

Abstract
Circulating von Willebrand factor (VWF) adopts a closed conformation that shields the platelet glycoprotein Ibα (GPIbα) binding site in the VWF-A1 domain. Immobilized at sites of vascular injury, VWF is activated by its interaction with collagen and the exertion of increased hemodynamic forces. Studies on native VWF strings and isolated A1 domains suggest the existence of multiple A1 binding states in different biophysical contexts. In this single-molecule study, we have used a biomembrane force probe (BFP) and a flow chamber to identify and characterize a collagen binding induced conformation with a higher affinity to platelet GPIbα. As force increases, our results show that collagen binding increases the stability of GPIbα bond with both VWF and isolated A1 domain. However, the collagen 2D binding affinity for VWF-A3 domain is 10 times of that for A1 domain, suggesting the initial VWF capture is mediated by A3–collagen interaction while A1–collagen regulates the subsequent VWF activation. Our results revealed the molecular mechanism of collagen-regulated, A1-mediated platelet adhesion enhancement. Characterization of different A1 states provides insights into binding heterogeneity of VWF in different scenarios of inflammation and thrombosis.
Keywords: Platelets, von Willebrand factor, Single bond, Glycoprotein Ibα, Microfluidics
Introduction
Platelets adhesion at sites of vascular activation or injury is synergistically orchestrated by biomechanical factors (flow and force) and biochemical factors (thrombogenic protein exposure and agonist release) [1-3]. At >500 s-1 shear rates, mostly seen in arteries, initial tethering and translocation of platelets to the vessel wall is primarily mediated by the interaction of the receptor complex glycoprotein (GP)Ib-IX to a multimeric adhesive protein – von Willebrand factor (VWF). This plasma protein is mostly seen to deposit at the injury-exposed extracellular matrix (ECM), particularly binding to collagen fibers, or anchor to locally stimulated endothelium [4-6].
The mature VWF monomer consists of a 2,050-residue subunit that contains multiple copies of A, C, and D type domains [7]. The A1 domain contains binding sites for GPIbα and collagen types I, III, and VI [8-12], while its homologous A3 domain only binds to collagen fibrils types I and III [13-15]. VWF multimers adopt a folded, globular conformation that shields the GPIbα binding sites in the A1 domain, preventing spontaneous binding to platelets in circulation (cf phase I, Fig. S1). The current view of VWF activation in physiological condition is that the increased shear stress at the vessel wall unfolds VWF upon its immobilization at sites of vascular injury via the A3–collagen interaction [7]. Recent in vitro biophysical studies using purified plasma (p)VWF and isolated A1 domain converge to a consensus on the role of mechanical force in VWF activation that includes two mechanisms: 1) elongational flow stretches globular auto-inhibited VWF into a globally extended conformation, revealed by microfluidic studies with VWF fibers [16-19]; 2) tensile force induces local conformational change within the A1 domain and upregulates its binding states, revealed by single-bond studies with recombinant A1 variants [20,21].
In addition to force, we previously demonstrated that the binding of A1 domain to collagen types I and III induces a conformational change in the A1 structure [11]. This suggests that collagen does more than merely anchors circulating pVWF. Therefore, we hypothesized that collagen directly modulates the force-dependent binding of A1 domain to GPIbα by inducing the transition of the A1 domain from a low to a higher binding state. Recently, we used a biomembrane force probe (BFP) to characterize distinct force-dependent kinetics of GPIbα dissociation from two widely used A1 constructs: 1238-A1 and 1261-A1 (N-termini starts at residues 1238 or 1261, representing N-longer or N-shorter A1 constructs, respectively). The inclusion of the N-terminal sequence Q1238-E1260, the segment between D3 and A1 domains, stabilizes the 1238-A1–GPIbα interaction against force by forming a catch bond (whose lifetime increases with increasing force) that enables stable platelet translocation on A1; whereas the exclusion of Q1238-E1260 weakens the 1261-A1–GPIbα interaction by forming a slip-only bond (whose lifetime decreases with increasing force) that does not support stable translocation of platelets under high shear [21].
Here we characterized the force-dependent kinetics of GPIbα dissociation from A1 of different N-terminal lengths and immobilization on different surfaces. Binding to collagen not only globally enhances the affinity for both 1238-A1 and 1261-A1, but also switch the slippery state of 1261-A1 into a catchy state. This finding sheds light to the binding state transition upon binding to a collagen surface and provides an explanation for a puzzle in VWF biology – the heterogeneous phenotypes of VWF binding in different contexts [6].
Results
We used a BFP to measure the adhesion frequency and bond lifetime between a VWF-or A1-coated glass bead (probe) attached to the apex of a micropipette-aspirated red blood cell (RBC) (Fig. 1A-C, left) and a GPIbα-bearing immobilized glass bead (target) aspirated by an apposing micropipette (Fig. 1A and C, right). As previously described [21], for better molecular orientation with purified protein, GPIb-IX was captured from platelet lysates by a monoclonal antibody (mAb) WM23 (Fig. 1B and C). After a controlled contact, the BFP detected the absence (Fig. 1D) or the presence (Fig. 1E) of adhesion from the force signal upon target retraction and calculated an adhesion frequency (Pa) from repeated tests (Fig. 1F). According to Poisson statistics [22], in rare (<20%) adhesion events, the bead was pulled by (most likely) a single A1–GPIbα bond, which was then held at a constant force until bond dissociation occurred.
Figure 1. BFP assay and control experiments.
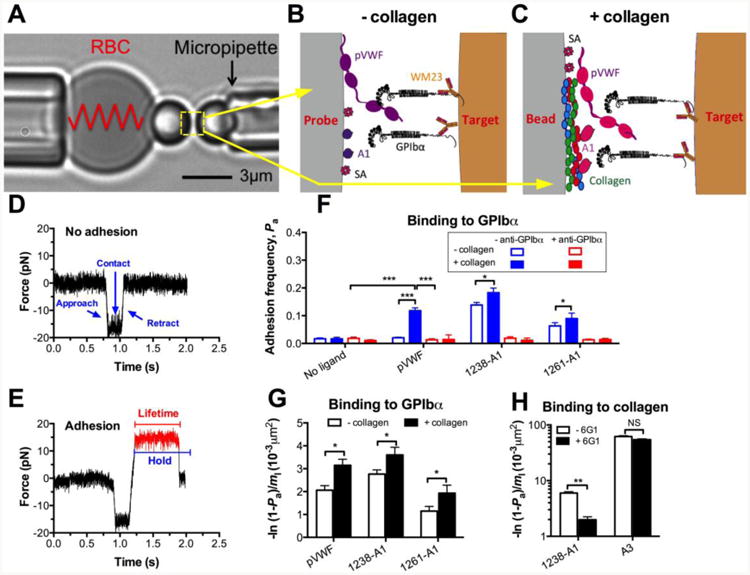
(A) BFP photomicrograph A micropipette-aspirated RBC with a bead (left, termed “probe”) attached to the apex formed a pico-force sensor, as depicted by a spring. It was aligned with another bead (right, termed “target”) aspirated by an apposing micropipette. (B and C) BFP functionalization. VWF-A1 or plasma VWF (pVWF) was coupled to the probe bead (left) via either direct linking to a plain surface, denote “- collagen” (B) or indirect linking to a collagen-bound surface, denote “+ collagen” (C). GPIb-IX was captured from platelet lysates by antibody WM23 covalently precoated on the target bead (right). Streptavidin (SA) was also covalently coupled to probe beads for attachment to biotinylated RBCs. (D and E) Force vs. time traces from two representative test cycles. A target bead was driven to approach a probe bead (∼0 pN), contacted for certain duration, retracted and ended the cycle if no adhesion (D) or held at a preset force (12 pN) until dissociation (signified by a force drop to zero) if adhesion was detected (indicated in red) (E), the duration of which was measured as bond lifetime (∼0.6 s). (F) Binding specificity. Adhesion frequencies (Pa) between the GPIbα targets and probes coated without or with indicated VWF ligands in the absence (blue) or presence (red) of 50 μg/ml anti-GPIbα (AK2) blocking mAb. (G) Average number of A1–GPIbα bonds per unit A1 site density [-ln(1 - Pa)/m1], which equals to the product of effective 2D affinity (AcKa) and GPIbα site density (mr). The same mr was used to test all VWF ligands. The results from “- collagen” (open) and “+ collagen” (closed) preparations are compared. (H) Collagen binding. Adhesion frequencies between the collagen III target and probes coated with 1238-A1 or A3 in the absence (open) or presence (closed) of 50 μg/ml anti-A1 (6G1) mAb. Each probe–target pair was tested repeatedly for 100 approach-contact-retract cycles to estimate a Pa. Five probe–target pairs were tested to obtain mean ± S.E.M. NS = not significant; * = p < 0.05; *** = p < 0.001, assessed by unpaired, two-tailed Student's t-test.
We previously showed that the isolated 1238-A1 simultaneously binds to collagen and GPIbα, supporting platelet adhesion under high-flow conditions [11]. To test the hypothesis that binding to collagen increases the A1 binding to GPIbα, we prepared the probe bead in two ways: 1) direct linking in which pVWF or A1 was covalently coupled to a glass bead [21,23,24], denoted as “- collagen”; and 2) indirect linking where pVWF or A1 was bound to human collagen (type III in this study) which was covalently pre-coupled to a glass bead, denoted as “+ collagen”. The binding specificity was established for both preparations. GPIbα-bearing targets adhered at significantly higher frequencies to probes coated with VWF ligands (pVWF, 1238-A1 and 1261-A1) than those not coated with VWF ligands. Addition of anti-GPIbα blocking mAb (AK2) abrogated adhesion (Fig. 1F). Consistently, VWF and A1 coated on “+ collagen” surfaces (Fig. 1F, closed) bound to GPIbα with higher Pa than those coated on “- collagen” surface (Fig. 1F, open). For each preparation method, we incubated the same amount of A1 with probe beads, and the flow cytometry with conformation-independent anti-A1 mAb (6G1, cf. Fig. 4A) showed comparable A1 coating site densities (m1; ∼55 μm-2).
Figure 4. Characterization of collagen binding induced A1 conformational change by antibodies.
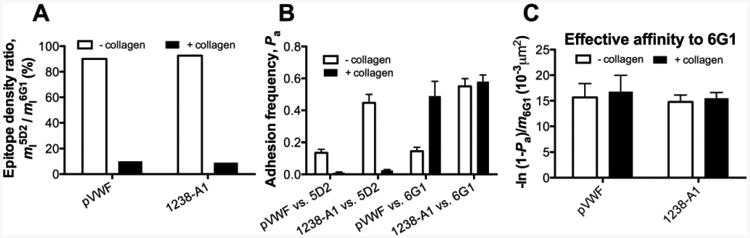
(A) Normalized A1 site densities detected by conformation specific anti-A1 mAbs. The ligand site densities for pVWF and A1 were measured by flow cytometry using 5D2 (m15D2), then normalized by the site densities measured with conformation-independent 6G1 (m16G1). The data were presented as m15D2/m16G1 in percentage. (B) Conformations probed by mAb coated targets. Adhesion frequencies (Pa) between the 5D2 or 6G1 coated target and probes coated with A1 or pVWF. (C) The effective binding affinities of pVWF and 1238-A1 to 6G1 by converting Pa6G1 (from panel B) to [-ln(1 - Pa6G1)/ m16G1]. The same keys were used as Fig. 1G.
Further, we converted Pa to normalized adhesion bonds by dividing the average number of adhesion bonds [-ln(1 - Pa)] by m1 [21]. This quantity also equals the product of the effective 2D affinity and the GPIbα site density mr that was kept constant in our experiments. The values for pVWF and A1 constructs prepared by “+ collagen” method was 53% and 30% higher than those prepared by “- collagen” method respectively (Fig. 1G). In addition, we replaced the GPIbα with the collagen on the target and found that both A1 and A3 domains bind directly to collagen (Fig. 1H). Matching our previous results [11], the specificity of A1–collagen interaction in BFP was ensured by the 6G1 blockade (Fig. 1H). Importantly, the affinity to collagen for A3 is 10 times of that for A1, confirming the A3 as the dominant collagen-binding domain. These results demonstrate that isolated A1 domain can directly bind to collagen across two-dimensional (2D) surfaces, and suggest that binding to collagen induces the A1 domain to adopt a high affinity conformation for GPIbα binding.
Collagen enhances VWF–GPIbα mediated platelet adhesion
It has been well accepted that the binding of platelets to collagen is mainly mediated by VWF at high wall shear rates (>500 s-1) [25-27]. We have also shown that collagen-bound A1 domain supports platelet adhesion under high-shear conditions [11]. To evaluate the role of collagen in a more physiologically relevant setting using a flow chamber [21,28], we immobilized pVWF on surfaces coated with (+) and without (-) collagen (Fig. 2A) and measured VWF-dependent platelet translocation over a range of wall shear stresses. To observe continuous platelet translocation mediated by VWF–GPIbα interaction alone, EDTA was added to prevent firm adhesion to VWF and collagen via integrins as previously described [25]. The translocation velocities of washed platelets on these substrates reproduce biphasic stress-dependent patterns confirming the translocation threshold phenomenon [25,29]. Although the same biphasic shear-dependent pattern was observed, “+ collagen” VWF surface supported more stable platelet translocation with lower velocities than “- collagen” VWF surface. This difference became more pronounced at higher wall shear stresses (>5 dyn/cm2) where rolling velocity transitioned from decreasing to increasing with increasing wall shear stress, which occurred at a higher wall shear stress on the “+ collagen” than “-collagen” VWF surface (Fig. 2A). As the VWF densities and spatial arrangements were confirmed to be similar on both surfaces (Fig. S2), the flow chamber data suggested that collagen enhance the VWF to a higher GPIbα binding state.
Figure 2. Enhancement of GPIbα binding to collagen-bound pVWF.
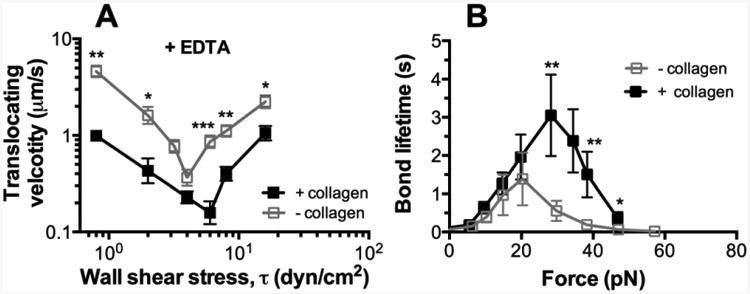
(A) Wall shear stress-dependent translocation velocity (mean ± S.E.M. of 20 platelets per point) on pVWF. 5 mM EDTA were added to the washed platelet perfusate. Data are presented as mean ± S.E.M. (B) Force-dependent of lifetime of GPIbα bonds with the pVWF. Lifetimes (mean ± S.E.M. of >20 measurements per point) of these bonds were measured by the force-clamp assay at each force. For each above assay, results by two immobilization methods were compared: directly coated pVWF (- collagen) and collagen-bound pVWF (+ collagen). Statistical analysis at each matched shear stress and force was performed to derive the p values. * = p < 0.05; ** = p < 0.01, *** = p < 0.001, assessed by unpaired, two-tailed Student's t-test.
To relate the observation of faster platelet translocation on “- collagen” than “+ collagen” VWF to possible differential GPIbα dissociation with the single-molecule setup (Fig. 1B and C), we used the BFP to measure GPIbα bond lifetimes under different forces. Again, biphasic catch-slip bonds [21,28] were observed for both collagen-bound and directly coated pVWF with longer bond lifetimes (especially at forces >20 pN) and a higher catch-slip transition force for the collagen-bound (∼30 pN) than directly coated (∼20 pN) pVWF (Fig. 2B). To ensure single-bond measurements, VWF-A1 site densities on the probe beads were titrated to ∼55 per μm2 such that the adhesion frequencies were below 20% (Fig. 1F) for both “- collagen” and “+ collagen” methods. The upshift of the lifetime curve (Fig. 2B) of GPIbα bonds with “+ collagen” VWF (relative to “- collagen” VWF) mirrors the downshift of the translation velocity curve (Fig. 2A), indicating that binding to collagen increases the VWF–GPIbα bond quality, manifested as a longer-lasting bond capable of supporting greater force. This data complements the earlier result that collagen increased the quantity of VWF–GPIbα bonds that are formed in higher frequency due to a higher affinity (Fig. 1G). Since translocation velocity are inversely related to bond number and bond lifetime – the greater the bond number and longer the bond lifetime, the slower the rolling velocity [28,30] – the two sets of complementary data obtained using the flow chamber and BFP show that the collagen-bound VWF binds GPIbα with a faster on-rate, a larger bond number, longer bond lifetimes, and greater bond strength, indicating that collagen directly induces the A1 domain to adopt a high binding conformation for GPIbα.
Collagen-captured A1 binds GPIbα with increased bond lifetime
To further examine the role of collagen in the context of VWF-A1–GPIbα binding, we functionalized the BFP with isolated A1 domains. Using the direct linking method (- collagen, Fig. 1B), we confirmed our previous results [21] that GPIbα forms triphasic slip-catch-slip bonds with 1238-A1 but monophasic slip-only bonds with 1261-A1 (Fig. 3A). Regardless of their different N-terminal sequence lengths, both A1 constructs showed quantitative changes with increased bond lifetimes at forces >17pN (compare Fig. 3A and B) when they were immobilized by a collagen bound surface (+ collagen, Fig. 1C). Most importantly, collagen capturing changed the 1261-A1–GPIbα bond qualitatively by converting it from a slip-only bond to a slip-catch-slip bond (Fig. 3B, open triangle). By comparison, collagen capturing only changed the 1238-A1–GPIbα bond quantitatively by upward stretching the bond lifetime vs. force curve at larger forces (>17pN) but kept the same slip-catch-slip pattern (Fig. 3B, open circle). Previously, we justified the use of 1238-A1 as a model system for VWF in terms of GPIbα binding at high forces (catch-slip force regime). Indeed, we reproduced the force-lifetime curve for GPIbα bond with pVWF (Fig. 2B, open square) that is indistinguishable to that with 1238-A1 (Fig. 3A, open circle) in the high force regime (>20 pN). However, the lifetime at the peak force for “+ collagen” pVWF (3.1 ± 1.0 s) doubles that for “+ collagen” 1238-A1 (1.5 ± 0.6 s), suggesting that the 1238-A1–GPIbα bond cannot fully recapitulate the pVWF–GPIbα interaction on a collagen surface.
Figure 3. Enhancement of GPIbα binding to collagen-bound VWF-A1 domains.
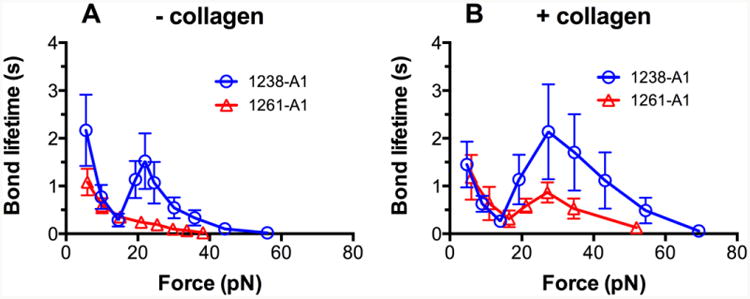
Plots of lifetime of vs. force of GPIbα bonds with “- collagen” (A) and “+ collagen” (B) VWF-A1 domains. For each A1 immobilization method, results of 1238-A1 (circle) and 1261-A1 (triangle) were compared. Lifetimes (mean ± S.E.M. of >20 measurements per point) of these bonds were measured by the force-clamp assay at each force.
Quantitative analysis of VWF-A1 domain bound to collagen using antibodies
Anti-A1 mAbs, such as 5D2 and 6G1, have been widely used to characterize A1 conformation dynamics in different contexts: subject to shear stress, bound to modulators (ristocetin and botrocetin) and altered by von Willebrand disease mutations [31-33]. Our previous results suggested that A1–collagen interaction induces a structural conformation in the A1 domain that abolishes the 5D2 but not the 6G1 epitope [11]. Here, to further characterize such a conformational change quantitatively, we first used the flow cytometry to measure the epitope densities of 5D2 (m15G2) and 6G1 (m16G1) on both “- collagen” and “+ collagen” coated A1 and pVWF beads. The 6G1 showed equivalent epitope densities (m16G1≈55 μm-2) for both conditions, indicating itself as a conformation-independent antibody. In contrast, 5D2 demonstrated poor epitope recognition (m15G2≈5 μm-2) on a collagen-bound surface, confirming itself as a conformation-dependent antibody. Therefore, we used the epitope density ratio, m15G2/m16G1, as a normalized measurement for collagen induced A1 conformational change. We found that m15G2/m16G1 reduced from 90% for “-collagen” condition to 9% for “+ collagen” condition consistently for both isolated A1 domain and full-length VWF (Fig. 4A). This result confirmed that the conformation of the A1 domain in VWF was affected by the binding of VWF to collagen.
In addition, we set up the BFP with the same beads and measured the adhesion frequencies between A1 or pVWF coated probe and 5D2 or 6G1 coated target. As expected, a “+ collagen” surface abolished the 5D2 interaction for both A1 and pVWF, but affected little of the 6G1 interaction for A1 alone (Fig. 4B). Surprisingly, the 6G1 interaction was even enhanced under “+ collagen” condition for full-length VWF. This data showed that the binding of full-length VWF to collagen results in the exposure of the cryptic epitope for antibody 6G1 that is located in the C-terminal flanking region of the A1 domain (sequence E1463-H1472) or outside the disulfide-domain structure [34]. Considering the site density effect, then we estimated the effective 6G1 binding affinity by converting Pa6G1 to-ln[(1 - Pa6G1)/m16G1]. Our data showed a similar level of 6G1 binding affinity for both full-length VWF and isolated A1 domain regardless of the coating method (Fig. 4C). This suggests that the A1 structure can adopt an open conformation without altering the recognition site for 6G1.
Discussion
The present study has added to our understanding in force-dependent pVWF activation. By comparing the force-dependent kinetics of GPIbα dissociation from directly adsorbed (Fig. 1B) and collagen-bound (Fig. 1C) VWF, we have shown that collagen not only anchors VWF but also upregulates its binding to GPIbα with an enhanced 2D effective affinity (a ratio of on-/off-rate, Fig. 1H). This collagen-induced binding enhancement was observed from both full-length VWF (Fig. 2) and isolated A1 domains (Fig. 3). Considering the similar pVWF–GPIbα bond lifetimes (which represents off-rate) at zero force for both coating surfaces (Fig. 2B), the 53% affinity increase upon binding to a collagen surface is due to the increased on-rate (Fig. 1G). Together with the dramatic conformational change in A1 quantified by conformation-dependent mAbs (Fig. 4), our results suggest that when VWF binds to collagen, A1 domain changes to a more open conformation with increased cellular on-rate, resulting in an overall higher GPIbα binding (Fig. 1G).
In contrast to the comparable site densities for the A1 coated beads, site density measurements for the pVWF coated beads showed that 6G1 epitope on a “+ collagen” surface (m16G1≈40 μm-2) is 4 times of that on a “- collagen” surface (m16G1≈10 μm-2). This result indicates that 6G1 epitope is exposed upon the binding of VWF to collagen unlike that of isolated A1 domain. Thus, this epitope is likely hidden by the neighboring D3 or A2 domains in full length VWF. It is important to acknowledge that there are other structural features in the full length VWF that directly influence on the binding of VWF to collagen. For example, our results demonstrate that the A3 has a much higher affinity for collagen than the A1, confirming that A3 domain contains the major collagen binding site in VWF. We speculate that the A3 domain plays the essential role in mediating the binding of the rapid flowing VWF to exposed collagen, changing the conformation of the neighboring A domains, thereby exposing the cryptic A1 domain which is shielded by either or both D3 and A2 domains (cf phase II, Fig. S1) [33,35,36].
In addition to the quantity enhancement, VWF–collagen interaction also showed a quality enhancement reflected by the increased effective affinity to GPIbα (Fig. 1G and cf. phase III, Fig. S1). Consistent with the wide belief of a closed conformation for pVWF, regardless of coating methods, pVWF binds to GPIbα with a lower zero-force affinity than 1238-A1 does (Fig. 1F). This suggests that binding to collagen cannot fully eliminate the auto-inhibition mechanism within the full-length VWF at zero force. However, when bound to a “+ collagen” surface, zero-force affinity increased by 53% for pVWF, higher than the 30% increase for A1. This discrepancy might be explained by the additional contribution from the A3 domain [37].
Moreover, we quantified the reactivity of two mAbs, 6G1 and 5D2, to A1 protein bound to both “- collagen” and “+ collagen” surfaces using two independent assays flow cytometry (Fig. 4A) and BFP (Fig. 4B). In comparison with 6G1, the conformation-dependent mAb 5D2 demonstrated the same level of epitope reduction for both A1 and pVWF on collagen-bound surfaces. On one hand, this demonstrates that A1 and pVWF indeed specifically bind to collagen on the probe surface; and that collagen induces the same A1 conformational change in both proteins (Fig. 4A). The ability of 6G1 to react with A1 and pVWF when they are bound to collagen confirmed our previous observation that the sequence does not overlap with the collagen contact site [11]. The 6G1 inhibitory effect (Fig. 1H) on A1–collagen axis is probably a consequence of steric hindrance.
When force increases, the observation of longer bond lifetimes at >20pN forces under “+ collagen” condition suggests that collagen directly alters the pVWF-GPIbα bond quality. In the absence of collagen, the lifetimes of GPIbα bonds with pVWF and 1238-A1 were similar at >20pN (compare Figs. 2B and 3A) as previously reported [21]. In the presence of collagen, by comparison, the lifetime vs. force curves were very different for GPIbα bonds with pVWF than 1238-A1 (compare Figs. 2B and 3B), supporting a cooperative role of A3 or other structural elements of the VWF in regulating A1–GPIbα interaction against the high-shear forces. The glycosylation sites in or around Q1238-E1260 could also in part contribute to differences between bacterial expressed recombinant fragments and pVWF [9,38]. Of note, the collagen affects the 1261-A1 in a similar way as it does to 1238-A1 except that it switch the intrinsic slippery state for 1261-A1 to a catchy state. Regardless of the collagen effect, the 1261-A1 consistently behaves with a lower binding phenotype (Fig. 3), suggesting the collagen regulation is distinct to the regulation by A1 N-terminal sequence (cf. phase IV, Fig. S1). In the future, the perspective new crystal structures of A1–collagen complex and 1238-A1 will help understand the intrinsic heterogeneity of A1 binding states.
Our findings can be summarized in a model that depicts the four phases of collagen dependent enhancement for VWF–GPIbα interaction under flow conditions (Fig. S1).
i) pVWF circulates in blood stream with an auto-inhibited close conformation.
ii) High affinity allows VWF-A3 to connect pVWF to collagen first, thereafter shear forces can unfold the bound VWF and expose A1 domains for interaction with collagen as well as platelet GPIbα.
iii) The subsequent interaction to collagen alters A1 itself into a conformation with a higher affinity for GPIbα.
iv) The further increasing force reduces the inhibitory effect of A1 N-terminal sequence and prolongs the bond lifetime, showing the catch-bond behavior.
We have demonstrated that the affinity upregulation is a combinatory enhancement in both bond quantity and bond quality. In the future, our single-bond experiment can be compared with perspective A1–collagen crystal structure as well as molecular dynamics simulation results to reveal more insights on collagen enhanced A1 binding state.
Materials and methods
Ethics statement
Blood samples were collected from healthy adults with their written consent in accordance with the protocol approved by the Georgia Institute of Technology Institutional Review Board specifically for this study.
Purification of platelets and red blood cells
Platelets and RBCs were isolated from 3 ml venous blood drawn from healthy donors. Whole blood was first collected in a 1:10 ACD buffer (6.25 g sodium citrate, 3.1 g citric acid anhidrous, 3.4 g D-glucose in 250 ml H2O, pH 6.7) and centrifuged at 150 g for 15 min at room temperature. Platelet-rich plasma (from the top layer) was then centrifuged at 900 g for 5 min. The platelet pellet was saved for making platelet lysates or washed platelets [21].
The RBCs (from the bottom layer) were biotinylated by covalently linking with biotin-PEG3500-SGA (JenKem USA, TX) with a 30 min incubation at room temperature [39]. To pre-swell RBCs for the force probe use in a buffer of physiological osmolarity, the RBCs were further incubated with nystatin (Sigma-Aldrich) in an N2 buffer (265.2 mM KCl, 38.8 mM NaCl, 0.94 mM KH2PO4, 4.74 mM Na2HPO4, 27 mM sucrose; pH 7.2, 588 mOsm) for 30 min at 0°C. The modified RBCs were washed twice with the N2 buffer and resuspended in the N2 buffer for the BFP experiments.
Proteins and antibodies
Recombinant wild-type (WT) monomeric VWF 1238-A1 (residues 1238-1471), 1261-A1 (residues 1261-1471) and A3 (residues 1687-1874) domains as well as the A1 N-terminal flanking region polypeptide (Lp, residues 1238-1260) were generated by E.coli as previously described [9,11,14]. Plasma VWF (Calbiochem, San Diego, CA), human placenta type III collagen (Sigma) and streptavidin (SA)-maleimide (Sigma) were purchased. Two anti-GPIbα mAbs were used: AK2 was purchased (Abcam, Cambridge, MA) and WM23 was a gift from Dr. Renhao Li (Emory University, Atlanta, GA). Mouse anti-human anti-A1 mAbs (5D2 and 6G1, IgG1 subtype) were gifts from Dr. Michael Berndt (Curtin University, WA, Australia). The epitope for 5D2 has not been defined and appear to be conformation-dependent; while the epitope for 6G1 has been mapped to a region at the C-terminus (E1463-H1472) of A1 where ristocetin binds [31]. PE-conjugated goat F(ab′)2 anti-mouse IgG (ab7002) was from Abcam (Cambridge, MA). All other reagents, if not specified, were purchased from Sigma.
Functionalization of glass beads
Proteins (pVWF, A1s, collagen III or Abs) were covalently modified with maleimide-PEG3500-NHS (MW ∼3500 Da; JenKem, TX) in carbonate/bicarbonate buffer (pH 8.5). To coat maleimized proteins on glass beads, 2-μm (diameter) silanized borosilicate beads (Thermo Scientific) were first covalently coupled with mercapto-propyl-trimethoxy silane (Sigma), followed by covalently linking to both streptavidin-maleimide (Sigma) and maleimide modified proteins in monobasic/dibasic phosphate buffer (pH 6.8) (Fig. 1B, left). After overnight incubation and resuspending in phosphate buffer with 1% HSA, beads were ready for immediate use in BFP experiments.
To immobilize full-length GPIb-IX complex on beads, WM23 mAb pre-coupled beads were incubated with platelet lysate in a rotor for 2-h at room temperature (Fig. 1B, right). After resuspending in phosphate buffer with 1% HSA, beads were ready for immediate use in BFP experiments. To study the collagen-bound pVWF and A1s, collagen pre-coupled beads were prepared as above. Unmodified pVWF and A1s were then captured to the collagen beads by a similar process to the GPIb capturing.
To measure VWF-A1 site density on the bead surface, pVWF- or A1-coated beads were incubated with the anti-A1 primary antibodies (5D2 or 6G1) at saturating concentration (10 μg/ml) at room temperature for 30 min, followed by a similar incubation process with the secondary antibody (PE conjugated anti-mouse IgG). Beads were washed twice between the incubations. The phosphate buffer with 1% HSA was used for all incubations and washings. After the final wash, bead fluorescent intensities were measured using a BD™ LSR II flow cytometer (BD Biosciences). The intensity results were compared with standard calibration beads (BD Quantibrite™ PE Beads; BD Biosciences) to determine total number of molecules per bead using a manufacturer-provided calculating form [40]. The site densities were calculated by dividing the total number of molecules per bead to the bead surface area, which was calculated from the bead diameters measured under the microscope (∼3.5 μm). To ensure the coating consistency, we repeated the measurements three times on different beads from the same preparation batch.
Of note, the possibility exists for some A1 proteins to be physically adsorbed next to each other as a result of their binding to the ‘+ collagen’ surface, an arrangement induced by the repetitive sequences found within collagen. However, we have shown by different means that this A1 protein does not form aggregates nor multimers in solution [41]. In addition, the similarly homogenous coating of VWF on ‘+ collagen’ as to ‘- collagen’ surfaces (Fig. S2), suggests that collagen does not significantly alter the A1 spatial arrangement.
BFP experiments
Our BFP experimental procedure used to study VWF–GPIbα interaction has been described [21]. In each test cycle, the target (GPIb captured bead) was driven to approach and contact the probe (pVWF or A1s coated bead) with an 18-pN compressive force for a certain contact time (0.2 s by default) to allow for bond formation and retracted for adhesion detection. During the retraction phase, an adhesion event was signified by tensile force (Fig. 1E), but no tensile force was detected in a non-adhesion event (Fig. 1D). For the adhesion frequency assay, adhesion and non-adhesion events were enumerated to calculate an adhesion frequency (Pa) in 100 repeated cycles for each probe–target pair. Since Pa depends on the site densities of receptors (mr) and ligands (m1) on the contact area, in order to compare the binding affinity of GPIbα to different VWF ligands, the normalized adhesion bonds per ligand density (Fig. 1G and H) were derived via formula -ln(1 -Pa)/m1 [21], representing the 2D effective affinities.
2D affinity describes the binding propensity between receptors and ligands on two opposing surfaces normalized by their site densities (assumed uniformly distributed). For second-order forward and first-order reverse binding reaction to form monomeric bonds, it equals the product of 2D affinity Ka = on-/off-rate and the contact area Ac [42]. Because Ac is difficult to measure, and is a constant as long as the bead size and the cell size are not dramatically changed, we combine the parameters Ka and Ac, and define AcKa as the effective 2D affinity. In our experiment, the effective 2D affinity (AcKa) was calculated from the measured adhesion frequency (Pa) and receptor (mr) and ligand (m1) densities. The equation used, AcKa = - ln(1 - Pa) /(mrm1). Consider the receptor densities (m1: GPIb) remain the same for all VWF ligands tested, we can write AcKa ∝ -ln(1 - Pa)/m1. Although the effective 2D affinity cannot directly reflect the value of the 2D affinity, it accurately reflects the relative affinity difference in different receptor-ligand systems.
In the force-clamp assay, the target was held at a desired force to wait for bond dissociation and returned to the original position to complete the cycle. Lifetime was measured from the instant when the force reached the desired level to the instant of bond dissociation (Fig. 1E, red trace). To ensure higher possibilities (>95%) for single-ligand bond events, we coated both A1 and GPIb at lower site densities (∼50 μm-2). Furthermore, we specifically excluded events with multiple bond signatures from analysis (i.e. events showing discrete slope decrease during force ramping phase or showing multiple-state unbinding in dissociation phase) [43].
Flow chamber assays
Platelet translocation velocity assays were performed as previously described [21,28]. Washed platelets were perfused in a PDMS channel (channel dimension: 500 μm (width) × 50 μm (height)) at various wall shear stresses as previously published [44]. For direct coatings (- collagen), pVWF (100 μg/ml) was physically absorbed on the plasma cleaned glass surface that forms the bottom of the channel. To prepare a ‘+ collagen’ surface, collagen (400 μg/ml) was pre-coated on the channel surface, which was then incubated with pVWF (100 μg/ml). For each step, mixture was incubated at room temperature for 1 h. The pVWF densities were confirmed to be constant for both preparation surfaces (Fig. S2). To abrogate all Ca2+ dependent platelet activation pathways and integrin dependent platelet adhesions on VWF, EDTA was mixed with the washed platelets before the perfusion. In addition, to exclude the later-stage platelet actions, i.e. granule secretion and VWF release, we only focused on the first 60s perfusion time window. The translocation velocity (μm/s), calculated as the total distance traveled by a surface-tethered platelet in 1 s, were measured with 9 frames per second video microscopy then frame-by-frame analyzed by ImageJ v1.48 software [40,45]. Each velocity represents an average of 20 platelets from 2 healthy donors.
Supplementary Material
Highlights.
v) Initial VWF capture on collagen is mediated by A3 domain;
vi) Collagen alters A1 itself into a conformation with a higher affinity for GPIbα;
vii) The length of A1 flanking region determines the accessibility of A1 domains to GPIbα;
viii) Force reduces the inhibitory effect from the A1 N-terminal sequence.
Acknowledgments
This work was supported by National Institute of Health grants HL091020 (C.Z.), HL072886 (M.A.C), and AI088023 (H.L.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare no competing financial interests.
Abbreviations used in this article
- BFP
biomembrane force probe
- GPIbα
Glycoprotein Ibα
- HSA
human serum albumin
- mAb
monoclonal antibody
- pVWF
plasma VWF
- SA
streptavidin
- VWF
von Willebrand factor
Footnotes
Addendum: L.J. and C.Z. designed experiments; M.A.C. provided the reagents; L.J. performed experiments and analyzed the data; Y.C., F.Z. and H.L. provided additional data. All authors contributed to writing the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson SP, Nesbitt WS, Westein E. Dynamics of platelet thrombus formation. J Thromb Haemost. 2009;7:17–20. doi: 10.1111/j.1538-7836.2009.03401.x. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nat Med. 2011;17:1423–36. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 3.Cosemans JMEM, Angelillo-Scherrer A, Mattheij NJA, Heemskerk JWM. The effects of arterial flow on platelet activation, thrombus growth, and stabilization. Cardiovascular Research. 2013;99:342–52. doi: 10.1093/cvr/cvt110. [DOI] [PubMed] [Google Scholar]
- 4.Ruggeri ZM. Von Willebrand factor: looking back and looking forward. Thromb Haemostasis. 2007;98:55–62. [PubMed] [Google Scholar]
- 5.Reininger AJ. VWF attributes--impact on thrombus formation. Thrombosis Research. 2008;122(Suppl 4):S9–13. doi: 10.1016/S0049-3848(08)70028-8. [DOI] [PubMed] [Google Scholar]
- 6.De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121:270–7. doi: 10.1182/blood-2012-07-442285. [DOI] [PubMed] [Google Scholar]
- 7.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014 doi: 10.1182/blood-2014-05-378638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohri H, Yoshioka A, Zimmerman TS, Ruggeri ZM. Isolation of the von Willebrand factor domain interacting with platelet glycoprotein Ib, heparin, and collagen and characterization of its three distinct functional sites. J Biol Chem. 1989;264:17361–7. [PubMed] [Google Scholar]
- 9.Cruz MA, Handin RI. The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. J Biol Chem. 1993;268:21238–45. [PubMed] [Google Scholar]
- 10.Cruz MA, Diacovo TG, Emsley J, Liddington R, Handin RI. Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J Biol Chem. 2000;275:19098–105. doi: 10.1074/jbc.M002292200. [DOI] [PubMed] [Google Scholar]
- 11.Morales LD, Martin C, Cruz MA. The interaction of von Willebrand factor-A1 domain with collagen: mutation G1324S (type 2M von Willebrand disease) impairs the conformational change in A1 domain induced by collagen. J Thromb Haemost. 2006;4:417–25. doi: 10.1111/j.1538-7836.2006.01742.x. [DOI] [PubMed] [Google Scholar]
- 12.Flood VH, Gill JC, Christopherson PA, Bellissimo DB, Friedman KD, Haberichter SL, et al. Critical von Willebrand factor A1 domain residues influence type VI collagen binding. Journal of Thrombosis and Haemostasis. 2012;10:1417–24. doi: 10.1111/j.1538-7836.2012.04746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalafatis M, Takahashi Y, Girma JP, Meyer D. Localization of a collagen-interactive domain of human von Willebrand factor between amino acid residues Gly 911 and Glu 1,365. Blood. 1987;70:1577–83. [PubMed] [Google Scholar]
- 14.Cruz MA, Yuan H, Lee JR, Wise RJ, Handin RI. Interaction of the von Willebrand factor (vWF) with collagen. Localization of the primary collagen-binding site by analysis of recombinant vWF a domain polypeptides. J Biol Chem. 1995;270:10822–7. doi: 10.1074/jbc.270.18.10822. [DOI] [PubMed] [Google Scholar]
- 15.Lankhof H, van Hoeij M, Schiphorst ME, Bracke M, Wu YP, IJsseldijk MJ, et al. A3 domain is essential for interaction of von Willebrand factor with collagen type III. Thrombosis and Haemostasis. 1996;75:950–8. [PubMed] [Google Scholar]
- 16.Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA. 2007;104:7899–903. doi: 10.1073/pnas.0608422104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barg A, Ossig R, Goerge T, Schneider MF, Schillers H, Oberleithner H, et al. Soluble plasma-derived von Willebrand factor assembles to a haemostatically active filamentous network. Thrombosis and Haemostasis. 2007;97:514–26. [PubMed] [Google Scholar]
- 18.Colace TV, Diamond SL. Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:105–13. doi: 10.1161/ATVBAHA.112.300522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Fallah MA, Huck V, Angerer JI, Reininger AJ, Schneider SW, et al. Blood-clotting-inspired reversible polymer-colloid composite assembly in flow. Nat Commun. 2013;4:1333. doi: 10.1038/ncomms2326. [DOI] [PubMed] [Google Scholar]
- 20.Auton M, Sedlák E, Marek J, Wu T, Zhu C, Cruz M. Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIb [alpha] Biophys J. 2009;97:618–27. doi: 10.1016/j.bpj.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju L, Dong JF, Cruz MA, Zhu C. The N-terminal flanking region of the A1 domain regulates the force-dependent binding of von Willebrand factor to platelet glycoprotein Ibα. J Biol Chem. 2013;288:32289–301. doi: 10.1074/jbc.M113.504001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesla S, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J. 1998;75:1553–72. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Lou J, Zhu C. Forcing switch from short- to intermediate- and long-lived states of the A domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem. 2010;285:35967–78. doi: 10.1074/jbc.M110.155770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Lou J, Evans EA, Zhu C. Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J Cell Biol. 2012;199:497–512. doi: 10.1083/jcb.201201091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 26.Ruggeri ZM, Dent JA, Saldívar E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood. 1999;94:172–8. [PubMed] [Google Scholar]
- 27.Wu YP, Vink T, Schiphorst M, van Zanten GH, IJsseldijk MJ, De Groot PG, et al. Platelet thrombus formation on collagen at high shear rates is mediated by von Willebrand factor-glycoprotein Ib interaction and inhibited by von Willebrand factor-glycoprotein IIb/IIIa interaction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1661–7. doi: 10.1161/01.atv.20.6.1661. [DOI] [PubMed] [Google Scholar]
- 28.Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, et al. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. 2008;118:3195–207. doi: 10.1172/JCI35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–97. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 30.Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol. 2004;166:913–23. doi: 10.1083/jcb.200403144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Luca M, Facey D, Favaloro E, Hertzberg M, Whisstock J, McNally T, et al. Structure and function of the von Willebrand factor A1 domain: analysis with monoclonal antibodies reveals distinct binding sites involved in recognition of the platelet membrane glycoprotein Ib-IX-V complex and ristocetin-dependent activation. Blood. 2000;95:164. [PubMed] [Google Scholar]
- 32.Dong JF, Berndt MC, Schade A, McIntire LV, Andrews RK, Lopez JA. Ristocetin-dependent, but not botrocetin-dependent, binding of von Willebrand factor to the platelet glycoprotein Ib-IX-V complex correlates with shear-dependent interactions. Blood. 2001;97:162–8. doi: 10.1182/blood.v97.1.162. [DOI] [PubMed] [Google Scholar]
- 33.Auton M, Sowa KE, Smith SM, Sedlák E, Vijayan KV, Cruz MA. Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibalpha and platelet activation under shear stress. J Biol Chem. 2010;285:22831–9. doi: 10.1074/jbc.M110.103358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley J, Cruz M, Handin R, Liddington R. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J Biol Chem. 1998;273:10396–401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
- 35.Ulrichts H, Udvardy M, Lenting PJ, Pareyn I, Vandeputte N, Vanhoorelbeke K, et al. Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. 2006;281:4699–707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 36.Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J Thromb Haemost. 2007;5:1363–70. doi: 10.1111/j.1538-7836.2007.02536.x. [DOI] [PubMed] [Google Scholar]
- 37.Obert B, Houllier A, Meyer D, Girma JP. Conformational changes in the A3 domain of von Willebrand factor modulate the interaction of the A1 domain with platelet glycoprotein Ib. Blood. 1999;93:1959–68. [PubMed] [Google Scholar]
- 38.Schulte am Esch J, Robson SC, Knoefel WT, Eisenberger CF, Peiper M, Rogiers X. Impact of O-linked glycosylation of the VWF-A1-domain flanking regions on platelet interaction. British Journal of Haematology. 2005;128:82–90. doi: 10.1111/j.1365-2141.2004.05253.x. [DOI] [PubMed] [Google Scholar]
- 39.Evans E, Ritchie K, Merkel R. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophys J. 1995;68:2580–7. doi: 10.1016/S0006-3495(95)80441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yago T, Leppänen A, Qiu H, Marcus WD, Nollert MU, Zhu C, et al. Distinct molecular and cellular contributions to stabilizing selectin-mediated rolling under flow. Journal of Cell Biology. 2002;158:787–99. doi: 10.1083/jcb.200204041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auton M, Zhu C, Cruz MA. The Mechanism of VWF-Mediated Platelet GPIbα Binding. Biophys J. 2010;99:1192–201. doi: 10.1016/j.bpj.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Zarnitsyna VI, Sarangapani KK, Huang J, Zhu C. Measuring Receptor-Ligand Binding Kinetics on Cell Surfaces: From Adhesion Frequency to Thermal Fluctuation Methods. Cel Mol Bioeng. 2008;1:276–88. doi: 10.1007/s12195-008-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Chen W, Zhu C. Molecular Force Spectroscopy on Cells. Annu Rev Phys Chem. 2015;66:150112150056003. doi: 10.1146/annurev-physchem-040214-121742. [DOI] [PubMed] [Google Scholar]
- 44.Lu H, Koo LY, Wang WM, Lauffenburger DA, Griffith LG, Jensen KF. Microfluidic shear devices for quantitative analysis of cell adhesion. Anal Chem. 2004;76:5257–64. doi: 10.1021/ac049837t. [DOI] [PubMed] [Google Scholar]
- 45.Ramachandran V, Nollert MU, Qiu H, Liu WJ, Cummings RD, Zhu C, et al. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc Natl Acad Sci USA. 1999;96:13771–6. doi: 10.1073/pnas.96.24.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.