence was quantified using an H1 plate reader (Biotek) and the final, normalized data fitted using a four-parameter asymmetric nonlinear regression model. Data is presented as mean ± s.e.m. (n = 3).
3.5. Assays measuring cellular viability
3.5.1. Calcein-AM uptake assay
The calcein-AM uptake assay was performed essentially as described previously for primary culture of cortical neurons (Burroughs et al., 2012). Briefly, cells in 96-well plates were loaded with 5 μM calcein-AM (Life Technologies, Carlsbad, CA) in growth media from a 5 mM stock dissolved in DMSO for 1 hour at 37 °C/5% CO2/95% humidity. Subsequently, cells were washed 2 × with PBS. Representative images of calcein fluorescence in ONHAs (Fig. 2B) were taken using a widefield DM-IL microscope (Leica Microsystems, Buffalo Grove, IL) equipped with a CL2000 metal-halide light source (Leica Microsystems, Buffalo Grove, IL) and a MicroPublisher camera (QImaging, Surrey, BC). A tBHP dose-dependent reduction in calcein fluorescence was observed microscopically (Fig. 2B, C). Fluorescence was quantified using the same settings as described above for the DCFDA assay. Fluorescence data was blanked to the first column containing calcein-AM without cells and plotted using Prism 5.0 (GraphPad, La Jolla, CA) and fitted using four-parameter asymmetric non-linear regression analysis (Fig. 2C). The half-maximal effect (EC50) was determined by fitting each data set individually using the same regression analysis and determining the mean value ± s.e.m. (115.2 ± 2.3 μM tBHP; n = 3 plates; Fig. 2C).
3.5.2. MTT assay
The MTT assay was performed essentially as described by us previously for C6 astroglia cells, HT-22 cells, and primary cortical neuron culture (Burroughs et al., 2012; Kaja et al., 2011; Kaja et al., 2015b). Briefly, the medium was aspirated from the cells and replaced with 100 μl of 1.2 mM MTT in HBSS with calcium and magnesium (Lonza, Walkersville, MD) supplemented with 10 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) at pH 7.3. Plates were incubated in a 37 °C oven for 2 hr. Media was aspirated and cells were lysed with 100 μl dimethylsulfoxide (DMSO) and gentle shaking. The conversion of MTT was quantified by measuring absorbance at 570 nm (A570) using a Synergy H1 plate reader (Biotek).
Data was exported to Microsoft Excel (Microsoft Corp.) for processing, and blanked against a MTT- and tBHP-free column. Data was then normalized to the control condition (0 μM tBHP but with MTT). Outlier exclusion was applied to data points (single wells) for each insult (tBHP concentration; column of 8 wells). On average, 2 data points were excluded per 96-well plate; the maximal number of outliers excluded per plate were 5 wells. Assuming a Gaussian distribution of the data, we excluded any data point 2 standard deviations from the columnar mean. Normalized data were grouped by treatment condition and exported to Prism software (Graphpad) for statistical analysis and plotting. Non-linear regression using a four-parameter logistic equation with variable Hill slope was performed separately for each biological replicate (96-well plate) to determine the mean IC50 values for tBHP for each pre-treatment condition (control, vehicle, and Trolox). Data was analyzed statistically using 2-Way analysis of variance (ANOVA) and the Bonferroni post-hoc test with pre-treatment condition (control, vehicle, or Trolox) and insult (tBHP concentration) as variables. Statistical significance was defined as P < 0.05. In ONHAs, the IC50 for tBHP was 194 ± 4 μM (n=5; Fig. 3A) and not statistically significantly affected by vehicle treatment with 0.1% ethanol (191 ± 5 μM; n=5; Fig. 3A). In contrast, pre-treatment with 100 μM Trolox shifted the IC50 significantly rightward to 383 ± 5 μM (n=5; P < 0.001; Fig. 3A).
Figure 3.
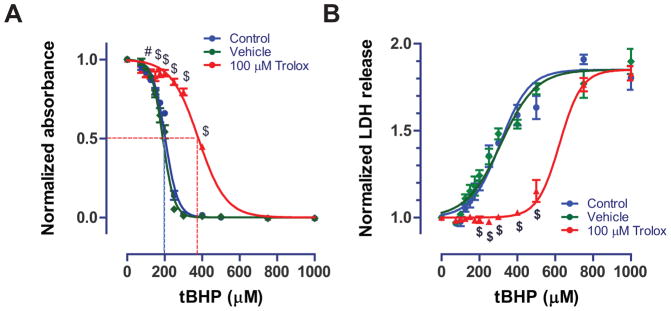
A. Dose-response curve for tBHP using the MTT assay. Trolox shifted the half-maximal effect (IC50) for tBHP rightward by approximately 180 μM. IC50 values obtained from the MTT assay were 194 μM, 191 μM, and 383 μM for control, vehicle (0.1% ethanol), and Trolox (100 μM), respectively. Dotted lines represent the IC50 value. Cells from passage 7 were used. B. Similar results were obtained when quantifying LDH release using our novel custom assay. The custom assay yielded reliable quantification of cellular LDH release, which increased in response to tBHP 1.85-fold. Trolox (100 μM) significantly shifted the half-maximal (EC50) values for tBHP (276, 281, and 550 μM for control, vehicle, and Trolox conditions). Cells from passage 7 were used. Data is shown as mean ± s.e.m. from five separate plates (n=5) and analyzed using ANOVA (P < 0.001). # P < 0.01, $ P < 0.001 based on a Bonferroni post-hoc test compared to the control condition (0 μM tBHP).
3.6. Assay measuring cell death: custom lactate dehydrogenase assay
We optimized a custom LDH release assay for ONHAs from the previously developed and validated protocol for glioprotection using C6 astroglioma cells (Kaja et al., 2015b). Following 5 hr exposure to tBHP, 50 μl of cell culture supernatant were transferred to a non-sterile, clear 96-well well plate (Nunc, Thermo Fisher Scientific, Waltham, MA). 50 μl Assay Buffer (2 mM iodonitrotetrazolium chloride, 3.2 mM β-nicotinamide adenine dinucleotide sodium salt, 160 mM lithium lactate, 7.5 μM 1-methoxyphenazine methosulfate in 0.2 M Tris-HCl, pH 8.2) were added. The Assay Buffer was prepared from stock solutions prepared in advance: Buffer A (2x; 4 mM INT in 0.2 M Tris-HCl, pH 8.2), Buffer B (2×; 6.4 mM NAD, 320 mM lithium lactate in 0.2 M Tris-HCl buffer, pH 8.2), and MPMS supplement (20,000×; 150 mM MPMS in 0.2 M Tris-HCl buffer, pH 8.2). Aliquots were stored frozen at −20 °C without loss of activity. Assay Buffer was prepared immediately prior to addition to supernatant samples.
Plates were incubated at room temperature in the dark for 1 hr. The reaction was stopped by addition of 50 μl 1 M acetic acid. LDH release was quantified by measuring absorbance at 490 nm (A490) using a Synergy H1 plate reader (Biotek, Winooski, VT). Data was exported to Microsoft Excel (Microsoft Corp.) for processing and normalized to the vehicle control condition (0 μM tBHP). Data analysis was performed analogously to that described for the MTT assay above.
We observed a maximal 1.85-fold increase of LDH levels after tBHP insult. The EC50 values for tBHP were 276 ± 31 μM, 281 ± 37 μM, and 550 ± 60 μM for control, vehicle and Trolox conditions respectively (n = 5; ANOVA P < 0.001; Fig. 3B). Vehicle (0.1% ethanol) had no statistically significant effect on LDH release while 100 μM Trolox shifted the EC50 significantly towards higher tBHP concentrations (P < 0.01).
4. Potential Pitfalls and Trouble Shooting
4.1. Purity of primary ONHA culture
Several methods for dissociation and culture of ONHAs have previously been described, mostly utilizing tissue from early postnatal or young adult rats (Kennedy and Lisak, 1980; Lukas and Wang, 2012; Mandal et al., 2010). While cultures from young animals often have significant contaminating cell types such as oligodendrocytes and pericytes (Kennedy and Lisak, 1980; Lukas and Wang, 2012), we obtained primary ONHA cultures of a single morphology. This can likely be attributed to two factors: 1.) The culture media used in our study (DMEM, 20% FBS) does not support oligodendrocyte or pericyte growth and these cells types are eliminated from the culture by passage 3; and 2.) various other cell types cannot be propagated from aged animals unlike those from early postnatal animals, in which final development and differentiation have not yet occurred. Furthermore, our detailed immunocytochemical analysis did not show immunoreactivity for markers of oligodendrocytes and fibroblasts, myelin basic protein and vimentin, respectively.
Based on these observations we recommend using cells of passage 4 or higher for experiments. We have tested cells up to 10 passages with no detectable change in morphological and/or functional properties.
Similarly, we have tested primary adult rat ONHA cultures from animals up to 9 months of age from different strains (Brown Norway, Wistar, Sprague Dawley) and did not find differences in yield, purity, morphology, or viability (data not shown). The choice of strain should, therefore, be based on the specific experimental question and compatibility with associated in vivo studies.
4.2. Choice of cell viability assays
There is a large number of cell viability assays that can be employed in studies of glioprotection or cytoprotection in general. These include the assays presented herein, which rely on intracellular enzyme function as well as vital dye stains such as trypan blue and neutral red (Chiba et al., 1998; Fotakis and Timbrell, 2006; Jauregui et al., 1981; Lobner, 2000; Weyermann et al., 2005). While vital dye stains such as trypan blue provide an easy visualization, enzymatic assays provide lower variability, greater accuracy, and ease of automation (Jauregui et al., 1981). Furthermore, different assays show selective sensitivity to different insults and cytoprotectants depending on their specific mechanisms of action (Chiba et al., 1998; Fotakis and Timbrell, 2006; Lobner, 2000; Weyermann et al., 2005). For instance, the LDH assay may yield misleading results if an insult only acts on intracellular pathways without compromising membrane integrity (Weyermann et al., 2005). Therefore, suitability of the assays must be determined for each insult and cytoprotectant. For primary ONHAs exposed to tBHP, as presented herein, calcein-AM uptake as well as LDH and MTT assays show comparable results, providing low-variability readouts for cell death in response to oxidative stress and cytoprotection by Trolox.
4.3. Assays measuring cellular viability: MTT assay
The MTT assay is a well-established assay for cell viability and proliferation (Stoddart, 2010). Several different protocols exist that utilize different reagents for cell lysis after incubation with MTT. We have tested DMSO, isopropanol, 0.1 M sodium dodecyl sulfate (SDS) with 10 mM HCl, and Triton X-100 and the achieved the best and most consistent results with DMSO. Use of isopropanol was discontinued due to significant fixation of the cells, resulting in heterogeneous distribution of the colored dye. Fixation was less with 0.1 M SDS with 10 mM HCl but even slight excess acidification can easily revert the reduced MTT dye to its uncolored form. There are also reports in the literature of lysis with solutions containing up to 50% dimethylformamide (Takahashi et al., 2002), however, we refrained from testing dimethylformamide given its toxicity profile. Optimizations should be performed when working with new cell types. Complete lysis and consistent homogeneity of the assay product is critical for consistent and reproducible data and users must optimize the vigor and time required for shaking the plates. For DMSO, shaking at 180 rpm for 2 – 3 minutes is sufficient for complete cell lysis.
The MTT assay can also be adapted such that the reagent is added to the complete media after insult treatment. The obvious advantage of this adaptation is to decrease the number of washes inflicted on the treated cells and maintenance of sterility. We, however, observed greater variability of the assay using this modification, likely due to one or more of the following parameters that can affect experimental outcomes: 1.) Any pretreatment drug or insult applied to the cells will still be present during the assay incubation time, effectively prolonging exposure and resulting in possible interference with assay components or the enzymatic reaction. 2.) Phenol red present in the complete media will greatly increase absorbance readings at 570 nm. Phenol red-free media may be used, but it should be noted that many suppliers of preformulated media have different formulations for media with or without phenol red, for example standard DMEM from Lonza (Walkersville, MD) includes glutamine while the phenol red free formulation is only available without glutamine.
Our protocol has the advantage of a thorough HBSS wash step in the form of the MTT assay buffer to remove any phenol red remaining from the complete media. This wash step removes chemicals added previously to induce or prevent cellular damage, thus stopping or slowing the specific treatment, and preserves the remaining cells under standard cell culture maintenance conditions suitable for assaying cell number in response to a specific insult.
4.4. Lactate dehydrogenase assay
4.4.1. Preparation of Assay Buffer and Execution of the Assay
Components of the assay buffer can be prepared in advance, scaled to users’ needs, and stored at −20 °C without detectable loss of activity. Assay Buffer components should be filtered to remove particulates that may lead to formation of aggregates/precipitates during the assay reaction. Buffer A solution should be clear to slightly yellow; heating or extensive mixing (> 15 min) during preparation can lead to the formation of a black precipitate that will interfere with the assay. In that case, a new solution should be prepared. Buffer B solution is clear when freshly prepared but may color to light yellow after a few days frozen. Sodium lactate can be used as an alternative for lithium lactate when preparing Buffer B without a detectable difference (data not shown). The MPMS supplement is a dark brown to purple solution, which may occasionally appear grainy, but this does not affect assay quality if the supplement is thoroughly mixed prior to addition to the Assay Reagent. Alternatively, the MPMS supplement can be mixed 1:1 with 0.2 M Tris-HCl, pH 8.2, centrifuged, and added at a concentration of 1:10,000 to Buffers A and B (75 mM). The Assay Buffer should be prepared immediately prior to performing the assay, as pre-mixing is known to generate fine yellow crystals or nucleations, likely due to a slow reaction of the MPMS with INT in both cell-free (Goodwin et al., 1995) and cellular systems (Scudiero et al., 1988).
When stopping the reaction, excess addition of acetic acid must be avoided. The reduced dye (purple) can spontaneously oxidize back to the initial reactant (light yellow) at pH < 4.0.
4.4.2. Need for appropriate sera and cell-free controls
Besides specific cell type properties, several other factors can impact the levels of LDH release. For instance, sera such as fetal bovine serum used in cell growth media often contain LDH and may contribute to high baseline LDH levels. Furthermore, it has not yet been clearly established how heat inactivation of sera affects the serum LDH activity. Therefore, it would be advisable to test the endogenous activity of serum by performing several cell-free serum and/or complete media controls. Typically, complete media LDH levels are similar to the LDH levels of untreated cells (data not shown).
4.4.3. Cell-specific optimization
We have previously validated our custom LDH assay in C6 astroglioma cells (Kaja et al., 2015b). Herein, we show that the assay can be adapted readily to other cell types, including primary cells. However, it is necessary to optimize the assay for specific cell types. This should include optimization of seeding density and adjustment for the time in culture prior to the assay, a time course analysis of LDH release in response to tBHP and/or other chemicals used to modify cellular viability, and a kinetic analysis in order to determine the linear range of the assay. For ONHAs, we determined the linear range of the LDH assay by reading the Assay Plate every 2 min for 8 hr (data not shown). Absorbance readings plateau at around 2 hr for high tBHP concentrations, and at around 5 hr for the control condition. We determined the slopes for the line of best fit for time period from 30 min to 1.5 hr in order to determine the linearity of the assay. The goodness of fit was R2 > 0.99 for all controls and tBHP conditions (data not shown).
4.4.4. Absorbance measurements
After addition of acetic acid, LDH plates can be quantified after briefly mixing on an orbital shaker. However, samples are stable when stored at room temperature overnight (data not shown), and frozen samples are stable for at least a week (data not shown; Nachlas et al., 1960).
5. Conclusion
We herein present a comprehensive set of protocols describing the primary culture of adult rat ONHAs and their use in plate reader-based assays for glioprotection. Our data provide important feasibility data for the use of primary rat ONHAs and plate reader-based assays in drug discovery targeting glaucoma and disorders of the ON and the optic nerve head. Furthermore, conversion of these novel standardized protocols to high-throughput and high-content platforms is readily attainable.
Highlights.
Novel method for primary adult rat optic nerve head astrocyte (ONHA) culture
Optimized immunocytochemical analysis of astrocyte markers in primary ONHA culture
Novel protocols for plate reader-based cell viability and proliferation assays using primary ONHA cultures
Validation of a novel custom lactate dehydrogenase assay in ONHAs
Acknowledgments
Research reported in this publication was supported by grants from the National Eye Institute (EY022774), the National Institute on Aging (AG010485, AG022550 and AG027956), the National Center for Research Resources and National Institute of General Medical Sciences (RR022570 and RR027093) of the National Institutes of Health (PK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support by the Felix and Carmen Sabates Missouri Endowed Chair in Vision Research, a Challenge Grant from Research to Prevent Blindness and the Vision Research Foundation of Kansas City (PK) is gratefully acknowledged. The authors thank Margaret, Richard and Sara Koulen for their generous support and encouragement.
List of abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- DCFDA
6-carboxy-2′, 7′ dichlorodihydrofluorescein diacetate
- DIV
days in vitro
- DMSO
dimethylsulfoxide
- EAAT1
excitatory amino acid transporter 1
- EtOH
ethanol
- GFAP
glial fibrillary acidic protein
- GLAST
glutamate/aspartate transporter
- HBSS
Hank’s Balanced Salt Solution
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- INT
iodonitrotetrazolium chloride
- IOP
intraocular pressure
- LDH
lactate dehydrogenase
- MPMS
1-methoxyphenazine methosulfate
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAD
beta-nicotinamide adenine dinucleotide sodium salt
- ON
optic nerve
- ONHA
optic nerve head astrocytes
- PBS
phosphate-buffered saline
- RGC
retinal ganglion cell
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- tBHP
tert-butylhydroperoxide
- Trolox
6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bettin P, Di Matteo F. Glaucoma: present challenges and future trends. Ophthalmic Research. 2013:50. doi: 10.1159/000348736. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–55. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Burroughs S, Duncan R, Rayudu P, Kandula P, Payne A, Clark J, Koulen P, Kaja S. Plate reader-based assays for measuring cell viability, neuroprotection and calcium in primary neuronal cultures. J Neurosci Methods. 2012:203. doi: 10.1016/j.jneumeth.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs S, Kaja S, Koulen P. Quantification of deficits in spatial visual function of mouse models for glaucoma. Investigative ophthalmology & visual science. 2011:52. doi: 10.1167/iovs.10-7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Goldberg J. Glaucoma 2.0: neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology. 2012:119. doi: 10.1016/j.ophtha.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Kawakami K, Tohyama K. Simultaneous evaluation of cell viability by neutral red, MTT and crystal violet staining assays of the same cells. Toxicology in vitro: an international journal published in association with BIBRA. 1998;12:251–8. doi: 10.1016/s0887-2333(97)00107-0. [DOI] [PubMed] [Google Scholar]
- Farkas RH, Grosskreutz CL. Apoptosis, neuroprotection, and retinal ganglion cell death: an overview. International ophthalmology clinics. 2001;41:111–30. doi: 10.1097/00004397-200101000-00011. [DOI] [PubMed] [Google Scholar]
- Foster PJ, Buhrmann R, Quigley HA, Johnson GJ. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. 2002;86:238–42. doi: 10.1136/bjo.86.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotakis G, Timbrell JA. In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett. 2006;160:171–7. doi: 10.1016/j.toxlet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Goodwin GW, Arteaga JR, Taegtmeyer H. Glycogen turnover in the isolated working rat heart. The Journal of biological chemistry. 1995;270:9234–40. doi: 10.1074/jbc.270.16.9234. [DOI] [PubMed] [Google Scholar]
- He Y, Baas PW. Growing and working with peripheral neurons. Methods Cell Biol. 2003;71:17–35. doi: 10.1016/s0091-679x(03)01002-1. [DOI] [PubMed] [Google Scholar]
- Heijl A, Leske MC, Bengtsson B, Hyman L, Hussein M. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002;120:1268–79. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- Hernandez M. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Progress in Retinal and Eye Research. 2000:19. doi: 10.1016/s1350-9462(99)00017-8. [DOI] [PubMed] [Google Scholar]
- Jauregui HO, Hayner NT, Driscoll JL, Williams-Holland R, Lipsky MH, Galletti PM. Trypan blue dye uptake and lactate dehydrogenase in adult rat hepatocytes--freshly isolated cells, cell suspensions, and primary monolayer cultures. In Vitro. 1981;17:1100–10. doi: 10.1007/BF02618612. [DOI] [PubMed] [Google Scholar]
- Kaja S, Duncan R, Longoria S, Hilgenberg J, Payne A, Desai N, Parikh R, Burroughs S, Gregg E, Goad D, Koulen P. Novel mechanism of increased Ca2+ release following oxidative stress in neuronal cells involves type 2 inositol-1,4,5-trisphosphate receptors. Neuroscience. 2011:175. doi: 10.1016/j.neuroscience.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Mafe O, Parikh R, Kandula P, Reddy C, Gregg E, Xin H, Mitchell P, Grillo M, Koulen P. Distribution and function of polycystin-2 in mouse retinal ganglion cells. Neuroscience. 2012:202. doi: 10.1016/j.neuroscience.2011.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Payne AJ, Patel KR, Naumchuk Y, Koulen P. Differential subcellular Ca(2+) signaling in a highly specialized subpopulation of astrocytes. Experimental Neurology. 2015a:265. doi: 10.1016/j.expneurol.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Payne AJ, Singh T, Ghuman JK, Sieck EK, Koulen P. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. Journal of Pharmacological and Toxicological Methods. 2015b:73. doi: 10.1016/j.vascn.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PG, Lisak RP. Astrocytes and oligodendrocytes in dissociated cell culture of adult rat optic nerve. Neurosci Lett. 1980;16:229–33. doi: 10.1016/0304-3940(80)90002-6. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE. The prevalence of age-related eye diseases and visual impairment in aging: current estimates. Invest Ophthalmol Vis Sci. 2013;54:ORSF5–ORSF13. doi: 10.1167/iovs.13-12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DA, Higginbotham EJ. Glaucoma and its treatment: a review. American journal of health-system pharmacy: AJHP: official journal of the American Society of Health-System Pharmacists. 2005;62:691–9. doi: 10.1093/ajhp/62.7.691. [DOI] [PubMed] [Google Scholar]
- Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods. 2000;96:147–52. doi: 10.1016/s0165-0270(99)00193-4. [DOI] [PubMed] [Google Scholar]
- Lukas TJ, Wang AL. Isolation and culture of astrocytes from the retina and optic nerve. Methods Mol Biol. 2012;814:105–15. doi: 10.1007/978-1-61779-452-0_8. [DOI] [PubMed] [Google Scholar]
- Mandal A, Shahidullah M, Delamere NA. Hydrostatic pressure-induced release of stored calcium in cultured rat optic nerve head astrocytes. Investigative ophthalmology & visual science. 2010;51:3129–38. doi: 10.1167/iovs.09-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi X, Zhang X, Feng Q, Lo A, Chung S, So K. Progressive retinal degeneration in transgenic mice with overexpression of endothelin-1 in vascular endothelial cells. Investigative ophthalmology & visual science. 2012:53. doi: 10.1167/iovs.12-9999. [DOI] [PubMed] [Google Scholar]
- Morrison J, Guo W, Johnson E. Pathophysiology of human glaucomatous optic nerve damage: insights from rodent models of glaucoma. Experimental eye research. 2011:93. doi: 10.1016/j.exer.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarieh M, Flammer J. New insights in the pathogenesis and treatment of normal tension glaucoma. New insights in the pathogenesis and treatment of normal tension glaucoma. 2013:13. doi: 10.1016/j.coph.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Nachlas MM, Margulies SI, Goldberg JD, Seligman AM. The determination of lactic dehydrogenase with a tetrazolium salt. Anal Biochem. 1960;1:317–26. doi: 10.1016/0003-2697(60)90029-4. [DOI] [PubMed] [Google Scholar]
- Nam Y, Brewer GJ, Wheeler BC. Development of astroglial cells in patterned neuronal cultures. J Biomater Sci Polym Ed. 2007;18:1091–100. doi: 10.1163/156856207781494430. [DOI] [PubMed] [Google Scholar]
- Noh YH, Kim KY, Shim MS, Choi SH, Choi S, Ellisman MH, Weinreb RN, Perkins GA, Ju WK. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013;4:e820. doi: 10.1038/cddis.2013.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai-Tatrai K, Xin H, Nguyen V, Szarka S, Blazics B, Prokai L, Koulen P. 17β-estradiol eye drops protect the retinal ganglion cell layer and preserve visual function in an in vivo model of glaucoma. Molecular pharmaceutics. 2013;10:3253–61. doi: 10.1021/mp400313u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Jakobs TC. The Time Course of Gene Expression during Reactive Gliosis in the Optic Nerve. PLoS One. 2013;8:e67094. doi: 10.1371/journal.pone.0067094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA. Glaucoma. Lancet. 2011;377:1367–77. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. The British journal of ophthalmology. 2006;90:262–7. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer research. 1988;48:4827–33. [PubMed] [Google Scholar]
- Takahashi S, Abe T, Gotoh J, Fukuuchi Y. Substrate-dependence of reduction of MTT: a tetrazolium dye differs in cultured astroglia and neurons. Neurochemistry international. 2002;40:441–8. doi: 10.1016/s0197-0186(01)00097-3. [DOI] [PubMed] [Google Scholar]
- Weyermann J, Lochmann D, Zimmer A. A practical note on the use of cytotoxicity assays. Int J Pharm. 2005;288:369–76. doi: 10.1016/j.ijpharm.2004.09.018. [DOI] [PubMed] [Google Scholar]