

Abstract
A gradual decline in insulin response is known to precede the onset of type 1 diabetes (T1D). To track age-related changes in the β-cell function of non-obese diabetic (NOD) mice, the most commonly used animal model for T1D, and to establish differences between those who do and do not become hyperglycemic, we performed a long-term longitudinal oral glucose tolerance test (OGTT) study (10–42 weeks) in combination with immunofluorescence imaging of islet morphology and cell proliferation. We observed a clear biphasic decline in insulin secretion (AUC0–30min) even in euglycemic animals. A first phase (10–28 weeks) consisted of a relatively rapid decline and paralleled diabetes development in the same cohort of animals. This was followed by a second phase (29–42 weeks) during which insulin secretion declined much slower while no additional animals became diabetic. Blood glucose profiles showed a corresponding, but less pronounced change: the area under the concentration curve (AUC0–150min) increased with age, and fit with a bilinear model indicated a rate-change in the trendline around 28 weeks. In control NOD scids, no such changes were observed. Islet morphology also changed with age as islets become surrounded by mononuclear infiltrates, and, in all mice, islets with immune cell infiltration around them showed increased β-cell proliferation. In conclusion, insulin secretion declines in a biphasic manner in all NOD mice. This trend, as well as increased β-cell proliferation, is present even in the NODs that never become diabetic, whereas, it is absent in control NOD scid mice.
Keywords: beta cell proliferation, NOD, glucose tolerance test, insulin secretion, Ki67, pancreatic islet
Introduction
Almost a century after the introduction of insulin, type 1 diabetes (T1D; juvenile-onset or insulin-dependent diabetes), in which the insulin producing β-cells of the pancreatic islets are destroyed by an autoimmune process (1–4), still represents a considerable therapeutic need. Not only because of the inconvenience of the ongoing need for administration of exogenous insulin, but also because chronic and degenerative complications still occur in a considerable fraction of T1D patients, despite all the improvements in management and care (5). The etiology, pathogenesis, and onset mechanism of T1D are complex. It is now well-recognized that an interplay of genetic predisposition, yet unknown environmental factors, and probably other stochastic events is needed (6). Nevertheless, the progressive loss of insulin producing β-cells is a central aspect of T1D (1–3, 7). The non-obese diabetic (NOD) mouse model (8–12) is a widely used prototypic model for human T1D that has contributed significantly to the study of this disease, and is by far the most commonly used animal model for T1D. NOD mice develop spontaneous diabetes in a manner that reproduces many crucial aspects of the human disease, such as the presence of islet-specific autoantibodies, inflammation of pancreatic islets, and dependence on MHC alleles. However, there are also important discrepancies like more severe insulitis, gender bias, and others (10, 12). Typically, around 60% to 90% of female NODs develop diabetes between 12 to 30 weeks of age whereas only 20% to 40% of males do – most likely due to roles played by sex hormones in the autoimmune process (13). Diabetes development in NODs can be altered by microbiota, and specific pathogen-free conditions are needed for such high incidence rates. It is also important to remember that while more than 200 successful therapeutic interventions have already been identified in NODs (10, 11), therapeutic success has been elusive in humans. An increasing number of therapeutic agents tested in large-scale human clinical trials have failed to halt the progressive decline of pancreatic β-cell function (14–16). Even the most successful interventions identified to date, such as cyclosporine, biologics targeting CD3 (teplizumab, otelixizumab) or CD20 (rituximab), and CTLA-4–B7 costimulatory blockade (abatacept), achieved only about a six-month delay in disease progression (14).
It is estimated that at the time of hyperglycemia onset, NOD mice have already lost 80–90% of their insulin-producing β-cell mass (17, 18) – a range likely to be similar to that in humans at the time of clinical manifestation of T1D (1, 19). In fact, prevention or reversal of T1D is particularly challenging because in addition to controlling the persistent autoimmunity against β-cells, restoring euglycemia is also difficult since β-cell mass is already extensively reduced at the time of onset. Hence, quantitative assessments of β-cell mass and function from the onset of islet autoimmunity through the manifestation of hyperglycemia to full diabetes are of crucial relevance to help identify high-risk individuals as early as possible and to clarify the optimal time for initiation of preventive immunotherapy. As might be expected, timing of the therapeutic intervention is important as even in NODs, the efficacy of the same treatment can vary depending on when it is initiated (11). In some cases, earlier administration led to better outcomes (e.g., anti-CD154), whereas in others, later administration did so (e.g., TNFα) (11).
Previous insulin secretion and β-cell mass studies focused almost exclusively on prediabetic NOD mice (e.g., <14 weeks (20) or <18 weeks (21), with the exception of an older work mainly examining the effects of insulitis (22)). Here, as part of a study evaluating possible effects on β-cell mass, our assessment continued through the period of diabetes development and far beyond (42 weeks) in all animals that remained normoglycemic. Such a longitudinal study with quantitative assessment of β-cell function may help elucidate the protective mechanisms by which some susceptible subjects can ultimately elude T1D.
Experimental Procedures
Animal care and treatment
Female non-obese diabetic (NOD) mice (NOD/MrkTac; 8–9 weeks old; n = 25) and matching NOD scid controls (n = 5) were obtained from Taconic (Hudson, NY). Starting from week 10, glycosuria was monitored twice a week. In animals that turned positive, blood glucose levels (glycemia) were monitored three times a week. Mice were followed for up to 42 weeks of age with oral glucose tolerance tests (OGTTs) administered every other week using a rotating schedule. Animals with elevated glucose levels (nonfasting glycemia >250 mg/dL) on two consecutive days were identified as hyperglycemic and were used for glucose tolerance testing as close to onset as possible. Following this, most hyperglycemic animals were implanted with sustained release insulin pellets and enrolled in different treatment studies; no data following insulin pellet implantations were used in this study. Animals with three consecutive readings of >250 mg/dL were categorized as diabetic and humanely euthanized for tissue analysis. At the end of the study, all animals were euthanized, and pancreata and other organ samples were collected and stored for analyses. All animal research was conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the United States National Institutes of Health, and all animal studies were carried out under protocols approved by the University of Miami Institutional Animal Care and Use Committee.
Oral glucose tolerance test
OGTTs were performed with a dose of 75 mg glucose (150 μL, 50%) administered by oral gavage to 16-h fasted mice, and blood samples (2–5 μL) were collected at predefined time intervals (0, 15, 30, 45, 60, 90, 120, and 150 min) from the tail vein for blood glucose assessment. Tests were administered every other week to a group of up to eight animals on a rotating basis; results shown are averages for each test with two to six NODs per test (navg = 4). Glucose levels were quantified using a glucose meter (OneTouch Ultra Blue; Lifescan, Inc., Milpitas, CA; 20–600 mg/dL). For insulin quantification, larger sample volumes are needed (50–100 μL), therefore, they were collected only at 0, 15, and 30 min by submandibular venipuncture using a lancet (Medipoint, Mineola, NY) as previously described (23). These time-points were selected as most likely to provide the highest insulin peak in the OGTT assay based on published data (24, 25). Insulin levels were assessed by ELISA (Ultrasensitive Mouse ELISA, Mercodia, Winston Salem, NC) using a 2.5-fold dilution and cubic spline regression for curve fitting (GraphPad Prism; GraphPad, La Jolla, CA). With this method, the lower limit of detection for insulin was 0.06 μg/L (ng/mL). Blood collection for the insulin assays represents an additional stress on the animals and somewhat delays the restoration of normal glucose levels. To assess this effect, we did parallel comparisons with blood collection following the regular protocol of glucose plus insulin (tail and submandibular vein) vs. glucose alone (tail vein) in NODs at 13 and 31 weeks of age (Figure S1, Supporting Information). While glucose normalization is clearly delayed by the insulin sampling, the glucose profiles were very consistent among assays; hence, they allow quantifiable comparisons of β-cell function and its change with time.
Immunofluorescence
Harvested tissues were immediately embedded in Tissue-Tek O.C.T. (Sakura Finetek, Torrance, CA) and snap frozen by placing the tissue mold in dry ice. Tissue sections (5–10 μm) were collected on glass slides and kept at −80°C until analysis. Samples were fixed for 10 min in 4% paraformaldehyde, washed three times with PBS 1X and permeabilized with PBS plus 0.1% TritonX100 for 1 h at room temperature. After blocking for at least 1 h in PBS with 10% Power Block (BioGenex, Fremont, CA) or normal goat serum as required by antibody detection, samples were incubated overnight at 4°C with the primary antibodies following the manufacturer’s recommendations. Triple immunofluorescence staining for insulin, glucagon, and Ki67 or CD45 was performed: insulin (1:100; Dako, Carpinteria, CA), glucagon (1:100; Dako), Ki67 (1:100; Abcam, Cambridge, MA), and CD45 (1:100; BD Bioscience, San Jose, CA). Sections were washed 5× with PBS plus 0.05 Tween20 and then incubated with the secondary antibodies, washed another 5× times with PBS plus 0.05 Tween20 and 2× with PBS 1X, and mounted for analysis via confocal microscopy. Secondary antibodies were AlexaFluor-conjugated: goat anti-mouse 568; goat anti-rabbit 488; goat anti-guinea pig 647 (1:800; Life Technologies, Carlsbad, CA). Nuclei were detected by DAPI staining (Life Technologies). After final washes, slides were mounted using Vectashield (Vector Laboratories, Burlingame, CA). Immunofluorescence imaging was performed at the Diabetes Research Institute Imaging Core. Images were captured using a Leica SP5 inverted confocal microscope with motorized stage; 3–5-layered z-stack (40× oil) for close-ups and tiled single-layer (5× air) for whole pancreatic sections. Representative images were merged and edited for contrast in Adobe Photoshop (Adobe Systems, Inc., San Jose, CA). Proliferation was quantified using MetaMorph imaging software (Molecular Devices, Sunnyvale, CA). The percentage of Ki67-positive β-cells was calculated as the number of Ki67+(nucleus) insulin+ cells divided by the total insulin+ cells. β-Cells were quantified using area-based estimates for multiple whole pancreatic sections from each animal. The fraction of the pancreas represented by β-cells was calculated using the Analyze Particles tool after thresholding for the corresponding colors in ImageJ (US National Institutes of Health, Bethesda, MD; (26)).
Data analysis
For concentration-time profiles, areas under the curve (AUCs) were calculated using the standard linear trapezoidal method (AUC0–150min for glucose and AUC0–30min for insulin, respectively) in Microsoft Excel. Data analyses including plotting of the Kaplan-Meier survival plots and statistical comparisons (one-way ANOVA with Tukey’s multiple comparison test) were done using GraphPad Prism 6.0 (GraphPad, La Jolla, CA). Fittings of the bilinear model (LinBiExp; y = f(t) = ηln[eα1(t–θc)/η + eα2(t–θc)/η] + χ) in Prism and model comparisons using the Akaike information criteria (AIC) as model selection criteria were done as described previously (27, 28).
Results
Effect of hyperglycemia onset on OGTT profiles
The response of (female) NOD mice to an oral glucose challenge was followed in a longitudinal study with repeated tests from 10 to 42 weeks of age in rotated groups of animals. In this cohort, 64% of animals (16 out of 25) developed diabetes (defined as three consecutive blood glucose readings of >250 mg/dL) by 28 weeks of age with no more new onsets through the end of the study at 42 weeks (see Figure 3A). This relatively low incidence rate as compared to others published (13) as well as ours with the same animals (29) is likely due to the ongoing manipulation required by the repeated OGTTs. For each test, glucose levels were monitored for up to 150 min. Insulin concentrations were monitored for up to 30 min, which was considered a good representation of first-phase response in an oral glucose test while still limiting the amount of blood needed to be collected. One of our goals was to perform as many OGTTs as possible in animals that just became hyperglycemic (BG > 250 mg/dL on two consecutive days; week 14–28) to compare these responses to the prediabetic ones in the same animals. Results shown in Figure 1 clearly indicate that at the onset of hyperglycemia, insulin responses are diminished with a much flatter response at 15 min. The difference between the peak at 15 min versus the baseline is no longer statistically significant, and the average area under the concentration curve (AUC0–30min) is decreased more than two-fold (from 29.3±15.2 to 13.9±7.7 μg·min/L). Corresponding blood glucose profiles are increased: concentrations measured at all time-points are higher (significantly higher after 30 min). The average glucose AUC0–150min is increased by more than 50% (from 45,782±7,303 to 69,879±6,581 mg·min/dL). Animals that became diabetic (hyperglycemic) lost their ability to normalize blood glucose levels and were not used for more OGTT evaluations in this present study. An illustrative example obtained in one such animal displaying the decline in insulin secretion and the corresponding ability to normalize blood glucose levels is shown in Figure S2, Supporting Information. While insulin responses flattened out, some insulin was usually detectable for some time (typically, several weeks) after hyperglycemia onset. We found no detectable insulin only in some animals that were diabetic for several weeks (data not shown).
Figure 3.

Decline of insulin secretion in euglycemic NOD mice parallels the onset of diabetes (hyperglycemia) within the same cohort of animals. (A) Kaplan-Meier survival curve showing the overall rate of diabetes onset within the NOD mice used (n = 25; female NOD, Taconic). (B) Change of insulin secretion (AUC0–30min, OGTT) in euglycemic NODs with age (purple diamonds, fitted with a bilinear curve). Data from age-matched NOD scid controls are also included (orange circles). (C) Change of blood glucose response (AUC0–150min, OGTT) in euglycemic NODs with age; same notation is used as in (B).
Figure 1.
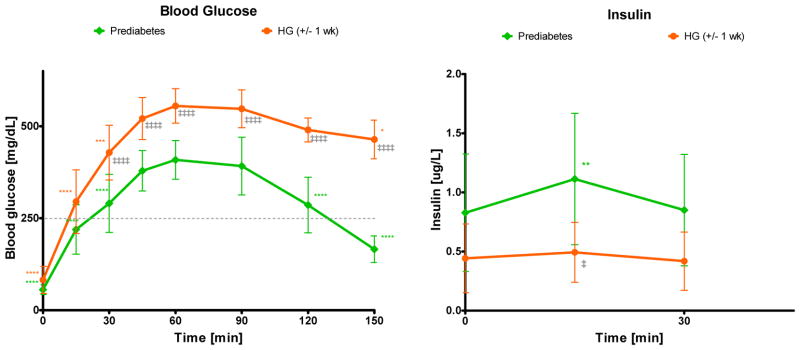
Blood glucose- (left) and insulin-time profiles (right) following an oral glucose tolerance test (OGTT) in NOD mice that have just become hyperglycemic (orange) compared to data in the same mice while they were prediabetic (green). Data (average ± SD) are for n = 9 animals in which OGTTs were performed around the onset of hyperglycemia (BG > 250 mg/dL on two consecutive days; week 14–28) and are compared to corresponding prediabetic responses in the same animals (week 10–16). OGTTs were performed with a dose of 75 mg glucose (150 μL, 50%) administered by oral gavage to 16-h fasted mice, and blood samples were collected at predefined time intervals for up to 150 min (30 min for insulin). Color-coded stars denote statistically significant differences vs. the tmax (60 min; glucose) and t0 (0 min; insulin) value of the same group (one-way repeated measures ANOVA with Tukey’s multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Double-daggers indicate statistically significant differences vs. the same time-point in the pre-diabetes group (one-way ANOVA with Tukey’s multiple comparison test; ‡p < 0.05, ‡‡‡‡p < 0.0001).
Effect of age on OGTT profiles
Insulin secretion and blood glucose data following an oral glucose challenge were collected from euglycemic mice on a rotating schedule for up to 42 weeks of age, well after the last animal became diabetic (27.7 wk). Average blood glucose- and insulin-time profiles obtained are shown in Figure 2 color-coded by age to better highlight the trends. There is an obvious declining trend in the insulin secretion of NOD mice with a noticeable, but not as pronounced increase in corresponding blood glucose values. Average profiles obtained in parallel from NOD scid mice are also included for comparison showing no trend and very little change with age (Figure 2, lower panel). Interestingly, in NODs, the decreasing trend is present not only for the group average, which might be caused by some of the animals gradually becoming pre-diabetic, but also in animals that ultimately do not became diabetic. A set of representative individual profiles is included in Figure S2.
Figure 2.

Change of blood glucose- (left) and insulin-time profiles (right) with age in a longitudinal OGTT study in NOD mice (top) and age-matched NOD scid controls (bottom). Data are the average of all non-diabetic (euglycemic) mice following an OGTT with a dose of 75 mg (150 μL, 50%) administered by oral gavage to 16-h fasted animals, and are color-coded from green through blue to purple with age.
Trends of overall change are much clearer in Figure 3B and C that show AUC data as a function of age for insulin and blood glucose, respectively. For insulin (Figure 3B), a first phase of relatively rapid decline up to about 28 weeks of age is followed by a second phase of much slower decrease up to the end of the study (42 weeks). To rigorously describe this biphasic behavior and quantitatively assess the time of the rate-change, we fitted the data with our bilinear model (LinBiExp). This model was specifically developed to fit data that have two different phases of linear behavior separated by a rate-change point (27, 28, 30, 31). Fitting showed that a first phase of relatively rapid decline (with an average slope of α1 = −1.80±0.40 μg·min/L/wk) comes to essentially a stop (α2 = −0.12±0.58 μg·min/L/wk) at a rate-change point located at 27.8±4.2 weeks. Notably, the first phase of rapid decline (even though measured exclusively in euglycemic mice) nicely parallels the overall diabetes development in the same cohort of animals as represented by the Kaplan-Meier survival curve (Figure 3A vs. B). Furthermore, the time of the last animal becoming diabetic (27.7 wk) corresponds with the time of the estimated rate-change in insulin secretion (27.8±4.2 wk) so that no other NOD mice became diabetic during the second phase of slow change in insulin AUCs. The bilinear model fits the data well accounting for almost 80% of the variability in the data (r2 = 0.79), which is better than the straight linear decrease model (r2 = 0.70; y = αt + χ) or even the power (r2 = 0.73; y = αtβ + χ) and exponential (r2 = 0.76; y = αeβt + χ) decrease models. Furthermore, the bilinear function not just fits better (as indicated by the r2 values), but is also more suitable according to statistically adequate measures for such comparisons; i.e., model selection criteria, such as the Akaike Information Criterion, AIC, which account for the different number of fitting parameters (27). The difference is small, but is indicative: AIC of 133.1 for LinBiExp vs. 135.5, 136.2, and 133.7 for the linear, power, and exponential functions, respectively (the lower AIC, the better).
These observations reinforce the idea of a relatively steady autoimmune-driven gradual erosion of β-cell function preceding the onset of diabetes. Similar destruction could be present in all susceptible individuals (here, NODs), but in some, is successfully counterbalanced by compensatory mechanisms that ultimately succeed in maintaining normoglycemia. Notably, the decrease of insulin secretion in NODs that never became diabetic follows a very similar biphasic trendline (Figure S3, Supporting Information). The AUC data are more scattered, as there are only few animals at each time-point, but the slopes and rate-change points obtained by fitting of the bilinear model are very similar (Figure S3). In control age-matched NOD scid mice, no such changes were observed as insulin secretion and blood glucose profiles remained steady throughout the study. At 10 weeks of age, NODs had considerably higher insulin responses (AUC0–30min) than NOD scids, but this steadily eroded resulting in diabetes in most animals by 28 weeks of age. In remaining NODs that did not become diabetic, the decline was slowed or halted.
Blood glucose profiles showed a corresponding, but less pronounced change: AUC0–150min increased with age, and there was a change in the trendline also around 28 weeks as indicated by fit with the LinBiExp bilinear model (rate-change point located at 28.3±7.5 weeks; Figure 3C). The biphasic character here is less pronounced, but fit with the bilinear model is still better than with the single linear one (r2 of 0.43 vs. 0.35). However, AIC as a model selection criterion no longer favors the bilinear model (396.1 vs. 394.7) due to the larger number of parameters.
Changes in islet composition and β-cell proliferation
Islet morphology changes with age in NOD mice (32, 33). Confocal microscopy (Figure 4) confirmed that in NOD mice, pancreatic islets begin to show peri-islet mononuclear infiltrates around 9–11 weeks of age, just before the onset of the first cases of hyperglycemia (Figure 3A). These peri-islet infiltrates are consistently present around islets in all older animals – even in those that do not become diabetic (Figure 4E). In agreement with the functional results of the OGTTs, quantification of β-cell mass by insulin immunoreactivity in whole pancreatic sections (Figure 5) also indicates that β-cells gradually deteriorate through this autoimmune process in NOD mice. While only very few insulin positive cells (<10% of the original mass) remain in the animals that become diabetic, in NODs that do not become diabetic, the decline stops, and a still functional mass is retained (Figure 6). Estimates of β-cell mass obtained here for young NODs (1.1 mg) as well as NOD scids (1.2 mg) nicely parallel previous results, for example, by Sreenan and co-workers (21) (1.09 and 1.30–1.60 mg for NODs and NOD scids, respectively) or Bock and co-workers (34) (1.1 mg for NODs).
Figure 4.

Changes in islet morphology and lymphocyte infiltration in NOD mice. Confocal microscopy images of representative pancreatic islets from young (C, D) and old non-diabetic (E) NOD mice as well as from corresponding control C57BL/6 (A) and NOD scid mice (B). Pancreatic sections were stained for DAPI (blue), insulin (red), glucagon (green), and CD45 (a lymphocyte marker, white).
Figure 5.

Pancreatic sections immunostained for insulin in different mice. Tiled confocal microscopy images of representative whole pancreatic sections from diabetic (D) as well as nondiabetic young (C) and old (E) NOD mice together with corresponding controls including C57BL/6 (A) and NOD scid (B) mice. Pancreatic sections were stained for DAPI (blue), insulin (red), and glucagon (green).
Figure 6.
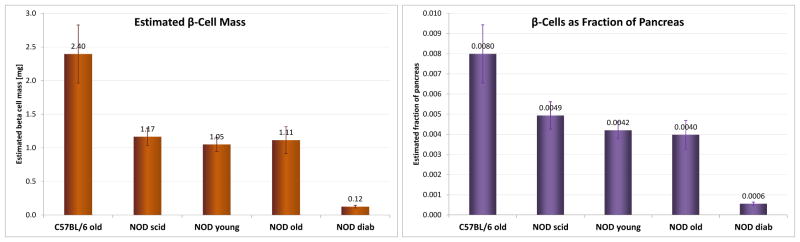
Estimated β-cell mass and prevalence. Total mass and prevalence (fraction of pancreas) estimates for β-cells in different NODs (diabetic as well as nondiabetic young/10 wk/ and old/>30 wk/ mice) with corresponding controls (normal C57BL/6/>30 wk/ and NOD scid mice/10 wk/) obtained from quantification of immunostained slides as described under experimental procedures. Data are the average of area-based estimates from all slides imaged and quantified for each group.
Proliferation of β-cells in NOD mice indicated by Ki67 staining (35) in insulin positive cells is considerably higher than in regular C57BL/6 mice (Figure 7, Figure 8). It is also higher than in NOD scids – in agreement with previous observations (36). Notably, β-cell proliferation in NODs seems to increase considerably with the onset of the autoimmune attack and the appearance of the peri-islet infiltrates: from 0.7–1.0% to 5–7% (Figure 7C vs. D, Figure 8). This could be an attempt to compensate for the ongoing erosion of β-cell mass, which is indicated by the continuing decrease in insulin secretion (Figure 3). It also agrees with observations that the presence of pathogenic cells induces increased proliferation (36, 37) (possibly by some T-cell secreted soluble factors as suggested recently (37)). In older NODs that do not become diabetic, this increased proliferation seems to be maintained albeit at somewhat lower levels (Figure 8) and could be one compensatory mechanism that maintains physiologically adequate insulin secretion in these animals. Remaining islets also show signs of restructuring (possibly due to formation of new insulin-producing β-cells) as in these islets, α-cell involution is noticeable: some glucagon positive α-cells are present within the β-cell core, whereas in normal mice, they are located at the periphery of the islets surrounding the β-cell core (Figure 7E, F vs. A, B, D) (38, 39). This could be a further indication of growth in these islets with the existing α-cells being slowly engulfed by the proliferating β-cells as a result of the proliferation process that tries to compensate for the already lost β-cell mass and restore physiological insulin response.
Figure 7.

Cell proliferation in pancreatic islets. Confocal microscopy images of representative pancreatic islets from young (C, D) and old non-diabetic (E, F) NOD mice as well as from corresponding controls including NOD scid (A) and C57BL/6 mice (B). Pancreatic sections were stained for DAPI (blue), insulin (red), glucagon (green), and Ki67 (a proliferation marker, white).
Figure 8.

Quantification of β-cell proliferation. Percent of Ki67+ β-cells in islets from different NOD mice and corresponding controls (normal C57BL/6 and NOD scid) was determined as described under experimental procedures. Data are the average of slides from at least three different animals for each group and were analyzed by one-way ANOVA with Tukey’s multiple comparison test. Asterisks denote statistically significant differences compared to NOD young (with no insulitis) (*p < 0.05, ***p < 0.001).
Discussion
The progressive loss of β-cell function is a central aspect of T1D. Thus, it is important to understand the timing of the physiological, cellular, and molecular changes through disease progression (1–3). Declining insulin responses have been observed in prediabetic NOD mice studied until the animals were 14 or 18 weeks of age (20, 21) – around the onset of hyperglycemia. Here, we found that the insulin response continues to decline during the period of diabetes onset – even in animals that never become diabetic. This decline then essentially ceases after a certain age (here, around 28 weeks). Animals were studied up to 42 weeks of age – almost 14 weeks after the last diabetes occurrence in this cohort of animals (week 27.7, Figure 3A). This study length seemed sufficient, especially considering the limited lifespan of the NOD scid mice (33–38 weeks), which were used as controls. A somewhat similar profile of gradual β-cell loss followed by plateauing in animals that did not become diabetic was found in a NOD mouse model with a luciferase optical reporter gene in their β-cells to enable their non-invasive longitudinal imaging. However, that work focused on the difference in overall decline (slope) between mice that did and did not become diabetic, and not on the biphasic nature of the decline (18).
It has been suggested that these NOD phenotypes follow a similar pattern to those seen in humans with T1D, further validating this animal model (20). Indeed, in humans, a steady (essentially linear) prediabetic decline was documented over three decades ago and well-studied since then (40, 41). More recently, a biphasic decline of β-cell function, as assessed by C-peptide production, following T1D diagnosis has been described – with a rapid first phase of decline during the first year followed by a noticeably slower decrease during the second year (42). This reinforces the possibility that similar mechanisms are at play: an initial steady immune destruction then slows down as the remaining β-cell mass decreases and can be better equilibrated by compensatory mechanisms such as β-cell proliferation. The possibility of β-cell hyperactivation before diabetes onset has been also raised (43). Here, we found no evidence for this since insulin responses declined with age in most NODs (Figure S2) with few exceptions.
The progressive decline of β-cell mass triggers compensatory mechanisms, including increased β-cell proliferation, which usually fail to fully compensate for the ongoing autoimmune destruction (21). We found that in NODs there is an increased β-cell proliferation that seems to persist even in the older animals that did not become diabetic (Figure 7, Figure 8). It stands to reason that in these older non-diabetic mice, this enhanced proliferation may be sufficient to slow the decline in insulin response resulting in the second slower phase of Figure 3B. The biphasic decline in β-cell function identified here could help delineate the protective mechanisms by which some of those susceptible to develop T1D ultimately elude the disease. The total insulin-producing β-cell mass is determined by a dynamic balance of proliferation, neogenesis, and apoptosis, and there is increasing evidence that β cells can proliferate and their function (i.e., insulin production and the maintenance of glucose homeostasis) can partly recover if the autoimmune attack is controlled (17, 37, 44–52). However, the capacity for regeneration is probably low, and it is even less in humans and large animals than in (young) rodents. It seems to mainly involve replication of β-cells and the islets growing in size rather than in number (44, 46, 53). Nonetheless, there is evidence of increased β-cell proliferation during the progression of diabetes in NOD mice (17, 21, 36) as well as in humans (54).
We found β-cell proliferation in all NOD mice to increase considerably in the presence of peri-islet infiltrates (from 0.7–1.0% to 5–7%; Figure 8). Otherwise, proliferation rates found here by Ki67 staining are in agreement with those published before (21, 36, 37, 45, 53, 54). For example, in pre-diabetic NODs, around 2% of β-cells were counted as proliferating by Ki67 staining and maybe somewhat less (around 0.5–1.0%) by BrdU staining (21, 36, 37). As compared to age-matched NOD scid mice, we also found β-cell proliferation in NODs to be higher. This is in agreement with a previous study (around 1.5% vs. 0.5%; Ki67), which also found rates in NODs to increase about two-fold during diabetes onset (to around 3%) (36). Our finding of significantly increased β-cell proliferation in islets with immune cell infiltration around them is consistent with a recent study suggesting that the presence of T cells (following splenocyte transfer) increases the proliferation rate of β-cells from 2% to almost 8% (in NOD.RAG1−/− mice) (37).
Elevated glucose levels might be one factor driving β-cell proliferation (21, 55). However, in the present study, we found increased proliferation in euglycemic NODs in infiltrated islets before the onset of hyperglycemia or even in animals that never became diabetic. Increased proliferation before the onset of hyperglycemia or independent of blood glucose levels has been observed by others as well (21, 37, 45). A recent study provided evidence that T cells can promote β-cell proliferation probably via some yet unidentified secreted soluble factors (37). In islets exhibiting insulitis, the proliferation of β-cells was found to positively correlate with the extent of lymphocyte infiltration (37). Along these lines, it has been suggested that immunosuppressive therapies are double-edged swords that can provide effective immune suppression, but they are also detrimental to β-cell proliferation and recovery from diabetes – possibly by reducing the number of lymphocytes present (45, 56). Indeed, in older NODs, all remaining islets are surrounded by peri-islet mononuclear infiltrates (possibly acting as tertiary lymphoid structures), and they show increased β-cell proliferation (Figure 7).
In conclusion, our long-term OGTT study in NOD mice revealed a biphasic decline of insulin secretion with a first phase of relatively rapid decline that parallels diabetes development within the same cohort of susceptible animals and a second phase of stagnation or much slower decline while no additional animals became diabetic. This trend, as well as increased β-cell proliferation in islets with immune cell infiltration around them, is present even in the NODs that never become diabetic, whereas, it is absent in control NOD scid mice.
Supplementary Material
Acknowledgments
Funding. This work was supported by funding from the NIH/NIAID (1R01AI101041), the Iacocca Foundation, and the Diabetes Research Institute Foundation.
Footnotes
Conflict of interest statement. The authors declare that they have no conflict of interest associated with this manuscript.
References
- 1.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 2.Daneman D. Type 1 diabetes. Lancet. 2006;367:847–858. doi: 10.1016/S0140-6736(06)68341-4. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, et al. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. 2006;55:1463–1469. doi: 10.2337/db05-1423. [DOI] [PubMed] [Google Scholar]
- 6.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 7.Morgan NG, Leete P, Foulis AK, Richardson SJ. Islet inflammation in human type 1 diabetes mellitus. IUBMB Life. 2014;66:723–734. doi: 10.1002/iub.1330. [DOI] [PubMed] [Google Scholar]
- 8.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 9.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 10.Roep BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: reevaluating the use of animal models of type 1 diabetes. Nat Rev Immunol. 2004;4:989–997. doi: 10.1038/nri1502. [DOI] [PubMed] [Google Scholar]
- 11.Shoda LK, Young DL, Ramanujan S, Whiting CC, Atkinson MA, et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity. 2005;23:115–126. doi: 10.1016/j.immuni.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Reed JC, Herold KC. Thinking bedside at the bench: the NOD mouse model of T1DM. Nat Rev Endocrinol. 2015 doi: 10.1038/nrendo.2014.236. ePub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. 2013;39:400–412. doi: 10.1016/j.immuni.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skyler JS, Ricordi C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes. 2011;60:1–8. doi: 10.2337/db10-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Herrath M, Peakman M, Roep B. Progress in immune-based therapies for type 1 diabetes. Clin Exp Immunol. 2013;172:186–202. doi: 10.1111/cei.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lernmark Å, Larsson HE. Immune therapy in type 1 diabetes mellitus. Nat Rev Endocrinol. 2013;9:92–103. doi: 10.1038/nrendo.2012.237. [DOI] [PubMed] [Google Scholar]
- 17.Alanentalo T, Hornblad A, Mayans S, Karin Nilsson A, Sharpe J, et al. Quantification and three-dimensional imaging of the insulitis-induced destruction of beta-cells in murine type 1 diabetes. Diabetes. 2010;59:1756–1764. doi: 10.2337/db09-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virostko J, Radhika A, Poffenberger G, Dula AN, Moore DJ, et al. Bioluminescence imaging reveals dynamics of beta cell loss in the non-obese diabetic (NOD) mouse model. PLoS ONE. 2013;8:e57784. doi: 10.1371/journal.pone.0057784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10(Suppl 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ize-Ludlow D, Lightfoot YL, Parker M, Xue S, Wasserfall C, et al. Progressive erosion of beta-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes. 2011;60:2086–2091. doi: 10.2337/db11-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, et al. Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes. 1999;48:989–996. doi: 10.2337/diabetes.48.5.989. [DOI] [PubMed] [Google Scholar]
- 22.Reddy S, Liu W, Thompson JM, Bibby NJ, Elliott RB. First phase insulin release in the non-obese diabetic mouse: correlation with insulitis, beta cell number and autoantibodies. Diabetes Res Clin Pract. 1992;17:17–25. doi: 10.1016/0168-8227(92)90039-t. [DOI] [PubMed] [Google Scholar]
- 23.Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. 2005;34:39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
- 24.Winzell MS, Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53(Suppl 3):S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 25.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchwald P. A general bilinear model to describe growth or decline time-profiles. Math Biosci. 2007;205:108–136. doi: 10.1016/j.mbs.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Buchwald P. Activity-limiting role of molecular size: size-dependency of maximum activity for P450 inhibition as revealed by qHTS data. Drug Metab Dispos. 2014;42:1785–1790. doi: 10.1124/dmd.114.059717. [DOI] [PubMed] [Google Scholar]
- 29.Cechin SR, Lopez-Ocejo O, Ricordi C, Buchwald P. Long-term reversal of hyperglycemia in experimental type 1 diabetes in NOD mice by inhibition of Smad7 with an antisense oligonucleotide. Diabetes. 2014;63 (S1):A426. [Google Scholar]
- 30.Horváth A, Rácz-Mónus A, Buchwald P, Sveiczer Á. Cell length growth in fission yeast: an analysis of its bilinear character and the nature of its rate change transition. FEMS Yeast Res. 2013;13:635–649. doi: 10.1111/1567-1364.12064. [DOI] [PubMed] [Google Scholar]
- 31.Das M, Drake T, Wiley DJ, Buchwald P, Vavylonis D, et al. Oscillatory dynamics of Cdc42 GTPase in the control of polarized growth. Science. 2012;337:239–243. doi: 10.1126/science.1218377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plesner A, Ten Holder JT, Verchere CB. Islet remodeling in female mice with spontaneous autoimmune and streptozotocin-induced diabetes. PLoS ONE. 2014;9:e102843. doi: 10.1371/journal.pone.0102843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pechhold K, Zhu X, Harrison VS, Lee J, Chakrabarty S, et al. Dynamic changes in pancreatic endocrine cell abundance, distribution, and function in antigen-induced and spontaneous autoimmune diabetes. Diabetes. 2009;58:1175–1184. doi: 10.2337/db08-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bock T, Pakkenberg B, Buschard K. Genetic background determines the size and structure of the endocrine pancreas. Diabetes. 2005;54:133–137. doi: 10.2337/diabetes.54.1.133. [DOI] [PubMed] [Google Scholar]
- 35.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 36.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, et al. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–1345. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 37.Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, et al. Soluble factors secreted by T-cells promote beta-cell proliferation. Diabetes. 2014;63:188–202. doi: 10.2337/db13-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, et al. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA. 2006;103:2334–2339. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Martin B, Cai H, Fiori JL, Egan JM, et al. Pancreas++: automated quantification of pancreatic islet cells in microscopy images. Front Physiol. 2012;3:482. doi: 10.3389/fphys.2012.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srikanta S, Ganda OP, Gleason RE, Jackson RA, Soeldner JS, et al. Pre-type I diabetes. Linear loss of beta cell response to intravenous glucose. Diabetes. 1984;33:717–720. doi: 10.2337/diab.33.8.717. [DOI] [PubMed] [Google Scholar]
- 41.Barker JM, McFann K, Harrison LC, Fourlanos S, Krischer J, et al. Pre-type 1 diabetes dysmetabolism: maximal sensitivity achieved with both oral and intravenous glucose tolerance testing. J Pediatr. 2007;150:31–36. doi: 10.1016/j.jpeds.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenbaum CJ, Beam CA, Boulware D, Gitelman SE, Gottlieb PA, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes. 2012;61:2066–2073. doi: 10.2337/db11-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amrani A, Durant S, Throsby M, Coulaud J, Dardenne M, et al. Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology. 1998;139:1115–1124. doi: 10.1210/endo.139.3.5823. [DOI] [PubMed] [Google Scholar]
- 44.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 45.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by β cell regeneration. J Clin Invest. 2007;117:2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab. 2007;3:758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- 47.Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes. 2008;57:2883–2888. doi: 10.2337/db07-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pechhold K, Koczwara K, Zhu X, Harrison VS, Walker G, et al. Blood glucose levels regulate pancreatic beta-cell proliferation during experimentally-induced and spontaneous autoimmune diabetes in mice. PLoS ONE. 2009;4:e4827. doi: 10.1371/journal.pone.0004827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, et al. Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, et al. Regulation of pancreatic beta-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes. 2006;55:3289–3298. doi: 10.2337/db06-0017. [DOI] [PubMed] [Google Scholar]
- 51.Lee SH, Hao E, Levine F. β-Cell replication and islet neogenesis following partial pancreatectomy. Islets. 2011;3:188–195. doi: 10.4161/isl.3.4.16338. [DOI] [PubMed] [Google Scholar]
- 52.Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. 2015 doi: 10.1038/nm.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia. 2010;53:2020–2028. doi: 10.1007/s00125-010-1817-6. [DOI] [PubMed] [Google Scholar]
- 55.Akirav EM, Baquero MT, Opare-Addo LW, Akirav M, Galvan E, et al. Glucose and inflammation control islet vascular density and beta-cell function in NOD mice: control of islet vasculature and vascular endothelial growth factor by glucose. Diabetes. 2011;60:876–883. doi: 10.2337/db10-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zahr E, Molano RD, Pileggi A, Ichii H, Jose SS, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation. 2007;84:1576–1583. doi: 10.1097/01.tp.0000296035.48728.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.