

Abstract
GDF15 (Growth and differentiation factor 15) is a secreted cytokine, a direct target of p53 and plays a role in cell proliferation and apoptosis. It is induced by oxidative stress and has anti-apoptotic effects. The role of GDF15 in hyperoxic lung injury is unknown. We tested the hypothesis that GDF15 will be induced in vitro, in a model of pulmonary oxygen toxicity, and will play a critical role in decreasing cell death and oxidative stress. BEAS-2B (human bronchial epithelial cells) and human pulmonary vascular endothelial cells (HPMEC) were exposed to hyperoxia, and expression of GDF15 and effect of GDF15 disruption on cell viability and oxidative stress was determined. Furthermore, we studied the effect of p53 knockdown on GDF15 expression. In vitro, both BEAS-2B and HPMEC cells showed a significant increase in GDF15 expression upon exposure to hyperoxia. After GDF15 knockdown, there was a significant decrease in cell viability and increase in oxidative stress compared to control cells transfected with siRNA with a scrambled sequence. Knockdown of p53 significantly decreased the induction of GDF15 by hyperoxia. In conclusion, we show that GDF15 has a pro-survival and anti-oxidant role in hyperoxia and that p53 plays a key role in its induction.
Keywords: Hyperoxia, BEAS-2B, HPMEC, Bronchopulmonary dysplasia, Pulmonary
Graphical abstract
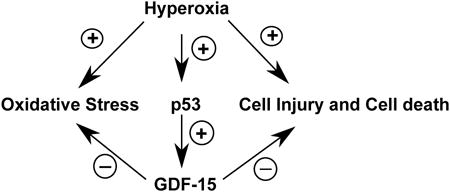
1. Introduction
Exposure to high concentrations of oxygen (hyperoxia) can cause or worsen lung injury in diseases such as acute respiratory distress syndrome (ARDS) and bronchopulmonary dysplasia (BPD) (Madurga et al., 2013). Hyperoxia creates an environment of increased oxidative stress and leads to the production of reactive oxygen species (ROS) (Saugstad, 2003) leading to cellular injury and death. The molecular mechanisms and pathways behind hyperoxic lung injury, including toxicity to various pulmonary cell types still need to be delineated.
GDF15 (also known as MIC-1, NAG-1, PLAB, PTGFB) is a divergent member of the TGF-β superfamily, which is widely distributed in mammalian tissues. It is recognized as a stress-responsive cytokine and its levels are elevated in diseases such as acute respiratory distress syndrome, pulmonary hypertension and heart failure (Clark et al., 2013; Kempf and Wollert, 2013; Nickel et al., 2011). The increased serum concentrations may indicate ongoing cellular injury or a protective response to cellular stress. Gdf15 is a part of the in vivo gene expression signature of oxidative stress (Han et al., 2008). GDF15 has been shown to have anti-inflammatory (Kempf et al., 2011; Kim et al., 2013; Preusch et al., 2013) and anti-apoptotic (Jin et al., 2012) effects. In the lungs, GDF15 is abundantly expressed in the plexiform lesions in patients with pulmonary hypertension and its levels are increased in the serum (Nickel et al., 2011). It is expressed and induced in response to hypoxia in human pulmonary vascular endothelial cells, and treatment with recombinant human GDF15 decreases apoptosis and improves cellular proliferation (Nickel et al., 2011). The goal of this study was to determine the expression and mechanistic role of GDF15 in human pulmonary epithelial and endothelial cells exposed to hyperoxia, in vitro. We tested the hypothesis that GDF15 will be induced in vitro in a model of pulmonary oxygen toxicity, and will play a critical role in decreasing apoptosis and oxidative stress in human pulmonary epithelial (BEAS-2B) and endothelial (HPMEC) cell lines exposed to hyperoxia. In the current study, we report that hyperoxia causes augmentation of GDF15 expression in both pulmonary epithelial and endothelial cells, which is accompanied by increase in cell survival and decrease in ROS generation, and that p53 plays a key role in the induction of GDF15 in hyperoxic conditions.
2. Materials and methods
2.1. Cell culture
The human bronchial epithelial cell line (BEAS-2B) was purchased from American Type Culture Collection (Rockville, MD). They were cultured in DMEM/F-12, 50/50, (Cell Gro, Manassas, VA) supplemented with 10% fetal bovine serum, 50 U penicillin/ml, and 50μg/ml streptomycin in a 5% CO2/95% air at 37°C. The HPMEC endothelial lung cells were obtained from ScienCell (Carlsbad, California). They were cultured in ECM media (ScienCell) supplemented with 25 ml of fetal bovine serum, 5 ml of endothelial cell growth supplement (ScienCell) and 5 ml of penicillin/streptomycin solution in a 5% CO2/95% air at 37°C. The plates for HPMEC were coated fibronectin, 2 μg/cm2, at 37°C overnight. All cells were routinely passaged every 3 - 4 days.
2.2. RNA Interference
p53 siRNA, GDF15 siRNA (GFP tagged) and control siRNA were obtained from Qiagen (Valencia, CA). Transfection was performed using Lipofectamine (Life Technologies, Carlsbad, CA). The final concentration of siRNA was 50nM, and 15 hours after transfection, the cells were used for the experiments.
2.3. Exposure of cells to hyperoxia
Hyperoxia experiments were conducted in a Plexiglas sealed chamber into which a mixture of 95% O2 and 5% CO2 was circulated continuously. The chamber was placed in a Forma Scientific water-jacketed incubator at 37 °C. Once the O2 level inside the chamber reached 95%, the cells were placed inside the chamber for the desired length of time. The control cell population was maintained at 5% CO2/95% air at 37°C. For each protocol, three or four independent experiments were performed
2.4. ELISA
The human GDF15 ELISA Kit (Cat No.155432) was obtained from AbCam (Cambridge, MA). Plates were placed in hyperoxia or room air and supernatant was collected at different time-points in triplicates and immediately placed at -81°C. 100uL of each sample was analyzed in duplicate according to the manufacturer recommendations. Plates were read immediately at 450 nm. TC20™ Automated Cell Counter (BioRad Laboratories, Hercules, CA) was used to obtain the number of total cells.
2.5. MTT Assay
The CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Inc. Madison, WI) was used for the MTT assay as per the manufacturer recommendations. BEAS-2B and HPMEC cells were seeded onto 96-well microplates at a density of 1×104 cells per well followed by exposure to room air or hyperoxia for up to 72 h. The absorbance at 490 nm was read using SpectraMax M3 microplate reader (Molecular Devices LLC, Sunnyvale, CA).
2.6. Measurement of ROS generation
The ROS-Glo™ H2O2 Assay (Promega Inc.) was used to measure the level of hydrogen peroxide (H2O2), a reactive oxygen species (ROS), directly in cell culture according to the manufacturer's recommendations. Cells were plated at a density of 100,000 cells/well on a 96 well format plate, and incubated overnight for attachment before transfection with respective siRNA. The plates were placed in hyperoxia or normoxia for 0 h, 4 h, 8 h, 12 h and 24 h. H2O2 substrate solution was added to the plates 6 h before read, and the plates were replaced in hyperoxia chamber. The detection solution was added to the plates 20 min before each read and incubated at room temperature and relative luminescence was recorded using SpectraMaxM3 microplate reader (Molecular Devices LLC, Sunnyvale, CA).
2.7. RT PCR
Total RNA was extracted using the RNeasy Mini Kit from (Qiagen Inc.) RNA concentration and quality was measured with the use of NanoDrop Spectrophotometer (Thermo Fisher Scientific, Waltham, USA). The QuantiTect Reverse Transcription Kit (Qiagen Inc.) was used for cDNA synthesis with integrated genomic DNA removal. RT- PCR was performed using the Quantifast SYBR Green kit (Qiagen). 18s rRNA was used as an internal control. The cycle for cDNA was 15 minutes at 42°C and denaturation at 95°C for 3 minutes. Amplification and detection were performed with Viia 7 RT-PCR System (Applied Biosystems). The thermal cycling step was for 40 cycles for denaturation at 95°C for 10 sec and 40 cycles at 60°C for 30 sec excluding initial heat activation at 95°C for 5 min. The ΔΔCt method was used to calculate the fold change in mRNA expression: ΔCt = Ct (target gene) - Ct (reference gene), ΔΔCt = ΔCt (treatment) - ΔCt (control), fold change = 2(-ΔΔCt). The primers used were purchased from Qiagen GDF15 Cat # QT 0082558, p53 Cat # QT 00060235, 18s rRNA Cat # QT 00199367.
2.8. Western Blot
Protein lysates were prepared using M-PER mammalian protein extraction reagent (Thermo Scientific). The protein concentration was measured using Bradford method. Total protein extract (20-30 μg) was resolved by SDS-PAGE and transferred to nitrocellulose membranes. Antibody to GDF15 (1:1000, rabbit monoclonal antibody, Cat. No 8479) and human recombinant GDF15 (used as positive control) were purchased from Cell Signaling Technologies (Beverly, MA, USA). The next day, membranes were washed with 1xTBST, and incubated with horseradish peroxidase (HRP)- conjugated anti-rabbit IgG secondary antibody for 1 h at room temperature. For loading controls, the membranes were stripped using a buffer containing SDS, Tris HCL and β-mercaptoethanol. Warmed buffer (50°C) was added to cover the membrane and incubated for 45 minutes with some agitation. After rinsing and washing extensively for 5 minutes in TBST the membrane was blocked and incubated with antibodies against β-actin. Protein bands were visualized with HyGLO™ Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific, NJ). Relative quantitation was performed using Image Studio Lite Software ™ with normalization against β-actin. The fold expression was indicated as the relative protein level.
2.9. Statistical Analysis
The statistical analyses were performed on three independent biological replicates. Results are reported as means ± standard error of the mean (SEM). Data were analyzed using 2-way analysis of variance (the main effects were: treatment group and hyperoxia duration), followed by Bonferroni posttests for comparisons against control conditions using GraphPad version 6. Significance was assigned for P < 0.05.
3. Results
3.1. Induction of GDF15 expression in pulmonary epithelial and endothelial cells in hyperoxia
GDF15 expression was measured at room air and following 24-72 h of hyperoxia exposure. There was a significant induction (upto 90-fold) of GDF15 expression at the mRNA level as measured by RT-PCR in BEAS-2B (1a) and HPMEC (1b) cells exposed to hyperoxia compared to room air controls. We performed ELISA (Figure 1c and 1d) of the cell culture supernatants at room air or after 0, 12, 24, 48, and 72 h of hyperoxia exposure, as GDF15 is a secreted protein. The values were normalized to total cell count. Similar trends were observed with significant induction of GDF15 production in response to hyperoxia in both the cell lines compared to respective room air values. There was a 4-fold increase in GDF15 levels compared to cells at room air. Figures 1e and f show the western blot densitometric analysis of GDF15 protein expression in cell lysates in BEAS-2B (1e) and HPMEC (1f) cell lines. The results were similar to those seen in ELISA experiments, with significant induction in the 13kD segment of GDF-15, which is the mature active form, in both cell lines with hyperoxia exposure. Figure 1g depicts a representative western blot from BEAS-2B cells showing induction of protein in hyperoxia compared to cells in room air. Similar results were seen in HPMEC (shown in supplementary figure 1).
Figure 1. Induction of GDF15 expression in pulmonary epithelial and endothelial cells in hyperoxia.

BEAS-2B (a,c,e,g) and HPMEC (b,d,f) cells exposed to room air (room air-5% CO2) and hyperoxia (95% O2-5% CO2) up to 72 hr. GDF15 expression at the mRNA level (a,b) and at the protein level (ELISA (c,d) and western blot (e-g) was measured. The protein band for GDF15 is the monomer (13kD). β-actin was used as the loading control for the western blot experiments. Values are means ± SEM of 3 independent biological replicates. Significant differences between corresponding room air and hyperoxia exposed cells are indicated by *P <0.05, **P <0.01 and ***P <0.001. Significant differences from 0 hr values are indicated by # P <0.05 and ### P < 0.001.
3.2. Knockdown of GDF15 abrogates the induction in hyperoxia
To achieve silencing of GDF15, we performed siRNA transfection of BEAS-2B and HPMEC cells using GDF15 siRNA, and used cells transfected with siRNA with a scrambled sequence as controls. Figure 2a and 2b demonstrate the successful knockdown of GDF15 mRNA expression with siRNA in BEAS-2B (2a) and HPMEC (2b) cells. As can be seen in figures 2c and 2d, cells transfected with GDF siRNA showed lesser induction in GDF15 mRNA upon exposure to hyperoxia compared to controls. Similar results were also obtained with ELISA (Figures 2e and f). In BEAS-2B (2e) cells, there was no increase in GDF-15 levels in cells transfected with GDF15 siRNA, followed by exposure to hyperoxia. In HPMEC cells (2f) though there was increase in levels in GDF15 siRNA transfected cells, the levels were higher in controls at each time point under hyperoxic conditions.
Figure 2. Knockdown of GDF15 abrogates GDF15 induction in hyperoxia.

BEAS-2B (a,c,e) and HPMEC (b,d,f) cells exposed to room air (room air-5% CO2) and 24, 48, or 72 h of hyperoxia (95% O2-5% CO2). GDF15 siRNA or negative control siRNA were transfected into BEAS-2B and HPMEC cells. Expression of GDF-15 was measured at the mRNA (a-d) and protein level (e,f) by ELISA. Values are means ± SEM of 3 independent biological replicates. Significant differences between GDF15 siRNA and negative control siRNA groups are indicated by *P <0.05, **P<0.01 and ***P <0.001.
3.3. Effects of GDF15 knockdown on cell viability and oxidative stress in BEAS-2B or HPMEC cells exposed to hyperoxia
To investigate whether GDF15 modulates hyperoxic lung injury, control or GDF15 siRNA transfected BEAS-2B and HPMEC cells were exposed to air or hyperoxia for up to 72 hr, following which the cells were harvested to determine cell viability (MTT assay) and H2O2 generation as a marker of oxidative stress. Hyperoxia significantly decreased cell viability (3a), and increased oxidative stress (3b) in GDF15 deficient BEAS-2B cells compared to cells transfected with siRNA with a scrambled sequence. In pulmonary endothelial cells, silencing of GDF15 similarly decreased cell viability (3c) and increased ROS generation compared to controls (3d). We confirmed the effects of GDF15 knockdown on cell viability and ROS generation with a second siRNA and results were similar. This is shown in supplementary figure 2.
3.4. Effect of p53 knockdown on GDF15 expression in BEAS-2B and HPMEC cells exposed to hyperoxia
To study the role of p53 in GDF15 induction in hyperoxia, we achieved p53 knockdown in BEAS-2B (Figure 4a) and HPMEC (Figure 4b). After silencing p53, the induction in GDF15 mRNA in BEAS-2B cells exposed to hyperoxia was significantly decreased by 94% when compared to controls (Figure 4c). GDF15 protein levels in cell culture supernatants (measured by ELISA), also showed significant attenuation of protein levels after p53 knockdown in BEAS-2B cells at 48 (by 40%) and 72 hr (by 33%) (Figure 4e). In HPMEC cells, p53 knockdown similarly abrogated the induction in GDF15 induction at the transcriptional level (Figure 4d) and translational level (Figure 4f) at 24, 48, and 72 hr by 60, 40, and 45% respectively.
Figure 4. p53 knockdown decreases GDF15 expression in BEAS-2B or HPMEC cells exposed to hyperoxia.

p53 siRNA or negative control siRNA were transfected into BEAS-2B and HPMEC cells. Cells exposed to room air (room air-5% CO2) and up to 72 hr of hyperoxia (95% O2-5% CO2). Figures 4a and 4b show successful p53 knockdown at the mRNA level in BEAS-2B (a) and HPMEC cells (b). Expression of GDF15 was measured at the mRNA (c, d) and protein level (e,f) in BEAS-2B (c, e) and HPMEC (d, f) cells. Values are means ± SEM of 3 independent biological replicates. Significant differences between p53 siRNA and negative control siRNA groups are indicated by *P <0.05, **P<0.01 and ***P <0.001.
4. Discussion
In the present study, we documented up regulation of GDF15, a secreted cytokine and a member of the TGF- β superfamily in pulmonary epithelial and endothelial cells after hyperoxia exposure. Using siRNA mediated gene silencing, we further established that loss of function of this gene leads to increased oxidative stress and decreased cell viability. In addition, we show that p53 plays a crucial role in hyperoxia induced GDF15 expression in pulmonary epithelial and endothelial cells. To the best of our knowledge, this is the first study showing that GDF15 is induced and may play a pro-survival and anti-oxidant role under hyperoxic conditions in human pulmonary epithelial and endothelial cells in vitro. These findings need to be further substantiated in relevant in vivo studies.
Exposure to supraphysiological concentrations of oxygen (hyperoxia) leads to lung injury in animal models and causes cell death in cultured cells (Buczynski et al., 2013; Hilgendorff et al., 2013; O'Reilly, 2001). It contributes to the development of diseases such as BPD in premature neonates and ARDS in children and adults. ROS generated during hyperoxia lead to damage of cellular components including DNA, lipid and protein ultimately leading to cell death. Being the first organ to encounter hyperoxia through inhalation, hyperoxia adversely affects all cell types in the lung, the endothelial cells being the most sensitive (O'Reilly, 2001). Both BEAS-2B (bronchial epithelial cell line) (Zhang et al., 2009) and HPMEC (human pulmonary microvascular endothelial cells) (Wright et al., 2010) have been used as models to study the effects of pulmonary oxygen toxicity in vitro.
GDF15 is a member of the TGF- β superfamily and is expressed in virtually all tissues. The exact biological functions of GDF15 are still poorly understood. GDF15 is synthesized as an inactive precursor that undergoes proteolytic processing involving removal of an N-terminal hydrophobic signal sequence followed by cleavage generating an active C-terminal domain that is secreted as a dimeric protein (Wang et al., 2013). It is up regulated in many pathological conditions, including inflammation, malignancy, and cardiovascular, pulmonary and renal disease (Nickel et al., 2011).
In situ hybridization studies in rats showed GDF15 expression in bronchial epithelial cells (Böttner et al., 1999). It is expressed and induced in response to hypoxia in human pulmonary vascular endothelial cells and treatment with recombinant human GDF15 decreased apoptosis and improved cellular proliferation. GDF15 is induced in animal models of lung injury (McGrath-Morrow et al., 2014; Zimmers et al., 2005). We have shown that GDF15 was among the top five genes induced in the murine lungs in vivo when subjected to hyperoxia (Lingappan et al., 2014).
GDF15 has been shown to have both pro- and anti-apoptotic effects depending on the tissue type and the cellular environment. It prevented low potassium induced death in cerebellar granule cells (Subramaniam et al., 2003) and had an anti-apoptotic effect on cultured cardiomyocytes, subjected to simulated ischemia/reperfusion and endothelial cells subjected to hypoxia (Kempf et al., 2006; Song et al., 2012) or high glucose stimulus (Li et al., 2013a). Agarwal et al. showed that application of exogenous GDF-15 or its constitutive expression from a cDNA provided remarkable protection of p53-null cells from apoptosis mediated by phosphonacetyl-l-aspartate, which blocks the synthesis of pyrimidines (Agarwal et al., 2006). GDF15 is a marker of ongoing oxidative stress and is consistently up regulated in such conditions. We show in our study that silencing of GDF15 increases oxidative stress in pulmonary epithelial and endothelial cells and point towards an anti-oxidant role of this protein. Li et al reported similar results in HUVEC cells exposed to high glucose conditions (Li et al., 2013b).
GDF15 is known to be a direct target of p53 (Kang et al., 2013; Kannan et al., 2000; Osada et al., 2007), which in turn is induced after hyperoxia exposure (Maniscalco et al., 2005; O'Reilly et al., 1998; Shenberger and Dixon, 1999). The interaction between GDF15 and p53 in response to hyperoxia exposure has not been investigated. Hyperoxia leads to DNA damage and induction of p53 and also causes the cell cycle arrest at the G1-S phase, which allows DNA repair to occur (O'Reilly, 2001). Agarwal et al observed that GDF15 mediated a p53-dependent protective arrest in S phase in response to starvation for DNA precursors (Agarwal et al., 2006). Zimmers et al on the other hand showed that in response to hyperoxic lung injury, induction in GDF-15 was unchanged in p53 null mice (Zimmers et al., 2005). We report herein that in BEAS-2B and HPMEC cells, silencing of p53 decreases the induction of GDF-15 at the transcriptional level indicating that increase in GDF-15 expression is at least partly mediated by p53 in hyperoxia in these cell types.
In summary, we demonstrate that hyperoxia induces GDF15 expression in human pulmonary epithelial and endothelial cells, which has a pro-survival and anti-oxidant effect in these cells. Also, the induction of GDF15 in response to hyperoxia is mediated through p53 in these cell lines. The present study provides an insight into the possible mechanism of protective effect of GDF15 in diseases such as BPD and ARDS, and future studies could lead to GDF15 as a target for prevention and/or treatment of these pulmonary diseases in humans.
Supplementary Material
Figure 3. GDF15 knockdown decreases cell viability and increases oxidative stress in BEAS-2B or HPMEC cells exposed to hyperoxia.

BEAS-2B (a,b) and HPMEC (c,d) cells were cultured in room air or hyperoxia were subjected to the MTT assay and ROS-Glo™ luminescent H2O2 assay. Values are means ± SEM of 3 independent biological replicates. Significant differences between GDF15 siRNA and negative control siRNA groups are indicated by *P <0.05, **P<0.01 and ***P <0.001. Significant differences between corresponding 0 hr values are indicated by # P <0.05 and ### P < 0.001.
Highlights.
GDF15 is induced in pulmonary epithelial and endothelial cells by hyperoxia.
Loss of GDF15 decreases cell viability and increases oxidative stress.
p53 plays a major role in the induction of GDF15 in hyperoxic conditions.
Acknowledgments
This work was supported in part by NIH grants HL-112516, HL-087174, ES009132, and ES-019689 to BM, and HL-127103 to KL.
Abbreviations
- BPD
Bronchopulmonary dysplasia
- ARDS
Acute Respiratory Distress Syndrome
- ROS
Reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal MK, Hastak K, Jackson MW, Breit SN, Stark GR, Agarwal ML. Macrophage inhibitory cytokine 1 mediates a p53-dependent protective arrest in S phase in response to starvation for DNA precursors. Proc Natl Acad Sci U S A. 2006;103:16278–16283. doi: 10.1073/pnas.0607210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttner M, Suter-Crazzolara C, Schober A, Unsicker K. Expression of a novel member of the TGF-beta superfamily, growth/differentiation factor-15/macrophage-inhibiting cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res. 1999;297:103–110. doi: 10.1007/s004410051337. [DOI] [PubMed] [Google Scholar]
- Buczynski BW, Maduekwe ET, O'Reilly MA. The role of hyperoxia in the pathogenesis of experimental BPD. Semin Perinatol. 2013;37:69–78. doi: 10.1053/j.semperi.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BJ, Bull TM, Benson AB, Stream AR, Macht M, Gaydos J, Meadows C, Burnham EL, Moss M the ARDS Network Investigators. Growth differentiation factor-15 and prognosis in acute respiratory distress syndrome: a retrospective cohort study. Crit Care. 2013;17:R92. doi: 10.1186/cc12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ES, Muller FL, Pérez VI, Qi W, Liang H, Xi L, Fu C, Doyle E, Hickey M, Cornell J, Epstein CJ, Roberts LJ, Van Remmen H, Richardson A. The in vivo gene expression signature of oxidative stress. Physiol Genomics. 2008;34:112–126. doi: 10.1152/physiolgenomics.00239.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorff A, Reiss I, Ehrhardt H, Eickelberg O, Alvira CM. Chronic Lung Disease in the Preterm Infant: Lessons Learned From Animal Models. American Journal of Respiratory Cell and Molecular Biology. 2013 doi: 10.1165/rcmb.2013-0014TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YJ, Lee JH, Kim YM, Oh GT, Lee H. Macrophage inhibitory cytokine-1 stimulates proliferation of human umbilical vein endothelial cells by up-regulating cyclins D1 and E through the PI3K/Akt-, ERK-, and JNK-dependent AP-1 and E2F activation signaling pathways. Cellular Signalling. 2012;24:1485–1495. doi: 10.1016/j.cellsig.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Kang SU, Lee BS, Lee SH, Baek SJ, Shin YS, Kim CH. Expression of NSAID-activated gene-1 by EGCG in head and neck cancer: involvement of ATM-dependent p53 expression. J Nutr Biochem. 2013;24:986–999. doi: 10.1016/j.jnutbio.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Kannan K, Amariglio N, Rechavi G, Givol D. Profile of gene expression regulated by induced p53: connection to the TGF-beta family. FEBS Lett. 2000;470:77–82. doi: 10.1016/s0014-5793(00)01291-6. [DOI] [PubMed] [Google Scholar]
- Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, Heineke J, Kotlarz D, Xu J, Molkentin JD, Niessen HW, Drexler H, Wollert KC. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circulation Research. 2006;98:351–360. doi: 10.1161/01.RES.0000202805.73038.48. [DOI] [PubMed] [Google Scholar]
- Kempf T, Wollert KC. Risk stratification in critically ill patients: GDF-15 scores in adult respiratory distress syndrome. Crit Care. 2013;17:173. doi: 10.1186/cc12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, Bolomini-Vittori M, Korf-Klingebiel M, Napp LC, Hansen B, Kanwischer A, Bavendiek U, Beutel G, Hapke M, Sauer MG, Laudanna C, Hogg N, Vestweber D, Wollert KC. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011;17:581–588. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kosak JP, Kim JK, Kissling G, Germolec DR, Zeldin DC, Bradbury JA, Baek SJ, Eling TE. NAG-1/GDF15 transgenic mouse has less white adipose tissue and a reduced inflammatory resp onse. Mediators Inflamm. 2013;2013:641851. doi: 10.1155/2013/641851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang L, Qin W, Zhang G, Yuan J, Wang F. Adaptive induction of growth differentiation factor 15 attenuates endothelial cell apoptosis in response to high glucose stimulus. PLoS ONE. 2013a;8:e65549. doi: 10.1371/journal.pone.0065549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang L, Qin W, Zhang G, Yuan J, Wang F. Adaptive Induction of Growth Differentiation Factor 15 Attenuates Endothelial Cell Apoptosis in Response to High Glucose Stimulus. PLoS ONE. 2013b;8:e65549. doi: 10.1371/journal.pone.0065549.g007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingappan K, Jiang W, Wang L, Couroucli XI, Moorthy B. Analysis of the transcriptome in hyperoxic lung injury and sex-specific alterations in gene expression. PLoS ONE. 2014;9:e101581. doi: 10.1371/journal.pone.0101581.t004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madurga A, Mizíková I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. AJP: Lung Cellular and Molecular Physiology. 2013;305:L893–905. doi: 10.1152/ajplung.00267.2013. [DOI] [PubMed] [Google Scholar]
- Maniscalco WM, Watkins RH, Roper JM, Staversky R, O'Reilly MA. Hyperoxic ventilated premature baboons have increased p53, oxidant DNA damage and decreased VEGF expression. Pediatr Res. 2005;58:549–556. doi: 10.1203/01.pdr.0000176923.79584.f7. [DOI] [PubMed] [Google Scholar]
- McGrath-Morrow SA, Lauer T, Collaco JM, Lopez A, Malhotra D, Alekseyev YO, Neptune E, Wise R, Biswal S. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine. 2014;65:4–9. doi: 10.1016/j.cyto.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel N, Jonigk D, Kempf T, Bockmeyer CL, Maegel L, Rische J, Laenger F, Lehmann U, Sauer C, Greer M, Welte T, Hoeper MM, Golpon HA. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res. 2011;12:62. doi: 10.1186/1465-9921-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly MA. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am J Physiol Lung Cell Mol Physiol. 2001;281:L291–305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- O'Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. American Journal of Respiratory Cell and Molecular Biology. 1998;18:43–50. doi: 10.1165/ajrcmb.18.1.2950m. [DOI] [PubMed] [Google Scholar]
- Osada M, Park HL, Park MJ, Liu JW, Wu G, Trink B, Sidransky D. A p53-type response element in the GDF15 promoter confers high specificity for p53 activation. Biochem Biophys Res Commun. 2007;354:913–918. doi: 10.1016/j.bbrc.2007.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusch MR, Baeuerle M, Albrecht C, Blessing E, Bischof M, Katus HA, Bea F. GDF-15 protects from macrophage accumulation in a mouse model of advanced atherosclerosis. Eur J Med Res. 2013;18:19. doi: 10.1186/2047-783X-18-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad OD. Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin Neonatol. 2003;8:39–49. doi: 10.1016/s1084-2756(02)00194-x. [DOI] [PubMed] [Google Scholar]
- Shenberger JS, Dixon PS. Oxygen induces S-phase growth arrest and increases p53 and p21(WAF1/CIP1) expression in human bronchial smooth-muscle cells. American Journal of Respiratory Cell and Molecular Biology. 1999;21:395–402. doi: 10.1165/ajrcmb.21.3.3604. [DOI] [PubMed] [Google Scholar]
- Song H, Yin D, Liu Z. GDF-15 promotes angiogenesis through modulating p53/HIF-1α signaling pathway in hypoxic human umbilical vein endothelial cells. Mol Biol Rep. 2012;39:4017–4022. doi: 10.1007/s11033-011-1182-7. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Strelau J, Unsicker K. Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of Akt and ERK pathways. J Biol Chem. 2003;278:8904–8912. doi: 10.1074/jbc.M210037200. [DOI] [PubMed] [Google Scholar]
- Wang X, Baek SJ, Eling TE. The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem Pharmacol. 2013;85:597–606. doi: 10.1016/j.bcp.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CJ, Agboke F, Chen F, La P, Yang G, Dennery PA. NO Inhibits Hyperoxia-Induced NF-κB Activation in Neonatal Pulmonary Microvascular Endothelial Cells. Pediatr Res. 2010;68:484–489. doi: 10.1203/PDR.0b013e3181f917b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lin L, Lee SJ, Mo L, Cao J, Ifedigbo E, Jin Y. Deletion of caveolin-1 protects hyperoxia-induced apoptosis via survivin-mediated pathways. AJP: Lung Cellular and Molecular Physiology. 2009;297:L945–L953. doi: 10.1152/ajplung.00081.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers TA, Jin X, Hsiao EC, McGrath SA, Esquela AF, Koniaris LG. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock. 2005;23:543–548. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.