

Abstract
Ligands of Peroxisome Proliferator Activated Receptor gamma (PPARγ) possess strong anti-fibrotic properties in the cornea and several other body tissues. In the cornea, we recently showed this class of molecules to prevent stromal myofibroblast differentiation partially by blocking the actions of p38 mitogen-activated protein kinase (MAPK). However, given the important role assigned to connective tissue growth factor (CTGF) in mediating corneal fibrosis, here we asked whether PPARγ ligands also act by affecting transforming growth factor-β (TGF-β 1-induced expression of CTGF in cultured corneal fibroblasts. Corneal keratocytes were isolated from young, adult cats and early passage cells were exposed to TGF-β1 with or without the PPARγ ligands Rosiglitazone, Troglitazone and 15d-PGJ2. Western blots were used to assay levels of CTGF and alpha smooth muscle actin (αSMA), a marker of myofibroblast differentiation. CTGF siRNA demonstrated a critical role for CTGF in TGF-β1-mediated myofibroblast differentiation, while exogenously applied CTGF potentiated the pro-fibrogenic effects of TGF-β1. TGF-β1-mediated increases in CTGF and αSMA expression were strongly inhibited by all three PPARγ ligands tested, and by a c-jun N-terminal kinase (JNK) inhibitor. However, while extracellular signal-regulated kinase (ERK) 1/2, protein kinase B (AKT) and p38 MAPK inhibitors also blocked TGF-β1-induced αSMA induction, they did not dampen TGF-β1-induced increases in levels of CTGF. Thus, we conclude that PPARγ ligands block TGF-β-induced increases in CTGF levels in cat corneal fibroblasts. They appear to do this in addition to their anti-fibrotic effect on p38 MAPK, providing a second intracellular pathway by which PPARγ ligands block αSMA induction.
Keywords: Rosiglitazone, Troglitazone, 15d-PGJ2, JNK, p38 MAPK, ERK, AKT
1. Introduction
Following damage to the corneal surface, surviving keratocytes are exposed to multiple wound healing modulators - including transforming growth factor-β (TGF-β - the strongest known pro-fibrotic agent (Jester et al 1996, Vesaluoma et al 1997). As a result, keratocytes adjacent to the damaged zone proliferate, migrate, transform into fibroblasts (Fini 1999) and then, into myofibroblasts (Jester et al 1987). Activated TGF-β receptors appear to regulate multiple genes that control the differentiation of fibroblasts into myofibroblasts, and the actions of myofibroblasts within their immediate extracellular matrix (ECM) environment (Desmouliere et al 1993, Jester et al 1995, Wilson 2012). One molecule thought to regulate many of the cell-ECM interactions during fibrosis and scar formation is connective tissue growth factor (CTGF) (Garrett et al 2004). TGF-β has been shown to directly increase CTGF mRNA and protein levels within cells (Blalock et al 2003, Igarashi et al 1996, Kikuchi et al 1995, Tall et al 2010).
CTGF is a major autocrine growth factor, which mediates the actions of TGF-β with respect to myofibroblast differentiation and behavior (Grotendorst 1997, Grotendorst et al 1996, Lipson et al 2012). CTGF is a secreted, cysteine-rich molecule approximately 36–38kD in size, which was initially identified in media conditioned by human umbilical vein endothelial cells, as a new fibroblast mitogen (Bradham et al 1991). CTGF is also known as CCN2, as it belongs to the CCN (cyr61, ctgf, neuroblastoma over-expressed gene (nov)) family, an important set of matrix-cellular regulatory factors involved in internal and external cell signaling (Moussad & Brigstock 2000). This family participates in angiogenesis, chondrogenesis, and wound healing (Perbal 2004), and appears to be a central mediator of tissue remodeling and fibrosis. TGF-β directly regulates the expression of CTGF in primary skin fibroblasts (Igarashi et al 1993), human pulmonary fibroblasts (Wang & Liu 2006), and in corneal fibroblasts (Folger et al 2001). Histological evaluation of corneal scar formation in pseudophakic bullous keratopathy showed an increase in CTGF expression and in the number of TGF-β-positive stromal cells (Liu et al 2012). Furthermore, CTGF protein and mRNA levels increased in rat cornea through day 21 after photorefractive keratectomy (PRK) (Blalock et al 2003). Therefore, CTGF appears to be an important effector molecule in both physiological and pathological processes associated with fibrosis (Wahab et al 2001) and may provide a new target for therapeutic interventions to mitigate fibrotic diseases (Moussad & Brigstock 2000).
In the cornea, fibrosis and scarring lead to vision loss, and remain without effective treatment. A solution would be a drug that selectively targets the TGF-β cascade, but with minimal side-effects. Our group and others have shown several ligands of PPARγ to be effective corneal anti-fibrotics in vitro (Jeon et al 2014, Kuriyan et al 2012, Pan et al 2009, Pan et al 2011) and in vivo (Huxlin et al 2013, Jeon et al 2014). PPARγ is a transcription factor belonging to a nuclear receptor superfamily that regulates important cellular functions, including metabolism, adipogenesis, proliferation, differentiation and inflammatory responses (Simpson-Haidaris et al 2010). A significant body of data suggests that PPARγ ligands can act as anti-fibrotics in a range of body tissues, including lung (Ferguson et al 2009, Lin et al 2010, Zhou et al 2012), skin (Ghosh et al 2004), kidneys (Liu et al 2011) and cornea (Huxlin et al 2013, Jeon et al 2014, Kuriyan et al 2012, Pan et al 2009, Pan et al 2011). In the cornea, two such ligands - Rosiglitazone and Troglitazone – were recently shown effective at control fibrosis after PRK, without significant side-effects (Huxlin et al 2013, Jeon et al 2014). Here, we asked whether PPARγ ligands exert part of their anti-fibrotic actions in the cornea by influencing TGF-β1-induced CTGF expression in stromal fibroblasts - a potentially novel mechanism of action for this promising group of molecules.
2. Materials and methods
All animal procedures were conducted according to the guidelines of the University of Rochester Committee on Animal Research (UCAR), the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the NIH Guide for the Care and Use of Laboratory Animals.
2.1. Isolation and culture of cat corneal fibroblasts
Primary feline corneal keratocytes were isolated as previously described (Huxlin et al 2013, Jeon et al 2014). In brief, fresh corneas were obtained immediately post-mortem from young, adult domestic short-hair cats (felis cattus). The corneal epithelium and endothelium were scraped off. After double enzyme digestion, the stroma was centrifuged at 500 rpm for 2 min at 4°C and the pellet removed. The resulting supernatant, containing mostly keratocytes, was centrifuged again at 3000 rpm for 10 min and was re-suspended in 2 ml of Fibroblast Growth Factor-containing medium (PromoCell GmbH, Germany), counted and seeded onto culture plates (Greiner Bio-one; Monroe, NC ). All experiments were performed in triplicate.
2.2. Measuring the effect of TGF-β1 on CTGF and αSMA expression
Passage 6–7 feline, corneal fibroblasts were seeded at a density of 1×105–2×105 cells/ 6-cm dish containing DMEM/F12 + 5% HS. The medium was changed to one containing DMEM/F12 + 1% HS for 1 day in order to promote cellular quiescence. Western blots were used to detect the expression of CTGF and αSMA relative to the expression of β-Tubulin as a loading control, as described previously (Huxlin et al 2013, Jeon et al 2014). Expression of αSMA was used as positive control to indicate myofibroblast differentiation. A 10% gel was run using 10–20 μg of cell lysates separated by electrophoresis, transferred to a nitrocellulose membrane and probed with primary antibodies - rabbit polyclonal anti-CTGF (1:1000, from ab6992 Abcam Inc. Cambridge, MA); mouse monoclonal anti-αSMA, (1:10,000, from Thermo Fisher Scientific, Pittsburgh, PA) and mouse monoclonal β-Tubulin (1:5000, from Santa Cruz Inc., Santa Cruz, CA). Primary antibodies were incubated overnight at 4°C, the membranes rinsed and secondary antibodies (anti-mouse IgG or anti-rabbit IgG-horse radish peroxidase, GE Healthcare; Piscataway, NJ) were applied and incubated for 1hr at room temperature. The membranes were scanned with a Chemi-doc machine (Bio-Rad, Hercules, CA) and the resulting images were imported into Image J (NIH) for measurement of relative protein expression.
2.3. Effect of CTGF siRNA on TGF-β1-induced CTGF and αSMA expression
To evaluate the role of CTGF in TGF-β1-induced myofibroblast differentiation, we used CTGF siRNA (5′-GAGAGACATTAACTCATTA-3′, cat. J-012633–13–05, GE Dharmacon) to knock-down expression of CTGF. Non-targeting siRNA (cat. D-001810-01-05, GE Dharmacon) was used as a negative control. Lipofectamine RNAiMAX (Invitrogen) reagent was used to perform siRNA transfections as per the manufacturer’s instructions. Briefly, 1×105 cells/6-well plate were seeded in DMEM/F12 with 5% HS. After attachment, cells were changed into reduced serum-containing media for 1 day, then transfected with CTGF siRNA or control siRNA. About 24 hours after transfection, cells were treated with recombinant human TGF-β1 (1 ng/ml, R&D Systems, Inc.; Minneapolis, MN). Two days later, cells were harvested and Western blots were used to estimate the expression of CTGF and αSMA relative to that of β-Tubulin.
2.4. Effect of exogenous CTGF on time-dependent αSMA expression
To assess whether exogenously-provided CTGF could induce the expression of pro-fibrotic molecules in corneal fibroblasts independent of TGF-β1, corneal fibroblasts were seeded at a density of 5×104cells/well in 6-well plates containing DMEM/F12 + 5% HS. After attachment, the medium was changed to one containing DMEM/F12 + 1% HS for 1 day in order to promote cellular quiescence. Cells were treated with 50ng of recombinant, C-terminus CTGF (Shenandoah Biotechnology, Warwick, PA) with or without 1ng/ml recombinant human TGF-β1 for indicated times. Western blots were used to quantify the expression of αSMA relative to that of β-Tubulin.
2.5. Effect of PPARγ ligands on TGF-β1-induced CTGF expression
We tested the effects of 3 different PPARγ ligands - Troglitazone, Rosiglitazone and 15-deoxy-delta12, 14-prostaglandin J2 (15d-PGJ2) - on CTGF expression after TGF-β1 stimulation. Cells were pre-treated with optimal doses of the three PPARγ ligands of interest, determined to decrease TGF-β1-induced expression of αSMA, COLI and FN in cultured feline corneal fibroblasts in a prior in vitro study using identical cell culture conditions (Jeon et al 2014). Either 15μM Troglitazone (Cayman; Ann Arbor MI), 75μM Rosiglitazone (Cayman; Ann Arbor MI), or 5μM 15d-PGJ2 (Enzo; Plymouth Meeting, PA) were applied to the cells in 1% HS in DMEM/F12 medium for 30 min. TGF-β1 (1 ng/ml) was added to the culture medium. Cells were harvested 1day later and Western blots were used to quantify expression of CTGF relative to that of β-Tubulin, as described earlier.
2.6. Effect of kinase inhibitors on TGF-β-induced CTGF and αSMA expression
In order to assess whether the molecular pathways mediating the effects of PPARγ ligands on CTGF were similar to those impacting myofibroblast differentiation, we used small-molecule kinase inhibitors to block four major, known, endogenous, pro-fibrotic protein kinases (Mu et al 2012). Specifically, we used the ERK inhibitor U0126 (Marampon et al 2011), the p38 MAPK inhibitor SB203580 (Barancik et al 2001), the AKT inhibitor LY294002 (Gharbi et al 2007) and the JNK inhibitor SP600125 (Wang et al 2007). All inhibitors were obtained from Calbiochem (San Diego, CA). Passage 6–7 cells were pre-treated for 30 mins with optimal doses of each kinase inhibitor (determined previously (Jeon et al 2014)), except for the JNK inhibitor, for which different doses had to be tested anew. All inhibitors were dissolved in 1% HS-DMEM/F12. After 30 mins, 1 ng/ml of TGF-β1 was added to the medium and the cells were incubated for 3 days, at which point Western blots were performed to assess the expression of CTGF relative to that of β-Tubulin. Expression of αSMA was used as a positive control for myofibroblast differentiation
2.7. Statistical analysis
In order to estimate differences in protein expression levels on Western blots, when three or more groups were compared, inter-group differences were tested with an ANOVA. When only two groups were compared, a two-sided Student’s t-test was performed. A probability of error of p<0.05 was considered statistically significant. All statistical tests were performed using the SPSS 20.0.0 software package (SPSS Inc., Chicago, Ill.).
3. Results
3.1. TGF-β1 increases expression of CTGF before αSMA
In the absence of TGF-β stimulation, basal levels of CTGF expression in cultured corneal fibroblasts were low but distinctly above zero (Fig. 1A, first lane), suggesting that CTGF was constitutively synthesized and expressed at low levels in culture. Addition of TGF-β1 to the culture medium induced a gradual increase in levels of CTGF protein relative to the untreated condition, starting within 1 hour and peaking at 2–3 days after onset of TGF-β1 treatment (Fig. 1A, B). Of note is the faster rate of increase of CTGF expression relative to that of αSMA over the first 24 hrs after TGF-β1 stimulation. During this time, αSMA levels remained almost flat, exhibiting the first significant increase in expression on the second day after TGF-β1 stimulation. A repeated measured ANOVA confirmed this, with a significant main effect of time in culture [F (8,32)=42.519, p<0.0001), a significant main effect of CTGF/αSMA expression [F (1,4)=9.752, p=0.035) and a significant interaction between the two [F (8,32)=5.426, p<0.0001).
Figure 1. Time-dependent TGF-β-induced expression of CTGF and αSMA in cultured cat corneal fibroblasts.
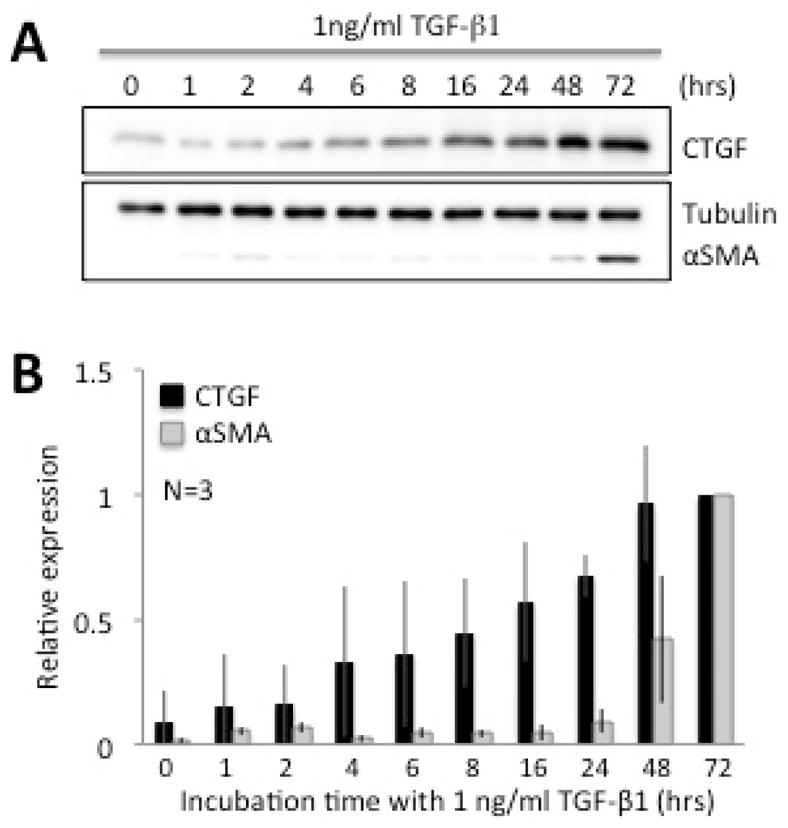
A. Representative western blots showing protein levels for CTGF and αSMA at different time-points (in hours) after TGF-β1 stimulation. β-Tubulin levels were assayed as a loading control. The basal level of CTGF was distinctly above zero. After 4 hours of culture, TGF-β1 started to affect the expression of CTGF, while αSMA expression remained too low to assay reliably until 48 hrs after stimulation. CTGF protein expression appeared to reach maximal levels between 48 and 72 hrs, while αSMA expression was greatest at 72 hrs, as previously reported for this cell culture system (Jeon et al 2014). β-Tubulin levels remained stable throughout. B. Plots of relative expression of CTGF and αSMA with respect to Tubulin expression at different time points after stimulation with 1ng/ml TGF-β. Values were normalized to the maximal levels attained (at 72 hrs for both CTGF and αSMA). Data shown are means ± SD, averaged over 3 experiments.
3.2. CTGF siRNA blocks TGF-β1-induced CTGF and αSMA expression
After 2 days in culture, corneal fibroblasts transfected with control siRNA, showed strong TGF- β1-induced increases in the expression of CTGF and αSMA relative to un-stimulated fibroblasts (Fig. 2A, B). In contrast, CTGF siRNA suppressed TGF-β1-induced CTGF expression by about 47±16% (mean ± SEM) relative to the control siRNA + TGF-β1 condition. This blockage of CTGF expression was associated with a 52±15% decrease in αSMA expression relative to control siRNA cells stimulated with TGF-β1 alone (Fig. 2A, B). In contrast, total β-tubulin levels were unaffected by addition of either TGF-β1- or siRNA (Fig. 2A). These data suggest that CTGF contributes significantly to TGF-β1-induced cat corneal myofibroblast differentiation.
Figure 2. Role of CTGF in TGF-ββ-induced corneal myofibroblast differentiation.
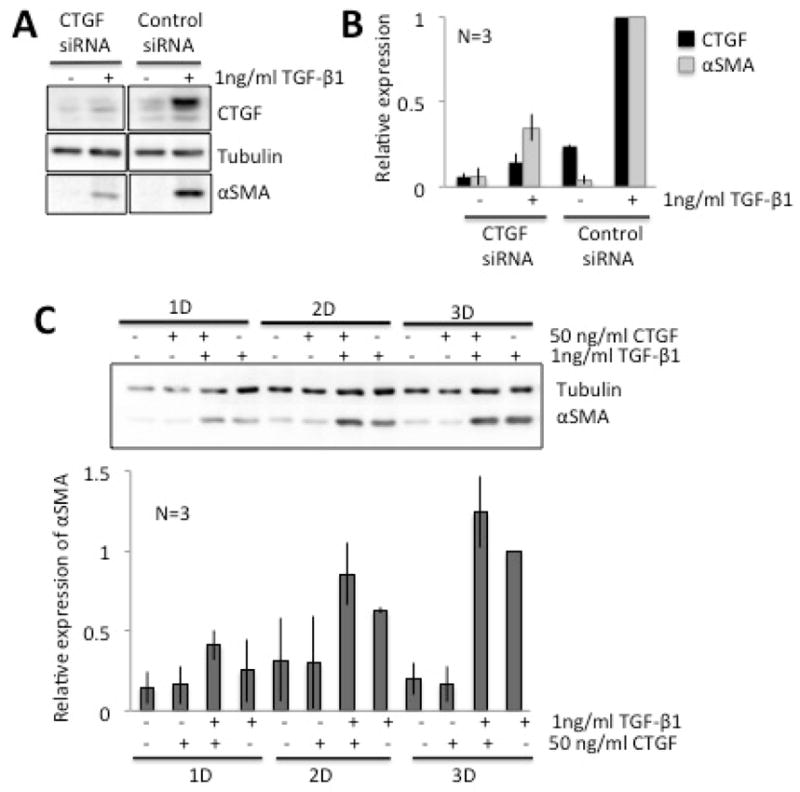
A. Representative Western blot showing protein levels for CTGF and αSMA in cells transfected with either CTGF siRNA or control (non-targeting) siRNA. β-Tubulin levels were assayed as a loading control. CTGF siRNA blocked the TGF-β-induced increases in CTGF and αSMA levels compared with control siRNA. β-tubulin levels remained stable throughout. B. Plot of relative densitometry data for CTGF and αSMA expression with respect to Tubulin, which was further normalized to values obtained in cells stimulated with 1ng/ml TGF-β following transfection with either CTGF or control siRNA (see sample blot in B). C. Representative Western blot showing protein levels for αSMA in cells treated with C-terminus, recombinant human CTGF with/without TGF-β. β-Tubulin levels were assayed as a loading control. Exogenously added CTGF potentiated the TGF- β-induced increase in αSMA levels compared with TGF-β alone, especially after 2 and 3 days in culture. β-tubulin levels remained stable throughout. The graph below is a plot of αSMA expression relative to β-tubulin expression, normalized to values obtained in cells stimulated with 1ng/ml TGF-β for 3 days. Data shown are means ± SD over 3 experiments.
3.3. CTGF potentiates the pro-fibrotic effects of TGF-β1
Next, we assessed whether exogenously supplied CTGF was able to induce corneal myofibroblast differentiation in the absence of TGF-β1 stimulation. As shown in Fig. 2C, CTGF alone did not increase expression of αSMA when used from 1–3 days in culture. Even the highest dose tested −200ng/ml - was unsuccessful at inducing myofibroblast differentiation (data not shown). However, combined exposure of corneal fibroblasts to 1 ng/ml TGF-β1 together with 50ng/ml of CTGF potentiated αSMA expression above levels seen following incubation with 1 ng/ml TGF-β1 alone. A repeated-measures ANOVA contrasting CTGF+TGF-β1 with TGF-β1 treatment across the 3 days of culture revealed a significant effect of time [F(2,17)=46.5, p<0.0001] and treatment [F(1, 17)=10, p=0.0341] but no significant interaction between time and treatment.
3.4. PPARγ ligands inhibit CTGF expression
All three PPARγ ligands tested (two synthetic ligands - Rosiglitazone, Troglitazone, and the natural ligand 15d-PGJ2) inhibited TGF-β1-induced increases in the expression of CTGF after 1 day in culture (Fig. 3A–C). In all cases, levels of CTGF returned back to baseline (i.e., pre-TGF-β1) levels. Importantly, these effects occurred at doses of the three PPARγ ligands, which were effective at blocking up-regulation of αSMA expression (Jeon et al 2014). In contrast, total β-tubulin levels were unaffected by addition of either TGF-β1 or PPARγ ligands (Fig. 3A–C).
Figure 3. Effect of PPARγ ligands on TGF-β1-induced CTGF expression in cultured corneal fibroblasts.
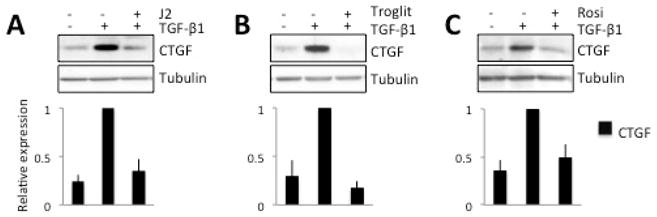
A. Representative Western blots showing protein levels for CTGF and β-tubulin, which was assayed as a loading control. After 1day in culture, 5μM 15d-PGJ2 (J2) blocked the TGF-β-induced increase in CTGF expression, while β-tubulin levels remained stable. These effects are confirmed in the plot of relative expression of CTGF normalized to densitometric values obtained in cells stimulated with 1ng/ml TGF-β. B. Representative blot illustrating the impact of 15μM Troglitazone (T), which also effectively blocked the TGF-β-induced increase in CTGF expression. The densitometry plot below confirms this result over 3 separate experiments. C. Representative blot illustrating the impact of 75μM Rosiglitazone (R) on the TGF-β-induced increase in CTGF expression after 1 day in culture. As in A and B, β-tubulin levels were unaffected. These effects are confirmed in the densitometry plot showing decreased relative expression of CTGF compared to the TGF-β-stimulated condition. In all cases, data shown are means ± SD, averaged over 3 experiments.
3.5. PPARγ ligand effects on CTGF are not mediated by p38 MAPK
Prior work in our laboratory showed PPARγ ligands to exert their anti-fibrotic effects in corneal fibroblasts by blocking phosphorylation of p38 via the p38 MAPK (Jeon et al 2014). To ascertain whether this or other signaling mechanism(s) were involved in regulation of CTGF levels, we used small-molecule inhibitors to block several key endogenous protein kinases, known to be responsible for phosphorylation of p38, Akt, ERK and JNK. We contrasted the impact of these kinase inhibitors on CTGF expression with the impact of one of the PPARγ ligands (75μM Rosiglitazone) previously shown to inhibit corneal fibrosis by inhibiting phosphorylation of p38 (Jeon et al 2014). As shown in Fig. 4A, basal CTGF levels were low but detectable in passage 6–7 corneal fibroblasts (Lane 1). Addition of 1 ng/ml TGF-β1 for 3 days increased both CTGF and αSMA expression (Lane 2, Fig. 3B). Pre-incubation with 75μM Rosiglitazone decreased the TGF-β-induced increases in CTGF and αSMA expression (Lane 3, Fig. 4A). While administration of p38, ERK and AKT kinase inhibitors decreased αSMA expression (as reported in our prior work (Jeon et al 2014)), none of them blocked the TGF-β1-induced increase in CTGF expression (Lanes 4–6, Fig. 4A). As a positive control, we tested an inhibitor of JNK. JNK signaling was previously shown to be critical for CTGF expression and scar formation in the cornea (Chang & Wu 2009, Chang & Wu 2010, Radhakrishnan et al 2012, Shi et al 2012). Consistent with this prior work, administration of SP600125, which inhibits phosphorylation of JNK, blocked TGF-β1-induced increases in the expression of both CTGF and αSMA in a dose-dependent manner (Figs. 4B, 5).
Figure 4. Effect of kinase inhibitors on TGF-β-induced expression of CTGF and αSMA.
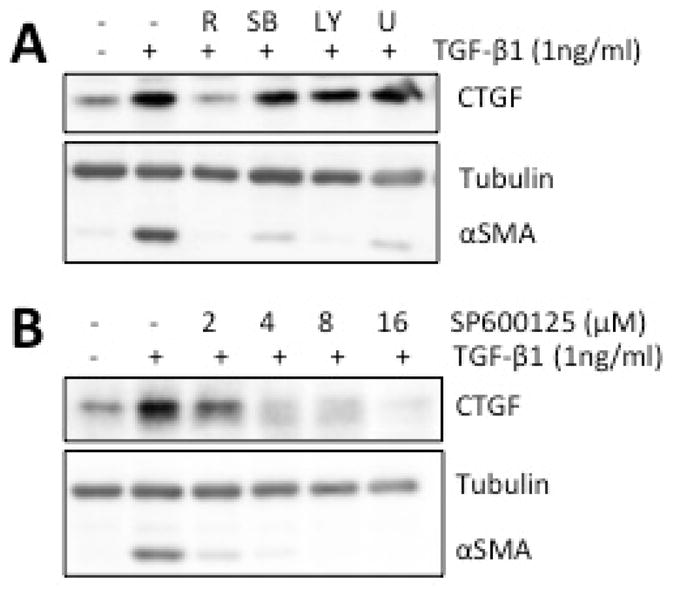
A. Representative Western blots showing the effect of incubating cultures with the PPARγ ligand Rosiglitazone (R, 75 μM) or different kinase inhibitors [for ERK: 10μM U0126 (U), for p38 MAPK: 6μM SB203580 (SB) and for AKT: 5μM LY294002 (LY)]. While Rosiglitazone and all 3 inhibitors blocked TGF-β-induced increases in the expression of αSMA, only Rosiglitazone effectively blocked CTGF induction. β-Tubulin levels were assayed as a loading control and remained unaffected by the different treatments. B. Representative Western blots showing the effect of different doses of the JNK inhibitor (SP600125) on TGF-β-induced increases in the expression of CTGF and αSMA in cultured corneal fibroblasts. β-Tubulin levels were assayed as a loading control. SP600125 blocked the expression of both CTGF and αSMA in a dose-dependent manner, although it appeared to inhibit αSMA at a lower dose than CTGF.
Figure 5. Schematic diagram of pro-fibrotic signaling pathways activated by TGF-ββ binding to its receptor in corneal fibroblasts.
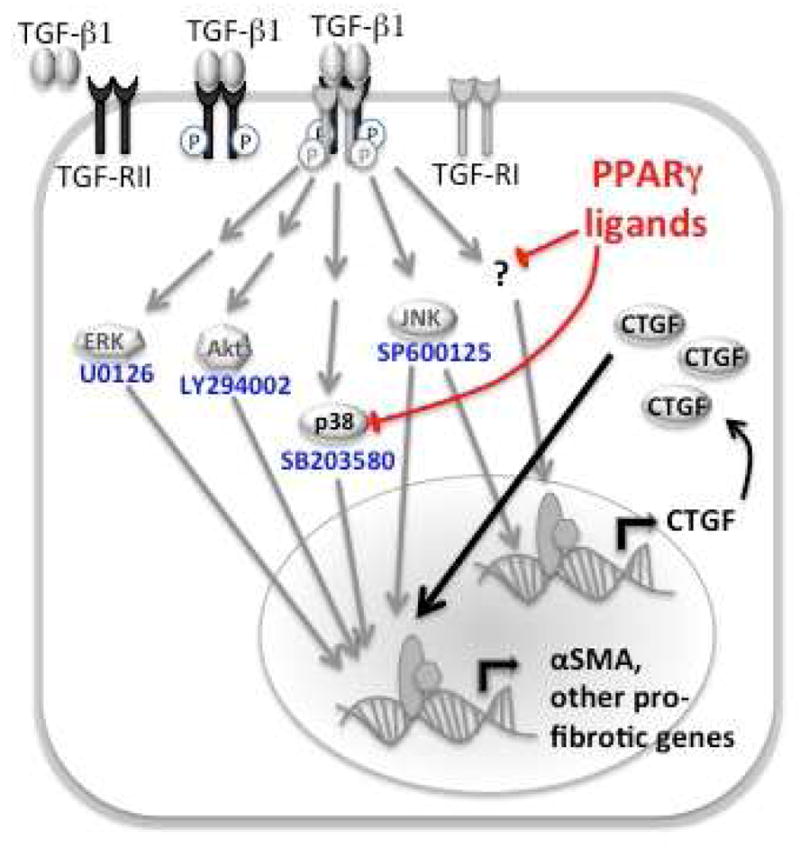
TGF-β1 increases αSMA expression through activation of ERK, Akt, p38, JNK and CTGF. PPARγ ligands block TGF-β1-induced up-regulation of CTGF, independently of p38 MAPK in corneal fibroblasts
4. Discussion
Corneal injury can lead to scar formation by way of corneal fibrosis, which involves the appearance of myofibroblasts and the improper deposition of ECM components (Wilson 2012). TGF-β is perhaps the most important, pro-fibrotic growth factor. It is known to stimulate myofibroblast differentiation and its associated phenotype, which includes the production of αSMA and ECM molecules. However, gaps in knowledge remain with respect to the molecular mechanisms of fibrosis in different body tissues, and how best to control them. Corneal scarring is a predominant affliction worldwide, for which there is no effective treatment without side-effects, and whose consequence when untreated and uncontrolled, can be blindness (Whitcher et al 2001). Finding a safe way of controlling fibrosis in the cornea would be of tremendous public health benefit.
CTGF has been recognized as a principal downstream mediator of TGF-β-induced fibroblast proliferation and differentiation into myofibroblasts (Grotendorst 1997, Grotendorst et al 1996, Lipson et al 2012). As summarized in Fig. 5, in corneal fibroblasts, TGF-β stimulation increases CTGF mRNA and protein levels (Tall et al 2010), and with respect to protein expression, we replicated this finding here. However, we also noted that TGF-β-induced increases in CTGF expression preceded increases in αSMA expression, supporting the notion that CTGF is produced in response to TGF-β as an immediate early gene product (Folger et al 2001, Grotendorst et al 1996). In addition, we confirmed that TGF-β regulates CTGF levels in cat corneal fibroblasts via JNK signaling (Fig. 5), consistent with prior studies in telomerase-immortalized human corneal stromal fibroblasts (Chang & Wu 2009, Chang & Wu 2010, Shi et al 2012) and in quiescent human corneal fibroblasts (Radhakrishnan et al 2012).
CTGF is a modular protein, containing up to four distinct functional domains that can bind diverse growth factors and cytokines (Leask & Abraham 2006). Therefore, this single molecule can modify a range of signal transduction pathways, depending on the particular partners or combination of partners with which it interacts. For example, CTGF binds TGF-β through its N-terminus domain, thus playing an important role in augmenting TGF-β activity (Leask & Abraham 2006). However, there is less information regarding CTGF’s other physiological interactions and effects, although epidermal growth factor (EGF) and insulin-like growth factor (IGF)-2 are both known to influence CTGF-mediated cell proliferation and differentiation (Grotendorst & Duncan 2005).
Curiously, while CTGF appears to control many cell-ECM interactions during fibrosis and scar formation, prior work showed that CTGF alone was not sufficient to induce myofibroblast differentiation, αSMA expression or collagen matrix contraction in human corneal fibroblast (Garrett et al 2004). We confirmed that CTGF alone was not sufficient to induce a pro-fibrotic response in cat corneal fibroblasts, but exogenously-supplied CTGF potentiated the increased expression of αSMA resulting from TGF-β1 stimulation. As a result, we decided to use siRNA to critically evaluate the role of CTGF in TGF-β1-induced myofibroblast differentiation of cat corneal fibroblasts. We found that CTGF knockdown blocked TGF-β1-induced increases in αSMA expression. Of relevance here, CTGF-deficient mouse embryonic fibroblasts exhibit a significant reduction in focal adhesion kinase, PI3kinase, Akt and ERK phosphorylation, in addition to delays in cell spreading and formation of actin stress fibers (Chen et al 2004). Moreover, loss of CTGF expression also impairs the ability of TGF-β to induce type 1 collagen and αSMA expression in such cells (Shi-wen et al 2006). All in all, these results support the notion that CTGF may be a key co-factor, required for TGF-β to induce the signals needed for myofibroblast differentiation. In the absence of CTGF, corneal fibroblasts appear to become refractory to pro-fibrotic stimuli like TGF-β.
One interesting point of note here is that we used two different methods to block the expression of CTGF (a JNK inhibitor and siRNA system) and found them to have a slightly different magnitude/rate of impact on αSMA expression. CTGF siRNA decreased αSMA expression by almost half (Fig. 2A), whereas the JNK inhibitor appeared to completely prevent TGF-β1-induced αSMA expression at very low doses (Fig. 4B). One interpretation of these findings is that the siRNA knockdown was less effective (and transient) as a blocker, and took some time to exert its effect after induction. In contrast, small molecule inhibitors with proven selectivity (such as the one used for JNK) can easily access all cells in a culture, and because they are membrane permeable, can diffuse very rapidly inside cells, providing inhibition fast, selectively and even irreversibly (if covalently bound to their targets).
Having established that CTGF plays an important role in corneal myofibroblast differentiation, our next question was: do PPARγ ligands exert some of their anti-fibrotic actions in the cornea because they prevent TGF-β1-induced increases in CTGF levels inside corneal fibroblasts? Recent work from our laboratory showed Rosiglitazone and Troglitazone – two synthetic PPARγ ligands - to effectively control fibrosis post-PRK without significant side-effects; as summarized in Fig. 5, in vitro, these ligands were shown to act at least partially, by inhibiting p38 MAPK (Huxlin et al 2013, Jeon et al 2014). Here, we report that PPARγ ligands also block TGF-β1-induced increases in CTGF levels in cultured corneal fibroblasts - an effect that appears to be independent of the ligands’ actions on p38 MAPK. Indeed, inhibiting this kinase did not change levels of CTGF in stimulated, cultured fibroblasts. One possibility is that PPARγ ligands block CTGF up-regulation by inhibiting JNK, as in adipocytes (Diaz-Delfin et al 2007). Another possibility (not mutually exclusive) is that PPARγ ligands decrease synthesis of CTGF receptors, thus reducing the uptake of this growth factor from the extracellular milieu. There are several possible candidates for this role, including the low-density lipoprotein receptor-related protein-1 (LRP1) (Kawata et al 2006, Wahab et al 2001) and the mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF-2-R) (Blalock et al 2012). Blalock and colleagues (2012) reported that M6P/IGF-2-R was the most likely receptor to regulate CTGF-stimulated proliferation of human corneal fibroblasts. In contrast, CTGF binding of LRP1 is the major event regulating ECM remodeling and fibrosis in cartilage (Kawata et al 2010). In fact, LRP1 mediates the uptake and intracellular transport of CTGF in chondrocytes (Wahab et al 2001) and in human pulmonary fibroblasts, TGF-β1 stimulation Increases the expression of both CTGF and LRP1 (Wang & Liu 2006). However, the nature of the CTGF receptor(s) in corneal fibroblasts is remains to be determined, and as such, we can only speculate as to whether PPARγ ligands are likely to regulate it.
In conclusion, the present results show that in cultured, feline, corneal fibroblasts, the expression of CTGF is rapidly increased by TGF-β1 stimulation. At least in part, this occurs via regulation of JNK signaling (Fig 5). PPARγ ligands blocked the up-regulation of CTGF at doses that prevented myofibroblast differentiation and increased expression of pro-fibrotic molecules such as αSMA, Fibronectin and Collagen 1. Details about the interactions between CTGF and other pro-fibrotic signaling pathways (Akt, ERK, p38 MAPK) remain to be elucidated, as do regulatory mechanism by which PPARγ ligands block TGF-β1-induced up-regulation of CTGF. However, because CTGF appears to be a critical mediator of TGF-β’s pro-fibrotic effects in the cornea, our findings present a novel mechanism of action by which PPARγ ligands exert their anti-fibrotic actions in an organ whose transparency is essential for our sense of sight.
Highlights.
TGF-β1 increases the expression of CTGF and αSMA in cultured cat corneal fibroblasts
CTGF siRNA blocks TGF-β1-induced myofibroblast differentiation in corneal fibroblasts
PPARγ ligands block TGF-β1-induced up-regulation of CTGF, independently of p38 MAPK
Acknowledgments
The authors thank Margaret DeMagistris for excellent technical assistance. This project was supported by the National Institute of Health (grants # EY015836, EY203239, EY017123, core grant P30 EY001319 to the Center for Visual Science) and an unrestricted grant to the University of Rochester’s Department of Ophthalmology from the Research to Prevent Blindness Foundation. KRH was a recipient of the Lew R. Wasserman Merit Award from the Research to Prevent Blindness Foundation.
Abbreviations
- CTGF
connective tissue growth factor
- CCN
cyr61, ctgf, neuroblastoma over-expressed gene
- Nov
neuroblastoma over-expressed gene
- ECM
extracellular matrix
- PRK
photorefractive keratectomy
- PPARγ
peroxisome proliferator activated receptor gamma
- 15d- PGJ2
15-deoxy-delta 12,14-prostaglandin J2
- UCAR
University of Rochester Committee on Animal Research
- TGF-β
transforming growth factor-beta
- αSMA
alpha smooth muscle actin
- MAPK
mitogen-activated protein kinase
- JNK
c-jun N-terminal kinase
- AKT
protein kinase B
- ERK
extracellular signal-regulated kinase
- FGF
fibroblast growth factor
- LPR1
low-density lipoprotein receptor-related protein-1
- M6P/IGF-2-R
mannose 6-phosphate/insulin-like growth factor2 receptor
- EGF
epidermal growth factor
- (IGF)-2
insulin-like growth factor
- MEFs
mouse embryonic fibroblasts
- FAK
focal adhesion kinase
Footnotes
The authors have no proprietary interest in any devices or pharmacologics used in this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barancik M, Bohacova V, Kvackajova J, Hudecova S, Krizanova O, Breier A. SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2001;14:29–36. doi: 10.1016/s0928-0987(01)00139-7. [DOI] [PubMed] [Google Scholar]
- Blalock TD, Duncan MR, Varela JC, Goldstein MH, Tuli SS, et al. Connective tissue growth factor expression and action in human corneal fibroblast cultures and rat corneas after photorefractive keratectomy. Invest Ophthalmol Vis Sci. 2003;44:1879–87. doi: 10.1167/iovs.02-0860. [DOI] [PubMed] [Google Scholar]
- Blalock TD, Gibson DJ, Duncan MR, Tuli SS, Grotendorst GR, Schultz GS. A connective tissue growth factor signaling receptor in corneal fibroblasts. Investigative ophthalmology & visual science. 2012;53:3387–94. doi: 10.1167/iovs.12-9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol. 1991;114:1285–94. doi: 10.1083/jcb.114.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Wu XY. The role of c-Jun N-terminal kinases 1/2 in transforming growth factor beta(1)-induced expression of connective tissue growth factor and scar formation in the cornea. J Int Med Res. 2009;37:727–36. doi: 10.1177/147323000903700316. [DOI] [PubMed] [Google Scholar]
- Chang Y, Wu XY. JNK1/2 siRNA inhibits transforming-growth factor-beta1-induced connective tissue growth factor expression and fibrotic function in THSFs. Molecular and cellular biochemistry. 2010;335:83–9. doi: 10.1007/s11010-009-0245-8. [DOI] [PubMed] [Google Scholar]
- Chen Y, Abraham DJ, Shi-Wen X, Pearson JD, Black CM, et al. CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Molecular biology of the cell. 2004;15:5635–46. doi: 10.1091/mbc.E04-06-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Delfin J, Morales M, Caelles C. Hypoglycemic action of thiazolidinediones/peroxisome proliferator-activated receptor gamma by inhibition of the c-Jun NH2-terminal kinase pathway. Diabetes. 2007;56:1865–71. doi: 10.2337/db06-1293. [DOI] [PubMed] [Google Scholar]
- Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, et al. Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 2009;41:722–30. doi: 10.1165/rcmb.2009-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fini ME. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res. 1999;18:529–51. doi: 10.1016/s1350-9462(98)00033-0. [DOI] [PubMed] [Google Scholar]
- Folger PA, Zekaria D, Grotendorst G, Masur SK. Transforming growth factor-beta-stimulated connective tissue growth factor expression during corneal myofibroblast differentiation. Investigative ophthalmology & visual science. 2001;42:2534–41. [PubMed] [Google Scholar]
- Garrett Q, Khaw PT, Blalock TD, Schultz GS, Grotendorst GR, Daniels JT. Involvement of CTGF in TGF-beta1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Invest Ophthalmol Vis Sci. 2004;45:1109–16. doi: 10.1167/iovs.03-0660. [DOI] [PubMed] [Google Scholar]
- Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, et al. Exploring the specificity of the PI3K family inhibitor LY294002. The Biochemical journal. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305–18. doi: 10.1002/art.20104. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8:171–9. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR, Duncan MR. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:729–38. doi: 10.1096/fj.04-3217com. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1996;7:469–80. [PubMed] [Google Scholar]
- Huxlin KR, Hindman HB, Jeon KI, Buhren J, MacRae S, et al. Topical rosiglitazone is an effective anti-scarring agent in the cornea. PLoS One. 2013;8:e70785. doi: 10.1371/journal.pone.0070785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. The Journal of investigative dermatology. 1996;106:729–33. doi: 10.1111/1523-1747.ep12345771. [DOI] [PubMed] [Google Scholar]
- Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4:637–45. doi: 10.1091/mbc.4.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon KI, Kulkarni A, Woeller CF, Phipps RP, Sime PJ, et al. Inhibitory effects of PPARgamma ligands on TGF-beta1-induced corneal myofibroblast transformation. The American journal of pathology. 2014;184:1429–45. doi: 10.1016/j.ajpath.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Induction of alpha-smooth muscle actin expression and myofibroblast transformation in cultured corneal keratocytes. Cornea. 1996;15:505–16. [PubMed] [Google Scholar]
- Jester JV, Petroll WM, Barry PA, Cavanagh HD. Expression of alpha-smooth muscle (alpha- SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci. 1995;36:809–19. [PubMed] [Google Scholar]
- Jester JV, Rodrigues MM, Herman IM. Characterization of avascular corneal wound healing fibroblasts. New insights into the myofibroblast. Am J Pathol. 1987;127:140–8. [PMC free article] [PubMed] [Google Scholar]
- Kawata K, Eguchi T, Kubota S, Kawaki H, Oka M, et al. Possible role of LRP1, a CCN2 receptor, in chondrocytes. Biochemical and biophysical research communications. 2006;345:552–9. doi: 10.1016/j.bbrc.2006.04.109. [DOI] [PubMed] [Google Scholar]
- Kawata K, Kubota S, Eguchi T, Moritani NH, Shimo T, et al. Role of the low-density lipoprotein receptor-related protein-1 in regulation of chondrocyte differentiation. J Cell Physiol. 2010;222:138–48. doi: 10.1002/jcp.21930. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Kadono T, Ihn H, Sato S, Igarashi A, et al. Growth regulation in scleroderma fibroblasts: increased response to transforming growth factor-beta 1. The Journal of investigative dermatology. 1995;105:128–32. doi: 10.1111/1523-1747.ep12313452. [DOI] [PubMed] [Google Scholar]
- Kuriyan AE, Lehmann GM, Kulkarni AA, Woeller CF, Feldon SE, et al. Electrophilic PPARg ligands inhibit corneal fibroblast to myofibroblast differentiation in vitro: A potentially novel therapy for corneal scarring. Experimental Eye Research. 2012;94:136–45. doi: 10.1016/j.exer.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. Journal of cell science. 2006;119:4803–10. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- Lin Q, Fang LP, Zhou WW, Liu XM. Rosiglitazone inhibits migration, proliferation, and phenotypic differentiation in cultured human lung fibroblasts. Experimental lung research. 2010;36:120–8. doi: 10.3109/01902140903214659. [DOI] [PubMed] [Google Scholar]
- Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis & tissue repair. 2012;5:S24. doi: 10.1186/1755-1536-5-S1-S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Xu Y, Sun D, Xie L. Histological evaluation of corneal scar formation in pseudophakic bullous keratopathy. PLoS One. 2012;7:e39201. doi: 10.1371/journal.pone.0039201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Dai B, Xu C, Fu L, Hua Z, Mei C. Rosiglitazone inhibits transforming growth factor-beta1 mediated fibrogenesis in ADPKD cyst-lining epithelial cells. PLoS One. 2011;6:e28915. doi: 10.1371/journal.pone.0028915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marampon F, Gravina GL, Di Rocco A, Bonfili P, Di Staso M, et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Molecular cancer therapeutics. 2011;10:159–68. doi: 10.1158/1535-7163.MCT-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussad EE, Brigstock DR. Connective tissue growth factor: what’s in a name? Molecular genetics and metabolism. 2000;71:276–92. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- Pan H, Chen J, Xu J, Chen M, Ma R. Antifibrotic effect by activation of peroxisome proliferator-activated receptor-gamma in corneal fibroblasts. Mol Vis. 2009;15:2279–86. [PMC free article] [PubMed] [Google Scholar]
- Pan HW, Xu JT, Chen JS. Pioglitazone inhibits TGFbeta induced keratocyte transformation to myofibroblast and extracellular matrix production. Mol Biol Rep. 2011;38:4501–8. doi: 10.1007/s11033-010-0581-5. [DOI] [PubMed] [Google Scholar]
- Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–4. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan SS, Blalock TD, Robinson PM, Secker G, Daniels J, et al. Effect of connective tissue growth factor on protein kinase expression and activity in human corneal fibroblasts. Invest Ophthalmol Vis Sci. 2012;53:8076–85. doi: 10.1167/iovs.12-10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Chang Y, Yang Y, Zhang Y, Yu FS, Wu X. Activation of JNK signaling mediates connective tissue growth factor expression and scar formation in corneal wound healing. PLoS One. 2012;7:e32128. doi: 10.1371/journal.pone.0032128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, et al. CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. The Journal of biological chemistry. 2006;281:10715–26. doi: 10.1074/jbc.M511343200. [DOI] [PubMed] [Google Scholar]
- Simpson-Haidaris PJ, Pollock SJ, Ramon S, Guo N, Woeller CF, et al. Anticancer Role of PPARgamma Agonists in Hematological Malignancies Found in the Vasculature, Marrow, and Eyes. PPAR Res. 2010;2010:814609. doi: 10.1155/2010/814609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall EG, Bernstein AM, Oliver N, Gray JL, Masur SK. TGF-beta-stimulated CTGF production enhanced by collagen and associated with biogenesis of a novel 31-kDa CTGF form in human corneal fibroblasts. Investigative ophthalmology & visual science. 2010;51:5002–11. doi: 10.1167/iovs.09-5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesaluoma M, Teppo AM, Gronhagen-Riska C, Tervo T. Release of TGF-beta 1 and VEGF in tears following photorefractive keratectomy. Curr Eye Res. 1997;16:19–25. doi: 10.1076/ceyr.16.1.19.5119. [DOI] [PubMed] [Google Scholar]
- Wahab NA, Brinkman H, Mason RM. Uptake and intracellular transport of the connective tissue growth factor: a potential mode of action. The Biochemical journal. 2001;359:89–97. doi: 10.1042/0264-6021:3590089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ji HX, Xing SH, Pei DS, Guan QH. SP600125, a selective JNK inhibitor, protects ischemic renal injury via suppressing the extrinsic pathways of apoptosis. Life sciences. 2007;80:2067–75. doi: 10.1016/j.lfs.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu XM. Expression of connective tissue growth factor and low density lipoprotein receptor related protein induced by transforming growth factor beta 1 in human pulmonary fibroblasts-1. Beijing da xue xue bao. Yi xue ban = Journal of Peking University. Health sciences. 2006;38:506–9. [PubMed] [Google Scholar]
- Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bull World Health Organ. 2001;79:214–21. [PMC free article] [PubMed] [Google Scholar]
- Wilson SE. Corneal myofibroblast biology and pathobiology: generation, persistence, and transparency. Exp Eye Res. 2012;99:78–88. doi: 10.1016/j.exer.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Buckley ST, Patel V, Liu Y, Luo J, et al. Troglitazone attenuates TGF-beta1-induced EMT in alveolar epithelial cells via a PPARgamma-independent mechanism. PLoS One. 2012;7:e38827. doi: 10.1371/journal.pone.0038827. [DOI] [PMC free article] [PubMed] [Google Scholar]