

SUMMARY
We developed Split DamID (SpDamID), a protein complementation version of DamID, to mark genomic DNA bound in vivo by interacting or juxtapositioned transcription factors. Inactive halves of DAM (DNA Adenine Methyltransferase) were fused to protein pairs to be queried Interaction or proximity enabled DAM reconstitution and methylation of adenine in GATC. Inducible SpDamID was used to analyze Notch-mediated transcriptional activation. We demonstrate that Notch complexes label RBP sites broadly across the genome, and show that a subset of these complexes that recruit MAML and p300 undergo changes in chromatin accessibility in response to Notch signaling. SpDamID differentiates between monomeric and dimeric binding thereby allowing for identification of half-site motifs used by Notch dimers. Motif enrichment of Notch enhancers coupled with SpDamID reveals co-targeting of regulatory sequences by Notch and Runx1. SpDamID represents a sensitive and powerful tool that enables dynamic analysis of combinatorial protein-DNA transactions at a genome-wide level.
Keywords: DamID, Chromatin, Notch, ChIP, transcription, method
INTRODUCTION
Transcriptional regulation involves combinatorial assembly of transcription factors (TFs) binding to specific sites, subsequent recruitment of general co-activator proteins, mediator complex and RNA polymerase II to initiate transcription (Lee and Young, 2013). Varied molecular mechanisms such as cooperative binding, alteration of nucleosomal dynamics, or changes in chromosomal architecture facilitate assembly of transcriptional complexes (Spitz and Furlong, 2012). Chromatin immunoprecipitation coupled with next-gen sequencing (ChIPseq) is the dominant method used to identify genomic sites bound by transcriptional regulators. ChIP has limitations (Park, 2009): it is dependent on highly specific antibodies; extensive washes needed to reduce background necessitate large amounts of starting material; formaldehyde can alter cellular structure and chromatin organization; and false positives may arise due to cross-linking bias favoring larger complexes and regions of open chromatin (Teytelman et al., 2013). Importantly, ChIP cannot distinguish enhancers regulated exclusively through cooperative binding of dimeric complexes from monomer-responsive enhancers (Arnett et al., 2010). Finally, the interrogation of TF assemblies requires sequential IP (Re-ChIP), which is more technically challenging (Furey, 2012). To address these shortcomings we developed a complementary approach to ChIPseq based on DamID, which uses fusions of the E. coli DNA Adenine Methyltransferase (DAM) to a protein of interest. DAM methylation of adenines within GATC sequence creates a site (GAmTC) cut by the restriction enzyme DpnI. To identify genomic DNA sites bound simultaneously by two proteins in vivo, we developed Split DamID (SpDamID), a protein complementation version of DamID (van Steensel and Henikoff, 2000), that enables analysis of genomic sites that are simultaneously bound by pairs of TFs as well as other proteins involved in the regulation of transcription in living cells. This complementation system identifies bound segments through DNA analyses similar to standard DamID while greatly reducing the background signals observed with DamID.
We tested SpDamID utility by interrogating the Notch pathway. The Notch signaling pathway is one of the major transcriptional pathways utilized throughout life in metazoans. Notch receptors are non-DNA binding proteins that undergo ligand-dependent proteolysis, releasing the intracellular portion (NICD) that enters the nucleus where it associates with RBPjκ (RBP), which provides DNA binding to the motif BVYGDGAD (Ong et al., 2006). The NICD/RBP complex has increased promoter occupancy compared to RBP (Krejci and Bray, 2007) (Figure 1A) and creates the binding surface for the adaptor mastermind (MAML). The RBP-NICD-MAML protein complex, henceforth the RNM (Kopan and Ilagan, 2009), recruits the general transcriptional co-activator p300 and the mediator complex, ultimately loading RNA polymerase II and initiating gene transcription. The ankyrin (ANK) domain of NICD mediates dimerization to allow cooperative binding at sequence paired sites (SPS) that contain RBP binding motifs in head to head orientation (Nam et al., 2007). Dimerization-mediated cooperative DNA binding allows use of non-consensus sites (Arnett et al., 2010) and has been shown to be important for T-cell development and disease (Liu et al., 2010). Transcriptional activation is coupled to NICD degradation through recruitment of the mediator complex that phosphorylates the Notch PEST domain permitting Fbw7 binding and NICD ubiquitination followed by proteosomal degradation (Kopan and Ilagan, 2009). This molecular sequence supports the hypotheses that dwell-time on chromatin increases as NICD joins RBP, and MAML joins this pair (Krejci and Bray, 2007) but decreases after p300 recruitment (Bars below Figure 1A).
Figure 1. SpDamID Proof-of-Concept Utilizing the Notch Signaling Pathway (see also Figure S1).

(A) The step-wise assembly of Notch transcriptional complexes. The hypothetical stability of the complex on DNA is indicated by the size of the gray bars below. Notch1 (N1) ChIP probes regions bound by all the complexes. SpDamID pairs identify only regions bound by a specific complexes (marked with +). (B) Crystal structure of Dam. The N-terminal D half (green), C-terminal AM half (red), and overlapping region (yellow) are shown. (C) DNA from mK4 cells expressing various proteins amplified by Ligation mediated PCR (panels labeled LMP, showing a library aliquot) or target specific PCR (panels labeled TSP, showing semi-quantitative PCR for Hes1 or Nrarp enhancer). Panels above the dotted line record enrichment from proteins expressed without DOX; panels below the dotted line record enrichment with DOX induced expression of the following proteins: Lane1, full-length Dam; Lane 2–3, individual halves of SpDam fused to either Notch (lane 2) or RBP (lanes 3); Lanes 4–7, co-expression of SpDam pairs fused to various Notch complex components or mutants as indicated. Lane 8, co-expression of unfused SpDam halves. Lane 9, non-transfected control. Lane 10, no DpnI and Lane 11, no ligase. All experiments were performed in the presence of 4OHT. (D) Expression of DN-MAML1 prevents recruitment of p300 to Notch, and thus blocks labeling of Nrarp by N1D/p300AM-Ert2. DN-MAML1 does not interfere with enrichment of Hdac9 by Mef2cD/p300AM-Ert2. (E) Target-specific PCRs on biological triplicate samples labeled by N1D/p300AM-Ert2 and Mef2cD/p300AM-Ert2 demonstrating specificity of Notch target sites in Hes1, Nrarp, and Cdh6, and the Mef2c targets Hdac9, Sgcg, and Cdkl5.
We fused Dam Haves (D and AM) Notch1 and its interacting partners (Notch1, RBP, MAML1, and p300). The interaction between Notch pathway constituents allowed reconstitution of DAM and methylation of adenines in the vicinity of the transcription complexes. Additionally, it allowed the labeling of enhancers co-bound sites by Notch and Runx1 showcasing the ability of SpDamID to identify regulatory elements bound by non-interacting TFs bridged by DNA. We further demonstrate the power of SpDamID by identifying Notch-dimer dependent enhancers and defining a new DNA binding motif at the half site used by Notch dimers. In conclusion, SpDamID will facilitate the in-depth characterization of transcription complexes and their dynamics in vivo, while utilizing orders of magnitude fewer cells (~10,000) than ChIPseq.
RESULTS
SpDamID Method Development
We designed partially overlapping complementary halves of DAM based on its crystal structure (Horton et al., 2006), retaining a surface-exposed helix from the DNA binding domain (Figure 1B, yellow) in both the N-terminal (D, green) and the C-terminal (AM, red; Figure 1B) halves (see (Luker et al., 2004)). The D and AM halves were fused to the amino or carboxy-termini Notch and RBP, to the general transcriptional co-activators (p300, MAML), and to Mef2c, a MAML partner unrelated to Notch (as a specificity control). We confirmed that the D and AM fusions to Notch, RBP and Mef2c proteins individually retained the ability to induce transcription of reporter constructs (Figure S1A, S1B and data not shown). Transient co-expression of SpDamID pairs from the bi-directional, Doxycycline (DOX)-responsive pBI Tet vector (Figure S1C and S1D) will test for reconstituted DAM activity in mK4 cells (mouse kidney progenitor cell line, (Valerius et al., 2002)) stably expressing the Tet-On tetracycline responsive transactivator rtTA. These cells were chosen because Notch proteins have important functions in kidney progenitor cells via poorly defined targets (Cheng et al., 2007; Liu et al., 2013), and to allow us to analyze the impact of protein expression level on the signal-to-noise ratio. To further improve signal-to-noise ratio and to allow temporal control of SpDamID, we fused the AM domain with the estrogen receptor hormone-binding domain (Ert2). Reconstitution of DAM activity was dependent on the presence of 4-hydroxytamoxifen (4OHT; Figure S1F and S1G). The presence of Ert2 did not convert RBP into a transcriptional activator in the presence of 4OHT (data not shown). Isolated genomic DNA was subjected to DpnI digestion followed by adaptor ligation and adaptor-mediated PCR (LMP) (Figure 1C, top panel). The lack of LMP products in the absence of DpnI cleavage or adapter ligation (Figure 1C, lanes 9–11) demonstrated that the observed PCR products were generated by Dam mediated methylation of genomic DNA. The LMP libraries were used as templates for semi-quantitative target specific PCR (TSP) reactions to determine whether SpDamID enriched for the Notch targets Hes1 and Nrarp (Figure 1C, middle and lower panels) relative to libraries generated by DAM (Figure 1C, lane 1), the individual fused halves (Figure 1C, lanes 2–3) or the unfused, co-expressed D and AM pair (Figure 1C, lane 8) which lack a full catalytic domain. Baseline expression of N1D/N1AM-Ert2 or N1D/RBPAM-Ert2 pairs in the presence of 4OHT produced strong enrichment for DpnI fragments containing Notch-responsive enhancers in Hes1 (Takebayashi et al., 1994) and Nrarp (Pirot et al., 2004) (Figure 1C, lanes 4–5). Enrichment for the Hes1 SPS-containing promoter or the Nrarp enhancer was lost when Notch dimer pairs contained a point mutation abolishing the dimer interface (N1RAD and N1RAAM-Ert2, (Arnett et al., 2010), Figure 1C Lane 6, upper panels) or when the high affinity domain for RBP binding was removed in N1ΔRAMD controls (Figure 1C lane 7, upper panels). Administration of DOX increased expression levels but did not affect the signal-to-noise ratio (Figure 1C, lower panels). The N1RAD and N1RAAM-Ert2 proteins lacking the dimer did not regain activity (Figure 1C, lane 6, lower panels). By contrast, N1ΔRAMD and RBPAM-Ert2 could now interact through the low affinity ANK domain (Lubman et al., 2007) to allow enrichment of the Hes1 and Nrarp enhancers (Figure 1C, lane 7, lower panels). Thus SpDamID enables the detection of interacting transcription factors or subunits of a multi-component complex at specific genomic sites in vivo across a wide range of protein concentrations.
We generated LMP libraries from multiple SpDamID pairs (N1D/N1AM-Ert2, N1D/RBPAM-Ert2, N1D/MAMLAM-Ert2 and N1D/p300AM-Ert2) and controls (D/AMErt2, N1RAD/N1RAAM-Ert2 and N1ΔRAMD/RBPAM-Ert2). We anticipated that the N1D/RBPAM-Ert2 pair would mark DNA bound by all complexes, the N1D/MAMLAM-Ert2 pair would mark DNA bound by all MAML containing complexes, and so on (Figure 1A). To confirm that DAM reconstitution strictly depends on the specific interaction between the fusion proteins we analyzed recruitment of p300 via MAML by Notch and Mef2c. We utilized a dominant negative Mastermind (DN-MAML) peptide encompassing AA1-74 (Maillard et al., 2004), coding for the Notch/MAML interaction domain. Mef2c interacts with MAML utilizing different domains then Notch to recruit p300. N1D/p300AM-Ert2 interaction at the Nrarp target sequence was dependent on OHT and inhibited by expression of DN-MAML (Figure 1D, left panel). By contrast, labeling of Hdac9 by the Mef2cD/p300AM-Ert2 pair was not affected by co-expression of DN-MAML. These data demonstrate that that SpDamID strictly depends on protein-protein interaction when only one partner binds to DNA.
Since Mef2c and Notch bind to different domains on MAML and p300, one could envision recruitment of Mef2c to Notch/RBP complexes and vice versa. To test for specificity of TF interactions with shared co-activators, we analyzed the recruitment of p300 by Notch1 or Mef2c on several of their known targets in biological triplicates (Figure 1E). N1D/p300AM-Ert2 methylated GATCs flanking known RBP binding sites in the Hes1 and Nrarp (−3kb) promoters, whereas Mef2cD/p300AM-Ert2 methylated GATCs flanking known binding sites in Hdac9 and Sgcg. Although we did not see evidence of methylation at nearby regions in the Hes1 (−8kb) and Nrarp (−6kb) promoters, we identified reproducible enrichment of DpnI digested fragments at −8kb, −10kb and −12kb relative to the transcription start site (TSS) within the Nrarp regulatory region. We also detected consistent methylation by Notch SpDamID near the TSS of the Notch target Cdh6 (Figure 1E, (Bielesz et al., 2010)) and Mef2c SpDamID labeled near the Mef2c target, Cdkl5 (Zweier et al., 2010). These results demonstrate that SpDamID can identify TF specific complexes on genomic sites in spite of utilization of shared co-activators.
In order to determine if SpDamID could work on small populations of cells as would be necessary for some in vivo applications, we performed SpDamID using decreasing amounts of DNA from the mK4 cells stably expressing N1D/RBPAM-Ert2 showing LMP library generation and enhancer enrichment in as little DNA as equivalent to 100 cells (Figure S1J).
Genomic Analysis of SpDamID Marked DNA Sequences
We next addressed whether SpDamID reproducibly marks TF-specific targets in a genome-wide manner using two methods: microarray hybridization and next generation sequencing (NGS). LMP libraries of DpnI released fragments were generated from biological triplicates of transiently transfected cells expressing Notch complexes (described above) and specificity controls (Mef2cD/MAMLAM-Ert2 and Mef2cD/p300AM-Ert2). SpDamID libraries were analyzed either by hybridization to NimbleGen 2.1M Deluxe Promoter arrays (Methods and array hybridization results described in supplemental information) or by NGS. For the latter, SpDamID libraries were sonicated to generate ~200bp fragments that were ligated to bar-coded sequencing adaptors, PCR re-amplified and subjected to NGS on Illumina HiSeq2500 (see Experimental Procedures for details). The libraries were read to a depth of ~10 million reads/library for each biological replicate from the same N1/RBP, N1/MAML, N1/p300 and N1/N1 dimer SpDamID libraries used for array hybridizations (along with the replicates of the D/AM controls and one sample each of the Mef2c/MAML and Mef2c/p300 pairs). The sequencing reads were mapped to DpnI fragments and enrichment was determined by comparing the sequencing reads in the experimental samples to the reads in controls. We required that a fragment had to be enriched in at least two out of the three biological replicates. To validate this scoring strategy, sequencing reads from each library were mapped to 5kb windows centered on the segments identified by the algorithm. The reads overlapped the algorithm-called fragments with strong cross-overlap between replicates from all Notch pairs (Figure S2B–C). Hierarchical clustering of the sequencing reads confirmed replicates were closely related to each other, to other Notch complexes, but not to Mef2c-labeled Dpn1 fragments in both NGS and hybridization experiments (Figure 2A and Figure S2A).
Figure 2. Genome-wide Analysis of SpDamID and Cooperative Labeling by Notch and Runx1 (see also Figure S2).

(A) Unsupervised hierarchical clustering of SpDamID-seq libraries. (B) The top 5 enriched TF motifs based on a HOMER analysis of N1D/p300AM-Ert2 SpDamID-seq. (C) The top 5 enriched motifs in the Mef2cD/p300AM-Ert2 SpDamID-seq library identified by Homer analysis. (D) Alignment of Runx1 ChIP in megakaryocytes with SpDamID and FAIRE identifies candidate enhancers with RBP and Runx1 binding within a 100bp window near the Smad3 and Id1 genes. (E) Biological duplicates of SpDamID libraries produced by Notch dimers and Runx1/Notch pairs but not by the Hif1a/Notch pair. The Notch dimers and Runx1/Notch pairs enriched for enhancers containing predicted binding sites for both factors within 100bp of each other (Smad3 +14kb, Id1 −4kb, Nrarp −8kb and Runx1 −80kb) but not for the Notch-only target (Hes1 −0.1kb). F) Heat maps of SpDamID and ChIP libraries centered on megakaryocyte Runx1 peaks present in FAIRE peaks from mK4 cells.
To test if DpnI fragments identified by SpDamID contain the RBP binding motif, we performed motif enrichment analyses using the HOMER motif discovery toolkit (Heinz et al., 2010) on the SpDamID libraries. HOMER analysis of the Notch SpDamID microarray (Figure S2D and S2E) or SpDamID-seq libraries (Figure 2B) identified the known RBP motif (BVYGDGAD) as being significantly enriched (N1D/RBP p<10−138, N1/MAML p<10−172, and N1/p300 p<10−182) ranking second to the AP1 motif consistent with the observation that AP1 sites are present in most in human enhancers irrespective of cell-type (Andersson et al., 2014). Similarly, the Mef2 motif was the top-ranking non-AP1 motif over-represented in segments identified by Mef2c/MAML (p<10−392) and Mef2c/p300 (p<10−292) (Figure 2C). The Mef2c motif was only weakly enriched in the libraries generated by the Notch pairs and the RBP motif was weakly enriched in the Mef2c libraries. Lastly, SpDamID can be leveraged to identify motifs utilized by dimers on a genome-wide basis. Segments methylated by N1D/MAMLAM-Ert2 and N1D/p300AM-Ert2 but not by N1D/N1AM-Ert2 would reflect binding of monomeric RNM complexes, whereas segments methylated by all three pairs would reflect binding of dimeric RNM complexes. Within each dimer “positive” segment, we identified the strongest RBP “traditional” motif (V$RBPJK_01) and extracted the DNA sequence 10–30 bases 3′ of the motif “anchors”. Sequences were extracted in an identical manner from our negative set. The HOMER toolkit was used to identify motifs distinguishing the “positive” from the “negative” dataset (Supplemental methods and Table 1). The ability to identify dimer dependent vs. independent binding events is a unique feature of SpDamID.
Table 1.
Top Ranking Notch Dimer Half Site Motifs Within 10–30bp Of A Canonical Half Site
Motif enrichment in segments labeled by N1D/N1AM-Ert2 dimers. Segments preferentially labeled by N1D/N1AM-Ert2 were identified by comparing all segments labeled by MAML-containing SpDamID pairs to those labeled only by N1D/MAMLAM-Ert2 and N1D/p300AM-ERT2 but not N1D/N1AM-Ert2. The analysis software (HOMER) uncovered a bi-partite site containing one canonical RBP motif next to either another canonical RBP site or novel half-site motifs.
| Rank | Top Half Site Motifs: | p-value | (Log) | % of Positive | % of Negative | Comments |
|---|---|---|---|---|---|---|
| 1 |
|
1e−6 | −1.86e01 | 16.67% | 1.24% | “Canonical/Cryptic” Hybrid |
| 2 |
|
1e−6 | −1.48e01 | 14.04% | 1.24% | “Canonical” |
| 3 |
|
1e−5 | −1.37e01 | 8.77% | 0% | “Cryptic” |
| 4 |
|
1e−4 | −1.09e01 | 7.02% | 0% | “Cryptic” |
| 5 |
|
1e−4 | −1.04e01 | 12.28% | 1.86% | “Cryptic” (Hes5-like) |
Runx1 and Notch do not interact physically (Terriente-Felix et al., 2013), yet an oPOSSUM Anchored Combination Site Analysis (Kwon et al., 2012) found the majority of the Notch SpDamID labeled fragments had Runx1 motifs within 100bp of a RBP binding site (Figure S2F), as was observed in Notch ChIP experiments (Terriente-Felix et al., 2013; Wang et al., 2014). The analysis also showed that 1 Runx1 ChIP sites identified in megakaryocytes (Pencovich et al., 2011) were labeled by Notch SpDamID within open chromatin identified by formaldehyde-assisted isolation of regulatory elements (FAIRE; (Giresi et al., 2007; Giresi and Lieb, 2009) (Figure 2D). Notably, we observed that Notch activation induced a 4.5 fold increase in Runx1 expression, suggesting the existence of a potential transcriptional feed-forward loop in which Notch induced expression of Runx1 allows cooperative regulation of downstream targets. These results prompted us to ask if SpDamID could identify binding of Notch in proximity to Runx1 on the same enhancers. Runx1D/N1AM-Ert2 produced strong enrichment for fragments containing sites for both factors (Figure 2E and data not shown). The methylation of segments containing sites for Runx1D/N1AM-Ert2 but not for Mef2cD/N1AM-Ert2 or Hif1AD/N1AM-Ert2 demonstrates the ability of SpDamID to detect simultaneous binding of non-interacting (or weakly interacting) TFs, provided they bind DNA in close proximity to each other. Colocalization of Notch SpDamID and Runx1 megakyrocytes ChIP peaks is evident in heat maps of SpDamID reads centered on megakaryocyte Runx1 peaks present in FAIRE peaks from mK4 cells. Labeling by Notch SpDamID was appreciably better than that observed with either Mef2C or D/AM (Figure 2F).
SpDamID Datasets Enrich For Notch-Regulated Genes
To ask if sites identified by SpDamID-seq enriched for regulatory modules we analyzed the fragments labeled in all the Notch SpDamID libraries with the Genomic Region Enrichment of Annotations Tool (GREAT). The Notch pathway emerged as the most significantly enriched signaling pathway in the PANTHER ontology database (p<4.1e−9; Figure S3A). Additionally, the most enriched structure in the MGI gene expression database was the kidney S-shaped body (p<2.1e−14; Figure S3A), a Notch-dependent structure induced from mK4-like cells in vivo (Cheng et al., 2007). These results confirm that SpDamID is enriching biologically relevant DNA regions bound by Notch complexes.
Next we asked how effectively SpDamID enriched for Notch regulated genes in mK4 cells. To assemble a stringent Notch-regulated transcript dataset, we profiled RNA from mK4 cells experiencing EGTA-induced activation of endogenous Notch receptors (Ilagan et al., 2011; Rand et al., 2000) using Illumina bead arrays. EGTA induces unfolding of the negative regulatory region, which undergoes cleavage by ADAM metalloprotease and γ-secretase to induce NICD release. We filtered out non-specific effects by requiring regulated transcripts to display sensitivity to 10 μM of the γ-secretase inhibitor DAPT (N-[N-(3,5-Difluorophenacetyl-L-alanyl)]-S-phenylglycine t-Butyl Ester) and included cycloheximide to avoid secondary transcriptional changes. Analyzing the expression data with Partek software identified 347 genes that displayed reproducible transcriptional responses to EGTA (FDR=0.05; p≤0.05) of which 122 are DAPT-sensitive (Table S1). We validated by qPCR a subset of the target list including Hes1, HeyL, Nrarp, Wisp2, Cdh6, Gper (Gpr30), and NGFb. These targets were upregulated in mK4 cells stably expressing constitutively active Notch (Notch1ΔE), activated when endogenous Notch was exposed to EGTA, but did not respond or were repressed in the presence of 10 μM DAPT or a dominant negative fragment of MamL1 (DN-MAML; Figures S3B–C).
To evaluate the enrichment for Notch-regulated genes in our SpDamID libraries, each of the SpDamID segments was assigned to the gene with the nearest TSS. We ranked the segments by their SpDamID scores and analyzed the data in bins of 400 (microarray data; Figure S3D) or 1000 (SpDamID-seq; Figure 3A) to determine the levels of enrichment for Notch-responsive genes by hypergeometric distribution analysis. Enrichment levels approached that expected by chance as scores decreased (Figure 3A). Even the highest scoring Notch segments failed to enrich for Mef2c targets (Figure S3E). Conversely, the highest scoring Mef2c/p300 segments failed to enrich for Notch transcriptional targets (Supplemental Figure 3D) while enriching for Mef2 targets obtained from the Gene Set Enrichment Analysis dataset (GSEA) (http://www.broadinstitute.org/gsea/index.jsp; (Subramanian et al., 2005); Figure S3E). Assessing the overlap between all the genes located near SpDamID-seq segments and Notch–regulated genes in mK4 cells revealed that most Notch-regulated genes were labeled by SpDamID pairs (Figure 3B). Hypergeometric distribution analysis confirmed highly significant enrichment (N1/RBP 1.82 fold OVEC, p<10−13; N1/MAML 2.14 fold OVEC, p<10−14; N1/p300 1.98 fold OVEC, p<10−12). These analyses indicate that the SpDamID successfully discriminated among regulated targets of Notch and Mef2c.
Figure 3. Comparison of SpDamID and Notch-Regulated Transcripts (see also Figure S3).
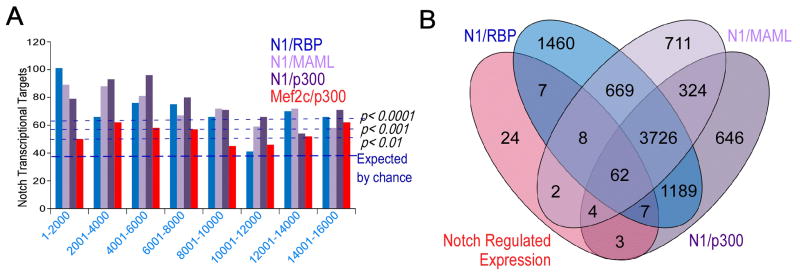
(A) Binning of segments identified by Notch SpDamID-seq show significant enrichment of Notch regulated targets. (B) Venn diagram detailing the overlap between Notch-regulated genes expressed in mK4 cells and the genes near a segment labeled by SpDamID-seq generated with the indicated Notch pairs.
SpDamID Identifies Notch-dependent Enhancers at the Nrarp and Cdh6 Loci
We next sought to confirm that SpDamID identified functional enhancer elements near Notch-regulated genes. We focused on the Notch-regulated genes Nrarp and Cdh6. The Nrarp locus is depicted in Figure 4A and Notch SpDamID identified potential enhancer elements at −3kb, −8kb and −12kb relative to the Nrarp TSS (Figure 4B) that overlapped with regulatory regions present in the Encode database (Figure 4C). Interestingly, only the −12kb site was not methylated by the N1D/N1AM-Ert2 dimer pair (Figure 4B), which could indicate that the putative enhancers at −3kb and −8kb were Notch dimer–dependent, but the −12kb enhancer was not. To test for Notch-dependent enhancer activity, we generated luciferase reporters driven by DpnI fragments containing the −3kb, −8kb and −12kb fragments. All three putative enhancers mediated strong transcriptional responses to activated Notch1 (N1ΔE) (Figure 4E). The −3kb and −8kb enhancers contain potential SPS dimer binding sites with one consensus RBP binding site located near a non-canonical site on the other strand (Arnett et al., 2010) (Figure 4D). By contrast, the −12kb region none of several putative RBP binding sites was in head-to-head orientation. Importantly, the dimer mutant N1ΔER1974A failed to activate the −3kb and −8kb reporters, but retained the ability to induce the −12kb reporter (Figure 4E). Mutagenesis of either the canonical or degenerate RBP site in the SPS within −3kb and −8kb enhancers significantly reduced their responsiveness to N1ΔE. By contrast, one site was sufficient to drive Notch-dependent transcription in the −12kb region. ChIP-PCR with either wild-type or the dimer-mutant Notch enriched for all three enhancer regions (Figure S4A), supporting their identification as Notch bound sites and highlighting a inability of ChIP to discriminate between sites bound by monomeric or dimeric Notch. Importantly, endogenous Nrarp was strongly expressed in mK4 cells stably expressing wild-type activated N1ΔE but not in cells that expressed the N1R1974AΔE dimer mutant (Figure S4B) confirming that Nrarp is a dimer-responsive locus.
Figure 4. Notch SpDamID Identification of Notch Dimer-Dependent and Dimer-Independent Enhancers Upstream of Nrarp (see also Figure S4).

(A) Scaled diagram of the Nrarp locus. (B) From the top: SpDamID score of DpnI segments is shown (highest score=darkest shade of gray). The coverage of the sub-genomic array is shown as a light yellow box. The order of the experiments N1/RBP cluster (immature complex), N1/MAML cluster (mature complex), and Mef2c samples. Below, a virtual DpnI digest is shown in alternating yellow and black boxes. Consensus RBP half-sites are displayed as lines below it. Note that the −12kb region was only labeled by the Notch-MAML and Notch-p300 pairs but not the Notch dimer pairs (pointer). (C) ENCODE DNase hypersensitive (DHS) sites from kidney and fibroblasts, along with the mammalian conservation score from the UCSC Genome Browser. (D) Conserved sequences surrounding the consensus RBP binding sites (colored red or blue depicting + or − strand, respectively) in the −12, −8 and −3 enhancers. Potential weak sites are shown with a dashed arrow. Mutated strong (S) or weak (W) sequences are shown below. (E) Luciferase reporter assays using DpnI fragments with or without indicated mutations in RBP binding sites and responding to either activated N1ΔE or a dimer-deficient N1ΔER1974A. Error bars indicate standard deviation.
Methylated segments in the Cdh6 locus overlap with sites ChIPed by Notch in mK4 cells (but not T-ALL cells) and were DNase I hypersensitive regions in kidney but not in fibroblasts, consistent with a tissue-specific enhancer (Figure 5A–C). We confirmed by ChIP-PCR that Notch bound to the conserved RBP site-containing region within intron1 of Cdh6 but not to a nearby region (Figure S5A). Notch-dependent, DAPT-sensitive regulation of Cdh6 mRNA and protein was observed in mK4 cells (Figure S3B, S3C, S5B, and S5C). To map the functional kidney-specific Cdh6 enhancer in vivo, we examined transgenic embryos harboring overlapping, LacZ-containing bacterial artificial chromosomes (BAC) fragments covering the Cdh6 locus (Inoue et al., 2008a; Inoue et al., 2008b, 2009). LacZ expression was detected in the developing kidney (Figure 5D) only when the BAC contained the enhancer identified by Notch SpDamID (segment “K”, Figure 5D) but not when it was absent. Collectively, these data confirm that Cdh6 is a direct Notch target during kidney development (Boyle et al., 2011; Cheng et al., 2007) and establish that SpDamID effectively identifies functionally relevant regions in Notch transcriptional targets.
Figure 5. Notch SpDamID Identification of a Regulatory Region of Cdh6 (see also Figure S5).

(A) Scaled diagram of Cdh6 locus. Below are Notch1 and RBP ChIP peaks from TLL (top two tracks) and Notch ChIP from mK4 cells (bottom track). Binding downstream of the TSS is only seen in mK4 cells. (B) Overlapping segments identified by mature Notch complexes (pointer shows poor labeling by immature complexes). (C) ENCODE data indicates a DHS and Pol2 ChIP signal in kidney cells, but not in fibroblasts, at this location. (D) BAC transgenic analysis across the Cdh6 locus drives LacZ expression and β-Galactosidase activity within embryonic renal epithelia only when region K is included.
Analysis of Immature and Mature N1 Complexes using SpDamID Versus ChIPseq
We compared ChIP, the dominant method for identifying protein-bound DNA, with SpDamID in the same cell-type to determine the relative performance of these complementary methods. We performed ChIPseq in mK4 cells stably expressing a DOX-inducible, Myc-tagged, activated Notch1 (N1ΔE, (Kopan et al., 1996)) expressing NICD1 at near physiological levels in the absence of DOX. We confirmed that the known SPS sites in Hes1 were immunoprecipitated and an upstream region was not (Experimental procedures and Figure S5A) before sequencing the immunoprecipitated DNA. In order to directly compare the Notch1 (N1) ChIPseq dataset to the SpDamID dataset, we mapped the ChIPseq reads onto the sequences of the inter-GATC segments included on the Nimblegen array and scored each segment by its average depth of ChIPseq reads (see Experimental Procedures). To be counted as ChIP-positive, a segment was required to have coverage ≥10-fold that of the input control. oPOSSUM analysis confirmed that N1 ChIPseq enriched for the RBP motif to a similar extent as Notch SpDamID, demonstrating similar specificity for the N1 ChIP and SpDamID (Figure S6A). The relationship between the GATC flanked segments containing N1 ChIP peaks and the recovered SpDamID fragments is shown in Figure 6A using a similar binning strategy as used in Figure 3A. Although the overlap of N1D/RBPAM-Ert2 fragments and N1 ChIP was statistically significant, the N1D/RBPAM-Ert2 score correlated poorly with the probability of a fragment being recovered by N1 ChIP. By contrast, higher scoring fragments enriched by N1D/MAMLAM-Ert2 or N1D/p300AM-Ert2 had a higher probability of being recovered by N1 ChIP (Figure 6A). The area under the receiver operating characteristic curve (AUROC) analysis found that SpDamID scores for N1D/MAMLAM-Ert2 (AUROC=0.78) and N1D/p300AM-Ert2 (AUROC=0.72) are predictive of N1 ChIP peaks containing RBP motifs but N1D/RBPAM-Ert2 scores (AUROC=0.55) were not (see Experimental Procedures). Heat maps of the SpDamID reads mapped to the N1 ChIP fragments confirmed that the mature complex labeling overlapped with N1 ChIPed sites to a greater extent than with the immature complex (Figure S6B).
Figure 6. Mature Notch SpDamID Enriches for Sites Displaying Dynamic Chromatin Remodeling in Response to Notch Signaling (see also Figure S6).

(A) Binning of SpDamID segments based on score shows significant overlap by Notch SpDamID but not Mef2c/p300 with the fragments recovered by Notch ChIP in mK4 cells. Correlation between SpDamID score and Notch ChIP enrichment score is shown in the upper right quadrant. (B) Titration of MAML1 disrupts N1D/RBPAM-Ert2 complementation and impairs DNA methylation, reflected by loss of LMP products. (C) QPCR detecting FAIRE sites in Hey1 (−60kb) and Wisp2 (−6kb) that open 4 hours after γ-secretase inhibitor removal are not opened in the presence of DN-MAML. (D) Heat maps showing the overlap of SpDamID and Notch1 ChIP reads within a 5kb window centered on all of the FAIRE peaks in cells expressing active Notch. (E) Depiction of the Wisp2 locus showing SpDamID-seq reads (D/AM in black, N1D/RBPAM-Ert2 in blue, N1D/MAMLAM-Ert2 in light purple, and N1D/p300AM-Ert2 in dark purple) and FAIRE-seq reads from control cells (black) or cells expressing active Notch (dark blue). ENCODE data of mouse kidney DHS or Pol2 binding shown in pink. DHS in mouse fibroblasts and the mammalian conservation score shown in black. The red boxes highlight Notch-dependent FAIRE peaks present in cells expressing active Notch that are labeled by mature Notch SpDamID complexes. (F) Heat maps showing overlap of SpDamID and Notch1 ChIP reads with Notch-dependent FAIRE sites.
A possible explanation for the difference in labeling of ChIPed fragments by N1D/RBPAM-Ert2 and N1D/MAMLAM-Ert2 complexes would be that the recruitment of MAML to the N1D/RBPAM-Ert2 pair resulted in loss of DAM complementation or produced a configuration that greatly limited access of DAM to DNA, preventing the N1D/RBPAM-Ert2 pair from labeling DNA bound by a mature, MAML-containing N1D/RBPAM-Ert2 complexes. To directly test this hypothesis we co-expressed increasing amounts of MAML with the N1D/RBPAM-Ert2 SpDamID pair. We observed a dose-dependent decrease in the ability of N1D/RBPAM-Ert2 to generate LMP libraries as MAML concentration increased, consistent with failure of DAM complementation (Figure 6B). This result indicated that all complexes methylated bound fragments in a similar fashion but with different efficiency and suggest that ChIPseq was biased towards the multi-member, mature complexes.
The comparison of SpDamID to ChIP allowed us to visualize the spread of methylation by DNA bound SpDam pairs. We plotted the frequency of methylation of every GATC located within a 10 kb window centered on a N1 ChIPed peak (Figure S6C). Roughly 80% of adenine methylation events occurred within a 1600bp footprint around a bound, reconstituted DAM as seen with DamID (van Steensel and Henikoff, 2000).
Mature Notch Complexes Enrich for Dynamic Sites that Open in Response to Notch
It has been observed that some sites in the genome are occupied only when Notch signaling is active (“dynamic sites”; (Bray and Bernard, 2010; Castel et al., 2013; Housden et al., 2013; Krejci et al., 2009; Wang et al., 2014; Wang et al., 2011). These dynamic sites show increased recoverability by RBP ChIP when NICD is present, leading to the proposal that the NICD/RBP complex acts as a pioneer factor to modify chromatin at these sites (Castel et al., 2013). These reports led us investigate chromatin opening in response to Notch binding via FAIRE. To confirm that dynamic sites reflected opening of chromatin at the Wisp2 and Hey1 promoters, we assessed the impact of DN-MAML, which stabilizes the Notch/RBP complex but prevents recruitment of p300, on formation of Notch-dependent FAIRE sites. QPCR confirmed that Notch-dependent FAIRE sites in the Wisp2 and Hey1 promoters appeared within 4 hours after DAPT washout in N1ΔE cells but not when DN-MAML was present (Figure 6C), confirming that Notch-dependent sites required p300.
SpDamID-seq reads from all complexes mapped to total FAIRE peaks to a similar degree, performing better than N1 ChIP (Figure 6D; details of putative regulatory sites in Wisp2 shown in 6E). Confining the analysis to Notch-dependent FAIRE sites demonstrated better overlap between N1/MAML and N1/p300 reads and Notch-dependent FAIRE sites relative to the N1/RBP dataset (increase relative to similarly normalized N1/RBP signal: N1/MAML, 52.4%, p=9.0×10−5; N1/p300, 57.9%, p=2.6×10−4; Figure 6F) that is clearly observable at individual loci as shown in the snapshot of the Wisp2 allele (Figure 6E). oPossum analysis showed that 1622 out of 2403 Notch dependent open chromatin identified by FAIRE in mK4 cells have a Runx1 motif within 100bp of a RBP motif (Z-score of 176.331). Similarly, “dynamic” RBP ChIPed sites identified in C2C12 cells (Castel et al., 2013) displayed greater labeling by mature complexes than by the N1/RBP immature complex (Figure S6F).
DISCUSSION
SpDamID Methylates DNA with Specificity and Low Background Signal
SpDamID is a protein-complementation version of DamID that enables identification of genomic sites bound by protein complexes. SpDamID satisfies the key validation criteria for a protein complementation assay: (i) DAM was split into two complementing N-terminal (D) and C-terminal (AM) halves. Specific signal was observed with relevant interacting fusions (e.g., NID/RBPAM), but not when D and AM were co-expressed on their own or when fused to noninteracting proteins (e.g., Mef2cD/N1AM); (ii) mutations expected to either reduce or inhibit the protein interaction (e.g., N1ΔRAMD/RBPAM-Ert2 or N1R1974AD/N1R1974AAM-Ert2) or interrupting dominant negative proteins (DN-MAML) have the expected corresponding effects on DAM complementation; (iii) fusion of SpDam halves does not prevent the TFs from activating their transcriptional targets, the Ert2 fusion did not alter the transcriptional activity of RBP, and the adenine methylation seems to have a negligible effect on transcription and (iv) specific and robust complementation was also observed with fragment swapping (NID/RBPAM, NID/NAM).
The strength of this approach is demonstrated by the ability of SpDamID to distinguish monomeric from dimeric binding, and detect DNA-bridged DAM complementation when noninteracting factors (Notch and Runx1) bind simultaneously to the same enhancer, a non-trivial tasks with antibody-based methodologies. When complementation is occluded by recruitment of additional members to the complex additional information can be obtained (differentiation mature from immature Notch complexes). Overall, these results demonstrate the utility of SpDamID, and the value of probing multiple protein pairs within a single transcription complex.
Compared to conventional ChIP-seq, SpDamID requires no antibodies and orders of magnitude less starting material (100–10,000 cells, Figure S1K), marking binding events that occurred in vivo within a defined temporal window while avoiding cross-linking artifacts inherent to ChIP. SpDamID can be applied to any protein pair of interest and in any cell type or species amenable to genome editing techniques to generate fusion of SpDam pairs to desired proteins. As an additional benefit, the use of protein complementation allowed SpDamID to resolve several deficiencies of DamID. SpDamID retained acceptable signal-to-noise ratio even at sustained high expression levels. The addition of an Ert2 domain to the SpDam system allowed temporal control of nuclear entry (with 4OHT; Figure S1E) to eliminate background labeling: SpDamID pairs stably integrated produced no background in the absence of tamoxifen for 100 generations, but labeled DNA upon addition of the drug.
SpDamID labeled segments displayed significant enrichment for the expected binding motifs and for TF-regulated target genes to a comparable levels observed with ChIP. With the proper design, SpDamID pairs can produce deeper insight into the assembly of transcription complexes. Crystal structure assisted placement of the D/AM halves can provide unique insights by allowing complementation only by specific sub-complexes differentiated by their confirmations.
SpDamID and ChIP Comparison Allows Higher Confidence Binding Site Identification
The application of SpDamID and ChIP in the same cells allows direct comparison of the sites enriched by different methods to identify bound sites with higher confidence and control for biases inherent to either methodology. There was significant yet incomplete overlap between SpDamID-positive and ChIP-positive datasets (Table S1) that extended to data obtained in distinct cell-types produced by other labs; the differences between SpDamID and ChIP opens interesting new questions as to the origin of the unique signatures of each method. Interestingly, many of the shared binding sites are not associated with Notch dependent transcriptional regulation of nearby genes, which suggests that a significant portion of protein:DNA interactions reflect a biochemical interaction whose biological relevance is yet to be understood. In order to discern the functional significance of a SpDamID labeled segment, the next step is the alteration of the potential regulatory sites in their native chromosomal context to determine their contribution to the transcriptional landscape, which has been greatly facilitated by the development of CRISPR technology (Crocker and Stern, 2013; Dowen et al., 2014).
SpDamID Potential for In Vivo Analyses
To evaluate the potential of SpDamID for in vivo applications, we tested background genomic DNA methylation associated with SpDamID by passaging mK4 cells stably expressing the DOX inducible N1D/RBPAM-Ert2 pair for 100 generations. We found no detectable methylation in the absence of DOX and 4OHT. Upon addition of DOX and 4OHT, Dam was reconstituted, allowing generation of LMP products (Figure S1H–J). We also generated p300AM-Ert2 knock-in mouse ES cells and confirmed 4OHT-dependent enhancer enrichment upon transfection with either N1D or Mef2cD (Figure S1L). These results affirm the potential of SpDamID for in vivo applications in vertebrate species.
In summary, the addition of SpDamID to the toolkit of genome analysis techniques complements existing technologies and opens new avenues of investigation. SpDamID identifies genomic bound simultaneously by protein pairs on the same chromosome segment even if they do not interact physically, provided they are bound close enough to allow self-association of the DAM halves. The genome-wide identification of DNA segments bound by TF pairs will facilitate analyses of cooperativity, which is thought to provide cell-type and temporal specificity of gene expression. Although cooperative binding of TFs at specific sites can be addressed by Re-ChIP (Furlan-Magaril et al., 2009), the Re-ChIP method is severely limited by the amount of material available that largely prevents the genome-wide analyses that are possible with SpDamID. Thus, SpDamID extends the capabilities of ChIP-seq, which typically infers combinatorial binding from shared regions in distinct ChIPs even though these regions may in fact represent sites bound only by a single factor in any given cell within the population. In combination with FAIRE, SpDamID can distinguish between formation of transcription complexes on poised chromatin versus the binding of preassembled complexes. The combination of SpDamID with functional validation using genome-editing technologies will facilitate deciphering the intricate nature of gene regulatory networks.
Limitations
The main limitation of SpDamID is that the initial LMP libraries are generated from DpnI fragments of varying sizes that are not necessarily amplified equivalently. Although our scoring algorithm corrects for differential PCR efficiency, future developments of methods to analyze just the ends of the DpnI fragments through the use type-III restriction enzymes in the adaptors will further assure complete genome coverage. Additionally, SpDamID utilizes fusions to proteins of interest that requires some genome modification in order to analyze endogenous proteins but this prospect has been greatly simplified through CRISPR technology that has allowed us to rapidly generate several cell lines with SpDamID knock-in fusions.
SpDamID DNA methylation resources
SpDamID methylation specificity being governed primarily by the TF “spokes” rather than by the shared “hubs” greatly simplifies the investigation of multi-component transcription complexes. The p300AM-Ert2 “hub” can effectively pair with a number of different TFs beyond Notch1 and Mef2c, including β-catenin, TCF4, p53, and the androgen receptor to reconstitute DAM and methylate GATC near target sites (data not shown). In order to facilitate rapid dissemination of SpDamID, we have generated a large collection of bi-directional DOX inducible vectors (Supplemental information) containing over 36 different TF-D fusions in combination with p300AM-Ert2 to allow DOX and 4OHT controlled SpDamID. Furthermore, we have generated a knock-in mouse ES cell line in which the AM-Ert2 coding ORF was inserted in frame with the C-terminus of the endogenous p300 gene. We observed no background adenine methylation in these cells after 10 passages and transfection with either N1D or Mef2cD produced SpDamID libraries and showed specific labeling of targets only in the presence of 4OHT. These cells can either be infected or genetically modified to express the TF-D fusion of interest in order to allow in vivo SpDamID analyses. It would be particularly interesting if the community generates ROSA-stop-TFD fusions that, in combination with p300AM-Ert2, could utilize existing Cre drivers to allow cell-type specific SpDamID analyses of TF binding in vivo beyond what is possible with ChIP.
EXPERIMENTAL PROCEDURES
Extended Experimental Procedures are located in the Supplemental Information
SpDamID analysis
mK4 TetOn cells were transiently transfected with bi-directional vectors expressing the SpDam pairs and treated with doxycycline and 4OHT as indicated. After 24hr of drug treatment, DNA was collected and then digested overnight with DpnI and then ligated to adapters that are used to generate libraries by ligation mediated PCR. Purified libraries wereused as the template for target specific PCR, labeled and hybridized onto NimbleGen promoter arrays, or sonicated to produce fragments for the SpDamID-seq analysis. A detailed description of the scoring algorithm developed to identify fragment enrichment in the promoter array or SpDamID-seq datasets is provided in the Extended Experimental Procedures.
Motif Identification
The DNA sequences of the SpDamID enriched fragments were extracted using Galaxy and utilized for TF motif enrichment analysis using HOMER. The oPOSSUM anchored motif analysis was performed by using the RBP motif identified by Ong et al. and searching for enriched motifs within 100bp of RBP motif.
Gene expression analysis
mK4 TetOn cells were plated at 90% confluence on 6 well plates. In 3 wells 10μM of DAPT was added after 20 hours. After 24hr the cells were washed with Hank’s balanced salt solution (HBSS) and incubated in HBSS containing 10μg/ml cyclohexamide and 500nM EGTA (or EGTA+DAPT). After 30 min the media was carefully aspirated and the cells incubated for an additional 3.5 hr in 10% FBS/DMEM, 10μM cyclohexamide and ± 10μM DAPT. 4hr after exposure to EGTA RNA was collected (Qiagen RNeasy) following the manufacturer’s recommendations. Hybridiztion to Illumina bead arrays was performed by the GTAC sequencing core at Washington University. Statistically significant transcriptional changes induced by the EGTA treatment only in DAPT-free samples were determined with Partek™ software. The significance of the observed enrichment for Notch-regulated transcripts in the SpDamID gene lists was determined using hypergeometric distribution analysis (R software version 3.0.1).
ChIP
mK4 TetON control cells or stably expressing N1ΔE-6myc were used for ChIP experiments using the 9E10 anti-myc antibody as detailed in the Extended Experimental Procedures. Bar-coded Illumina libraries were generated from the ChIP material and subjected to next-generation sequencing on an Illumina HiSeq2000.
FAIRE
mK4 TetON control cells or stably expressing N1ΔE-6myc were cross-linked with 1% formaldehyde for 10 minutes, which was followed by nuclear isolation and sonication. The FAIRE sites were recovered by Phenol:Chloroform IsomylAlcohol extraction of open chromatin, which was quantified and used to generate Illumina next-generation sequencing libraries for analysis with the Illumina HiSeq2000. MAC peak calling was used to call FAIRE peaks in the controls relative to input and in the N1ΔE-6myc FAIRE libraries relative to control.
SpDamID-seq Read Heat Map Analysis
SpDamID-seq reads were mapped to the genomic locations of the FAIRE peaks (either total FAIRE sites or Notch-dependent FAIRE sites that open in response to Notch) or the RBP ChIP peaks that were induced by Notch signaling in C2C12 cells (Castel et al., 2013) using the bam coverage tool in the deeptools package of Galaxy. Heat maps and graphs of the SpDamID read coverage of these sites were generated using the computeMatrix and heatmapper tools of the deeptools package.
Establishment and analysis of Cdh6 bacterial artificial chromosome (BAC) transgenic mouse lines
Stable transgenic mouse lines with E. coli beta-galactosidase reporter modified Cdh6-BAC were generated as described previously (Inoue et al., 2008a). Kidneys from E14.5 mice were dissected out and stained for beta-galactosidase as described in Supplementary information. Whole mount images were taken under the binocular microscope equipped with a CCD camera (Leica MZ8).
Antibody and Primer lists
All of the antibodies and primers utilized are provided in the Extended Experimental Procedures.
Supplementary Material
Acknowledgments
This work was supported by funding from the American Recovery and Reinvestment Act Grant 1RC1NS068277-01 (RK) and National Institutes of Health grants R01CA163653-01A1 (RK). We thank Bas van Steensel for the original DamID plasmids. We are grateful for the material assistance provided by Gary Stormo (funded by F32GM083604). We thank Maxene Ilagan for helpful discussions, Brian Gebelein and Harinder Singh for critically reading the manuscript and Alexandra Ragsdale, Anjum Rangwala and Yu Wue for technical support.
Footnotes
ACCESSION NUMBER
The GEO accession number for the NGS and promoter array data is GSE70402.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett KL, Hass M, McArthur DG, Ilagan MX, Aster JC, Kopan R, Blacklow SC. Structural and mechanistic insights into cooperative assembly of dimeric Notch transcription complexes. Nat Struct Mol Biol. 2010;17:1312–1317. doi: 10.1038/nsmb.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040–4054. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle SC, Kim M, Valerius MT, McMahon AP, Kopan R. Notch pathway activation can replace the requirement for Wnt4 and Wnt9b in mesenchymal-to-epithelial transition of nephron stem cells. Development. 2011;138:4245–4254. doi: 10.1242/dev.070433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray S, Bernard F. Notch targets and their regulation. Curr Top Dev Biol. 2010;92:253–275. doi: 10.1016/S0070-2153(10)92008-5. [DOI] [PubMed] [Google Scholar]
- Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013;27:1059–1071. doi: 10.1101/gad.211912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134:801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker J, Stern DL. TALE-mediated modulation of transcriptional enhancers in vivo. Nat Methods. 2013;10:762–767. doi: 10.1038/nmeth.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schuijers J, Lee TI, Zhao K, et al. Control of cell identity genes occurs in insulated neighborhoods in Mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan-Magaril M, Rincon-Arano H, Recillas-Targa F. Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. Methods Mol Biol. 2009;543:253–266. doi: 10.1007/978-1-60327-015-1_17. [DOI] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JR, Liebert K, Bekes M, Jeltsch A, Cheng X. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol. 2006;358:559–570. doi: 10.1016/j.jmb.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Fu AQ, Krejci A, Bernard F, Fischer B, Tavare S, Russell S, Bray SJ. Transcriptional dynamics elicited by a short pulse of notch activation involves feed-forward regulation by E(spl)/Hes genes. PLoS Genet. 2013;9:e1003162. doi: 10.1371/journal.pgen.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan MX, Lim S, Fulbright M, Piwnica-Worms D, Kopan R. Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci Signal. 2011;4:rs7. doi: 10.1126/scisignal.2001656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Inoue YU, Asami J, Izumi H, Nakamura S, Krumlauf R. Analysis of mouse Cdh6 gene regulation by transgenesis of modified bacterial artificial chromosomes. Dev Biol. 2008a;315:506–520. doi: 10.1016/j.ydbio.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Inoue YU, Asami J, Inoue T. Cadherin-6 gene regulatory patterns in the postnatal mouse brain. Mol Cell Neurosci. 2008b;39:95–104. doi: 10.1016/j.mcn.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Inoue YU, Asami J, Inoue T. Genetic labeling of mouse rhombomeres by Cadherin-6::EGFP-BAC transgenesis underscores the role of cadherins in hindbrain compartmentalization. Neurosci Res. 2009;63:2–9. doi: 10.1016/j.neures.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: Importance of proteolytic processing and its regulation by the extracellular domain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to notch activation: signaling crosstalk and incoherent logic. Sci Signal. 2009;2:ra1. doi: 10.1126/scisignal.2000140. [DOI] [PubMed] [Google Scholar]
- Krejci A, Bray S. Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 2007;21:1322–1327. doi: 10.1101/gad.424607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3. 2012;2:987–1002. doi: 10.1534/g3.112.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chi AW, Arnett KL, Chiang MY, Xu L, Shestova O, Wang H, Li YM, Bhandoola A, Aster JC, et al. Notch dimerization is required for leukemogenesis and T-cell development. Genes Dev. 2010;24:2395–2407. doi: 10.1101/gad.1975210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen S, Boyle S, Ilagan MX, Zhu Y, Zhang A, Kopan R. The Extracellular Domain of Notch2 Increases its Cell surface Abundance and Ligand Responsiveness During Kidney. Dev Cell. 2013;25:585–598. doi: 10.1016/j.devcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman OY, Ilagan MX, Kopan R, Barrick D. Quantitative Dissection of the Notch:CSL Interaction: Insights into the Notch-mediated Transcriptional Switch. J Mol Biol. 2007;365:577–589. doi: 10.1016/j.jmb.2006.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC, Pear WS. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004;104:1696–1702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- Nam Y, Sliz P, Pear WS, Aster JC, Blacklow SC. Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc Natl Acad Sci U S A. 2007;104:2103–2108. doi: 10.1073/pnas.0611092104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong C, Cheng H, Chang LW, Ohtsuka T, Kageyama R, Stormo DG, Kopan R. Target selectivity of vertebrate Notch proteins: collaboration between discrete domains and CSL binding site architecture determine activation probability. J Biol Chem. 2006;281:5106–5119. doi: 10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencovich N, Jaschek R, Tanay A, Groner Y. Dynamic combinatorial interactions of RUNX1 and cooperating partners regulates megakaryocytic differentiation in cell line models. Blood. 2011;117:e1–14. doi: 10.1182/blood-2010-07-295113. [DOI] [PubMed] [Google Scholar]
- Pirot P, Grunsven LA, Marine JC, Huylebroeck D, Bellefroid EJ. Direct regulation of the Nrarp gene promoter by the Notch signaling pathway. Biochem Biophys Res Commun. 2004;322:526–534. doi: 10.1016/j.bbrc.2004.07.157. [DOI] [PubMed] [Google Scholar]
- Rand DM, Grimm MLM, Artavanis-Tsakonas S, Patriub V, Blacklow CS, Sklar CJ, Aster CJ. Calcium depletion dissociates and activates heterodimeric Notch receptors. Molecular and Cellular Biology. 2000;20:1825–1835. doi: 10.1128/mcb.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. Journal of Biological Chemistry. 1994;269:5150–5156. [PubMed] [Google Scholar]
- Terriente-Felix A, Li J, Collins S, Mulligan A, Reekie I, Bernard F, Krejci A, Bray S. Notch cooperates with Lozenge/Runx to lock haemocytes into a differentiation programme. Development. 2013;140:926–937. doi: 10.1242/dev.086785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerius MT, Patterson LT, Witte DP, Potter SS. Microarray analysis of novel cell lines representing two stages of metanephric mesenchyme differentiation. Mech Dev. 2002;110:151–164. doi: 10.1016/s0925-4773(01)00581-0. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, Aster JC. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A. 2014;111:705–710. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, Bijlsma EK, Holder SE, Zenker M, Rossier E, et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat. 2010;31:722–733. doi: 10.1002/humu.21253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.