

Abstract
The melanocortin-3 receptor (MC3R) is a member of family A G protein-coupled receptors (GPCRs). The MC3R remains the most enigmatic of the melanocortin receptors with regard to its physiological functions, especially the role in energy homeostasis. The N/DPxxY motif and the eighth helix (helix 8) in the carboxyl terminus of GPCRs have been identified to be important for receptor expression, ligand binding, signal transduction and internalization. To gain a better understanding of the structure-function relationship of MC3R, we performed systematic study of all the 20 residues in this domain using alanine-scanning mutagenesis. We showed that although all mutants were expressed normally on cell surface, eleven residues were important for ligand binding and one was indispensable for downstream cAMP generation. F347A showed constitutive activity in cAMP signaling while all the other mutants had normal basal activities. We studied the signaling capacity of nine mutants in the ERK1/2 signaling pathway. All of these mutants showed normal basal ERK1/2 phosphorylation levels. The pERK1/2 levels of six binding- or signaling-defective mutants were enhanced upon agonist stimulation. The unbalanced cAMP and pERK1/2 signaling pathways suggested the existence of biased signaling in MC3R mutants. In summary, we showed that the DPLIY motif and Helix 8 was important for MC3R activation and signal transduction. Our data led to a better understanding of the structure-function relationship of MC3R.
Keywords: Melanocortin-3 receptor, G protein-coupled receptor, Constitutively active mutant, Biased signaling, Energy homeostasis
Introduction
The melanocortin-3 receptor (MC3R), a member of family A G protein-coupled receptors (GPCRs) (Gantz et al. 1993; Roselli-Rehfuss et al. 1993), has received increasing attention with regard to its multiple physiological functions (reviewed in (Renquist et al. 2011)). The MC3R is primarily expressed in hypothalamus, especially in the arcuate nucleus, the ventromedial nucleus and the posterior hypothalamic region (Jegou et al. 2000). It is also expressed in several peripheral tissues, including the placenta, gut, heart, kidney, and peritoneal macrophages (Chhajlani 1996; Gantz et al. 1993; Getting et al. 2003; Ni et al. 2006). Based on its wide distribution, the MC3R has been shown to be involved in regulating cardiovascular function (Mioni et al. 2003; Versteeg et al. 1998), natriuresis (Chandramohan et al. 2009; Ni et al. 2006), and inflammation (Catania et al. 2004; Getting et al. 2006; Getting et al. 2008).
The MC3R, together with melanocortin-4 receptor (MC4R), another member of melanocortin receptor family expressed in the central nervous system, has been considered as a potential regulator of energy homeostasis. But unlike the MC4R, which is a well-known mediator of leptin action (Cone 1999) and is crucial for both food intake and energy expenditure regulation (Huszar et al. 1997) (reviewed in (Tao 2010a)), the MC3R is shown to be primarily involved in affecting feed efficiency rather than mediating food intake or energy expenditure (Butler et al. 2000; Chen et al. 2000). The MC4R plays an undisputed role in human obesity pathogenesis since mutations in MC4R have been characterized as the most common monogenic form of obesity in human (Farooqi et al. 2003; Hinney et al. 2013; Tao 2009). However, the role of MC3R in human obesity pathogenesis is more controversial (reviewed in (Tao 2010b)), although some MC3R mutations (such as I183N and I335S) have been recognized as possible genetic contributors for morbid obesity (Lee et al. 2007; Lee et al. 2002; Mencarelli et al. 2008; Rached et al. 2004; Tao 2007; Tao and Segaloff 2004; Yang et al. 2015; Yang and Tao 2012).
The MC3R is a typical GPCR consisting of 7 transmembrane helices (TMs) with an extracellular N-terminus and intracellular C-terminus. The currently known crystal structures of typical family A GPCRs reveal the existence of an eighth helix (Helix 8) (Mustafi and Palczewski 2009; Rosenbaum et al. 2009), which initiates just after the highly conserved N/DPxxY motif (Asn/Asp-Pro-Xaa-Xaa-Tyr) in TM7 (DPLIY in the MC3R) and terminates either with the anchorage into the plasma membrane by acylation of cysteine residues, or with kinks produced by proline residues. There are only a few GPCRs that do not have this helix in the crystal structures (Zhang et al. 2015). To date, the functional importance of the N/DPxxY motif and Helix 8 has been emerging in GPCR expression, conformational switch upon GPCR activation, G protein coupling, and GPCR internalization (Barak et al. 1995; Delos Santos et al. 2006; Fritze et al. 2003; Prioleau et al. 2002; Swift et al. 2006; Tetsuka et al. 2004; Wess et al. 1993).
However, no systematic study of the DPLIY motif and Helix 8 of MC3R has been reported. In order to gain a better understanding of the structure-function relationship of the human MC3R (hMC3R), we investigated the function of each residue in these two domains of the receptor using alanine-scanning mutagenesis. We generated 20 mutants and studied the cell surface expression, ligand binding and signaling properties of the mutant receptors. Since MC3R activation has also been reported to stimulate ERK1/2 phosphorylation (Begriche et al. 2012; Chai et al. 2007) (one report suggested that the MC3R does not activate ERK1/2 (Daniels et al. 2003)), and we and others recently reported biased signaling in the MC3R (Huang and Tao 2014; Montero-Melendez et al. 2015; Yang et al. 2015), the ERK1/2 signaling pathway of the hMC3R was also investigated.
Materials and methods
Materials
[Nle4, d-Phe7]-α-melanocyte stimulation hormone (NDP-MSH) was purchased from Bachem (King of Prussia, PA, USA). 125I-NDP-MSH was iodinated as previously described (Mo et al. 2012). Radiolabeled cAMP was iodinated in our lab with chloramine T method (Tao et al. 2010).
Site-directed mutagenesis
The wide-type (WT) hMC3R tagged at the N-terminus with 3×HA tag was obtained from Missouri S&T cDNA Resource Center (http://www.cDNA.org). Mutations were generated from the WT receptor by QuikChange™ site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using primers listed in Table 1. Plasmid DNAs were purified by IsoPure Maxi Prep Kit (Denville Scientific, Metuchen, NJ, USA). DNA sequencing was performed by the DNA Sequencing Facility of University of Chicago Cancer Research Center (Chicago, IL, USA) to confirm the presence of intended mutations and nonexistence of unintended mutations.
Table 1.
Forward primer sequences used for site-directed mutagenesis studies of hMC3R. The mutated codons are underlined.
| Constructs |
Primer sequences |
|---|---|
| D332A | CAACTCCGTCATCGCCCCACTCATCTACG |
| P333A | CTCCGTCATCGACGCACTCATCTACGC |
| L334A | CTCCGTCATCGACCCAGCCATCTACGCTTTCCGG |
| I335A | CATCGACCCACTCGCCTACGCTTTCC |
| Y336A | CATCGACCCACTCATCGCCGCTTTCCGGAGCCTG |
| A337G | CCACTCATCTACGGTTTCCGGAGCCTG |
| F338A | CCCACTCATCTACGCTGCCCGGAGCCTGGAATTG |
| R339A | CTCATCTACGCTTTCGCGAGCCTGGAATTGCG |
| S340A | CATCTACGCTTTCCGGGCCCTGGAATTGCGCAAC |
| L341A | CTACGCTTTCCGGAGCGCGGAATTGCGCAACACC |
| E342A | CGCTTTCCGGAGCCTGGCATTGCGCAACACCTTTAG |
| L343A | CTTTCCGGAGCCTGGAAGCGCGCAACACCTTTAGGG |
| R344A | CGGAGCCTGGAATTGGCCAACACCTTTAGGGAG |
| N345A | GGAGCCTGGAATTGCGCGCCACCTTTAGGGAGATTC |
| T346A | CTGGAATTGCGCAACGCCTTTAGGGAGATTC |
| F347A | GAATTGCGCAACACCGCTAGGGAGATTCTCTG |
| R348A | GCGCAACACCTTTGCGGAGATTCTCTGTG |
| E349A | CAACACCTTTAGGGCGATTCTCTGTGGC |
| I350A | CACCTTTAGGGAGGCTCTCTGTGGCTGC |
| L351A |
CACCTTTAGGGAGATTGCCTGTGGCTGCAACGGC |
Cell culture and DNA transfection
Human embryonic kidney (HEK) 293T cells, purchased from American Type Culture Collection (Manassas, VA, USA), were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% newborn calf serum. The cells were plated into 6-well clusters (or 100mm dishes for western blot) pre-coated with 0.1% gelatin and transfected with purified plasmids at 50-70% confluency using calcium phosphate precipitation method (Chen and Okayama 1987). Flow cytometry assay, ligand banding, and signaling studies were performed approximately 48h after transient transfection.
Quantification of MC3R cell surface expression by flow cytometry
48 h after transfection, HEK 293T cells were washed with filtered phosphate buffered saline for immunohistochemistry (PBS-IH) (Tao and Segaloff 2003) and fixed by 4% paraformaldehyde in PBS-IH for 30 min. After blocking with PBS-IH containing 5% BSA for 1h, the cells were then incubated with the primary antibody anti-HA.11 (Covance, Princeton, NJ, USA), which was diluted 1:100 in PBS-IH containing 5% BSA, for another 1 h. The cells were washed once with 0.5% BSA in PBS-IH and then incubated with the secondary antibody Alexa Fluor 488-labeled goat anti-mouse IgG (Invitrogen) for 1 h. The cell surface expression of WT and mutant hMC3Rs was analyzed by Accuri flow cytometer (Accuri Cytometers, Ann Arbor, MI, USA). The expression levels of the mutant hMC3Rs were calculated as the percentage of WT hMC3R expression using the formula: [mutant - pcDNA3.1] / [WT - pcDNA3.1] ×100%, where the pcDNA3.1 were used as control for background staining (Wang et al. 2008; Yang and Tao 2012).
Ligand binding assays
48 h after transfection, HEK 293T cells were washed twice with warm Waymouth's MB752/1 media (Sigma-Aldrich) containing 1mg/ml bovine serum albumin (Waymouth/BSA). The cells were incubated with Waymouth/BSA containing 100,000 cpm of 125I-NDP-MSH with or without different concentrations of unlabeled NDP-MSH (from 10-12 to 10-6 M) at 37°C for 1 h. The reaction was terminated by washing twice with cold Hank's balanced salt solution on ice. The cells were lysed by 100 μl 0.5N NaOH, collected using cotton swabs, and detected in gamma counter.
Ligand stimulated cAMP production
48 h after transfection, HEK 293T cells were washed twice with warm Waymouth/BSA and then incubated with fresh Waymouth/BSA containing 0.5mM isobutylmethylxanthine (Sigma-Aldrich) at 37°C for 15 min. Then different concentrations of NDP-MSH were added into each well to make the final volume to be 1 ml and final concentration ranging from 10-12 to 10-6 M. After 1 h incubation at 37°C, the reaction was terminated on ice and the intracellular cAMP was extracted by adding 0.5 N perchloric acid containing 180 μg/ml theophylline and 0.72 M KOH/0.6 M KHCO3 into each well. Cyclic AMP concentrations were determined by radioimmunoassay as described in detail before (Tao et al. 2010; Yang and Tao 2012).
Protein preparation and western blot
24h after transfection, HEK 293T cells were starved in Waymouth/BSA at 37°C for 24h. After treated with or without 1 μM NDP-MSH for 5 min at 37°C on the second day, the cells were transferred directly on ice, washed twice using cold 0G (150 mM NaCl and 20 mM Hepes, pH 7.4), and then scraped into lysis buffer (0G containing 0.5% NP-40, 2 mM EDTA, 1 mM Na3VO4, and 1 mM NaF). Total protein concentrations were determined by Bradford protein assay and 35 μg protein samples were separated on 10% SDA-PAGE gel and then blotted onto PVDF membranes. After blocking in 10% nonfat dry milk (containing 0.2% Tween-20) for at least 4 h at room temperature, the membranes were then immunoblotted with rabbit anti-pERK1/2 antibody (Cell Signaling, Beverly, MA, USA) 1:2000 and mouse anti-β-tubulin antibody (Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA, USA) 1:5000 diluted in Tris-buffered saline containing Tween 20 (TBST) with 5% BSA overnight at 4 °C. The membranes were then probed with HRP-conjugated secondary antibody, donkey anti-rabbit (Jackson ImmunoResearch, West Grove, PA, USA) 1:1500 and donkey anti-mouse IgG (Jackson ImmunoResearch) 1:5000 diluted in 10% nonfat dry milk for 2 h at room temperature. Specific bands were visualized using ECL reagent (Thermo Scientific, Rockford, IL, USA) and then analyzed using Image J software (NIH, Bethesda, MD, USA).
Statistical analysis
GraphPad Prism 4.0 software (San Diego, CA, USA) was used to calculate the ligand binding parameters including maximal binding (Bmax) and IC50 as well as the cAMP signaling parameters including maximal response (Rmax) and EC50. The significance of differences in binding and signaling parameters, receptor cell surface expression levels, and pERK1/2 activities between the WT and mutant hMC3Rs were determined by Student's t test using GraphPad Prism 4.0.
Results
NDP-MSH, an analog of endogenous α-MSH (Sawyer et al. 1980), has been shown to be a superpotent agonist for MC3R and was used in present study. To determine the function of each residue of the DPLIY motif and Helix 8 of MC3R, we generated 20 mutants using alanine-scanning mutagenesis (Table 1, Fig. 1). Since the mutant I335A has been studied previously (Tao 2007), its ligand binding and signaling property studies are not included herein.
Fig. 1.
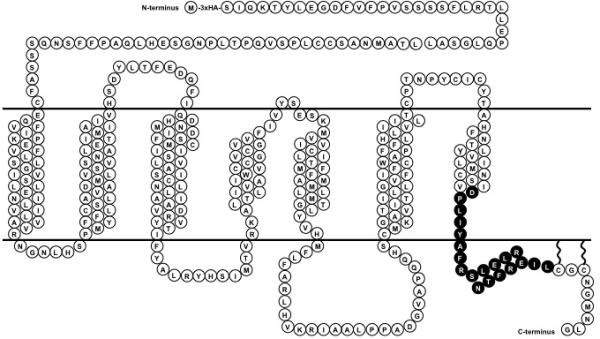
Schematic model of the hMC3R with the 20 residues characterized in the present study highlighted.
Cell surface expression of the WT and mutant hMC3Rs
Retention of the mutant receptors in the endoplasmic reticulum (ER) resulting from failing to pass the quality control system has been recognized as the major defect of inactivating mutations in numerous GPCRs (reviewed in (Tao 2006; Tao and Conn 2014)). To investigate the cell surface expression of the mutants, flow cytometry was performed. As shown in Fig. 2, all mutants were expressed normally on cell surface and no significant differences were observed between the WT hMC3R and the mutants. The data for I335A here were in agreement with previous results using confocal microscopy (Tao 2007). To confirm the results, we further tested the HA-tagged I335S and D158E. Our data were consistent with previous reports that I335S is intracellularly retained (with only ~5% WT hMC3R expression at the cell surface) while D158E is partially retained with decreased cell surface expression compared to the WT hMC3R (Fig. 2) (Tao 2007; Wang et al. 2008).
Fig. 2.
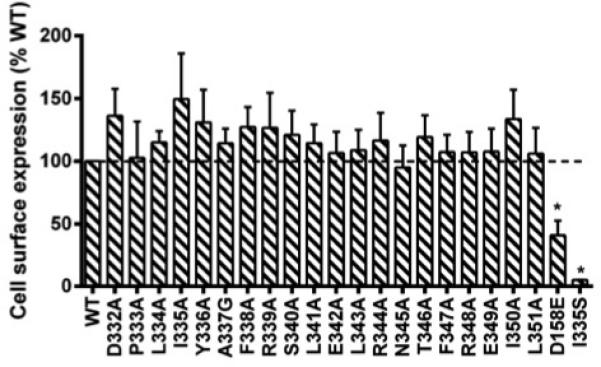
Cell surface expression of the WT and mutant hMC3Rs by flow cytometry. The results are expressed as % of cell surface expression level of WT hMC3R after correction of the nonspecific staining in cells transiently transfected with the empty vector as described in Materials and methods. Values are mean ± SEM of at least three independent experiments. Star (*) indicates significantly different from WT hMC3R (P < 0.05).
Ligand binding of the WT and mutant hMC3Rs
125I-NDP-MSH was used as the radioligand in the binding assay to compete with different concentrations of unlabeled NDP-MSH. The result showed that P333A, L343A, and R344A had no detectable binding. Nine mutants (L334A, Y336A, S340A, E342A, F347A, R348A, E349A, I350A, and L351A) showed decreased IC50 while only one mutant, L341, had increased IC50. There were no significant differences between the IC50s of all the other mutants and that of the WT hMC3R (Table 2, Fig. 3).
Table 2.
The ligand binding and signaling properties of WT and mutant hMC3Rs.
| hMC3R | NDP-MSH binding |
NDP-MSH-stimulated cAMP |
|
|---|---|---|---|
| IC50 (nM) | EC50 (nM) | Rmax (100%) | |
| WT | 2.63 ± 0.27 | 0.47 ± 0.05 | 100 |
| D332A | 1.59 ± 0.27 | 0.43 ± 0.04 | 98.50 ± 18.45 |
| P333A | N/Ad | N/Ad | N/Ad |
| L334A | 1.09 ± 0.07b | 0.39 ± 0.10 | 79.50 ± 12.12 |
| Y336A | 0.79 ± 0.10b | N/A | N/A |
| A337G | 2.32 ± 0.42 | 0.57 ± 0.17 | 98.25 ± 11.27 |
| F338A | 3.17 ± 0.79 | 0.90 ± 0.24a | 94.75 ± 15.58 |
| R339A | 2.38 ± 0.28 | 2.34 ± 0.37c | 48.33 ± 20.00a |
| S340A | 0.84 ± 0.12b | 0.77 ± 0.22 | 52.29 ± 13.18a |
| L341A | 4.81 ± 1.13a | 0.50 ± 0.07 | 102.50 ±19.31 |
| E342A | 0.58 ±0.02c | N/Ad | N/Ad |
| L343A | N/Ad | N/Ad | N/Ad |
| R344A | N/Ad | N/Ad | N/Ad |
| N345A | 1.89 ± 0.17 | 0.36 ± 0.08 | 107.00 ± 12.13 |
| T346A | 2.08 ± 0.18 | 0.48 ± 0.16 | 91.80 ± 8.89 |
| F347A | 1.09 ± 0.19b | 0.25 ± 0.06a | 67.25 ± 8.38a |
| R348A | 0.91 ± 0.04b | 0.42 ± 0.11 | 53.40 ± 5.62b |
| E349A | 1.21 ± 0.18a | 0.47 ± 0.16 | 103.00 ± 23.34 |
| I350A | 0.61 ± 0.07b | 0.33 ± 0.07 | 15.75 ± 1.55c |
| L351A | 0.64 ± 0.03b | 0.32 ± 0.10 | 17.50 ± 2.02c |
Values are expressed as the mean ± SEM of at least four independent experiments.
The Rmax of WT hMC3R was 2386.00 ± 239.95 pmol/106 cells upon NDP-MSH stimulation.
Significantly different from WT hMC3R, p<0.05.
Significantly different from WT hMC3R, p<0.01.
Significantly different from WT hMC3R, p<0.001.
Not detectable.
Fig. 3.
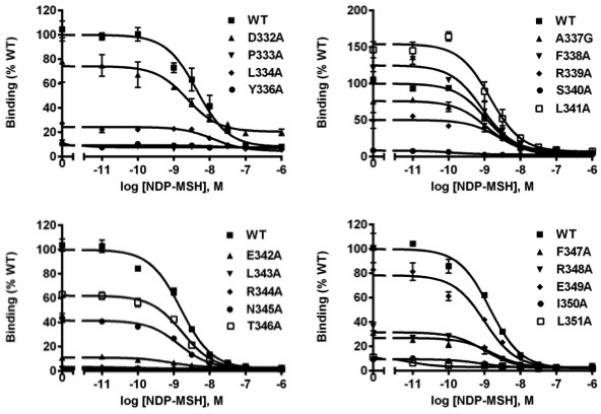
Ligand binding properties of the WT and mutant hMC3Rs with NDP-MSH as the ligand. HEK293T cells were transiently transfected with WT and mutant constructs, and the binding properties were measured 48h later by displacing the binding of 125I-NDP-MSH using different concentrations of unlabeled NDP-MSH as described in Materials and methods. Data are expressed as % of WT binding ± range from duplicate measurements within one experiment. All experiments were performed as least three times independently.
As shown in Fig. 4, 9 mutants (D332A, L344A, Y336A, S340A, E342A, F347A, R348A, I350A, and L351A) had significantly decreased maximal binding compared with WT hMC3R while one mutant (F338A) showed significantly, although very slightly, increased maximal binding. All the other mutants had similar maximal binding as the WT hMC3R.
Fig. 4.
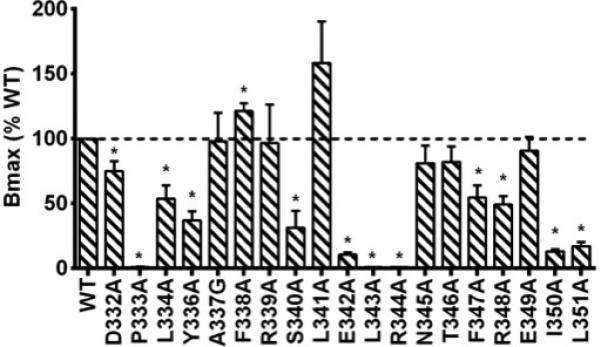
Total specific binding of WT and mutant hMC3Rs with NDP-MSH as the ligand. Data are mean ± SEM of at least three independent experiments. Star (*) indicates significantly different from WT hMC3R (P < 0.05).
Signaling properties of the WT and mutant hMC3Rs
Whether the hMC3R variants could respond to NDP-MSH stimulation with enhanced cAMP generation was then investigated. As expected, NDP-MSH could stimulate dose-dependent increase of intracellular cAMP in HEK293T cells transfected with WT hMC3R. As shown in Table 2, two mutants (Y336A and E342A), in addition to three binding-defective mutants (P333A, L343A, and R344A), had no measurable signaling. F338A and R339A showed increased EC50 while F347A displayed reduced EC50. No significant differences were observed between the EC50s of all the other mutants and that of the WT hMC3R.
Our data of maximal response in Fig. 5 also showed that six mutants (R339A, S340A, F347A, R348A, I350A, and L351A) had significantly decreased maximal response compared with WT hMC3R. All the other mutants had apparent normal signaling.
Fig. 5.
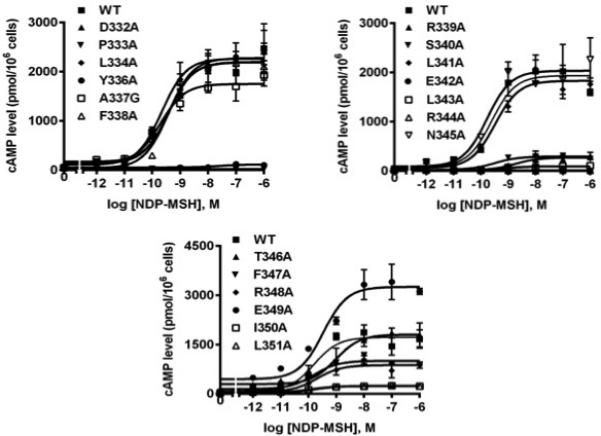
Signaling properties of the WT and mutant hMC3Rs with NDP-MSH as the ligand. HEK293T cells were transiently transfected with WT and mutant constructs, and intracellular cAMP levels were measured by RIA after stimulation with different concentrations of NDP-MSH as described in Materials and methods. Data are mean ± SEM from triplicate measurements within one experiment. All experiments were performed as least three times independently.
Constitutive activity of hMC3Rs
Unlike the human MC4R, which has been previously shown to be constitutively active (Nijenhuis et al. 2001; Tao 2014), the hMC3R has been recognized with little or no constitutive activity (Tao 2007). In present study, the constitutive activities of the 19 mutants of hMC3R (I335A not included) were analyzed. We demonstrated that the F347A had significantly increased basal activity compared with the WT hMC3R. As shown in Fig. 6A, the nearly 4.6 times elevation of basal activity demonstrated that the F347A was a constitutively active mutant, consistent with our previous report (Tao et al. 2010). All the other mutants had similar basal activities compared with that of the WT hMC3R. The dose-response curve in Fig. 6B highlights that F347A, although constitutively active, had reduced maximal response compared with the WT hMC3R.
Fig. 6.
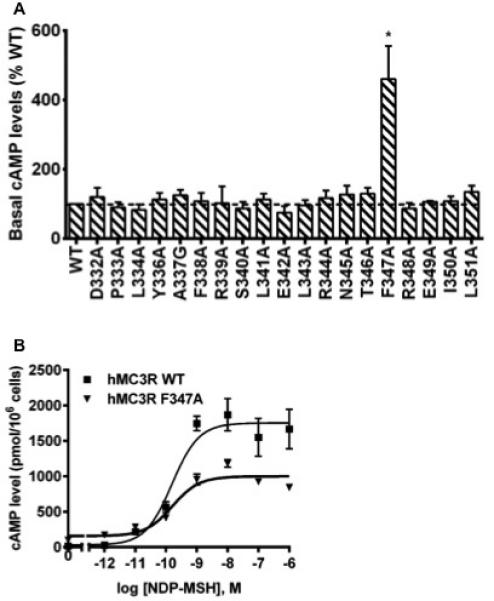
Basal activities of WT and mutant hMC3Rs. (A) HEK293T cells were transiently transfected with WT and mutant constructs, and intracellular cAMP levels were measured without ligand stimulation. Data are mean ± SEM of at least four independent experiments. The basal cAMP level of WT hMC3R was 25.79 ± 2.69 pmol/106 cells. Star (*) indicates significantly different from WT hMC3R (P < 0.05). (B) A representative dose-response curve of WT and F347A hMC3Rs. Similar results were obtained in at least three independent experiments.
ERK1/2 signaling of the WT and mutant hMC3Rs
In order to further investigate the ERK1/2 signaling pathway, especially the newly-identified biased signaling of hMC3R, the WT hMC3R together with nine mutants, including one constitutively active mutant (F347A) and eight binding- or signaling-defective mutants (P333A, I335A, Y336A, E342A, L343A, R344A, I350A, and L351A) which led to less than 20% the WT receptor cAMP production, were studied using western blots. Empty vector pcDNA3.1 and WT hMC3R without 3xHA tag were also studied to excluding non-specific stimulation.
Our results showed that the basal pERK1/2 levels in cells transfected with either WT MC3Rs (with or without 3xHA-tag) were similar as that of cells transfected with the empty vector (Fig. 7B). Upon 1 μM NDP-MSH stimulation, WT MC3Rs showed significant ERK1/2 phosphorylation but no ERK1/2 activation was detected in cells transfected with empty vector. There was no significant difference between the pERK1/2 levels of HA-tagged and non-tagged WT MC3Rs (Fig. 7C).
Fig. 7.
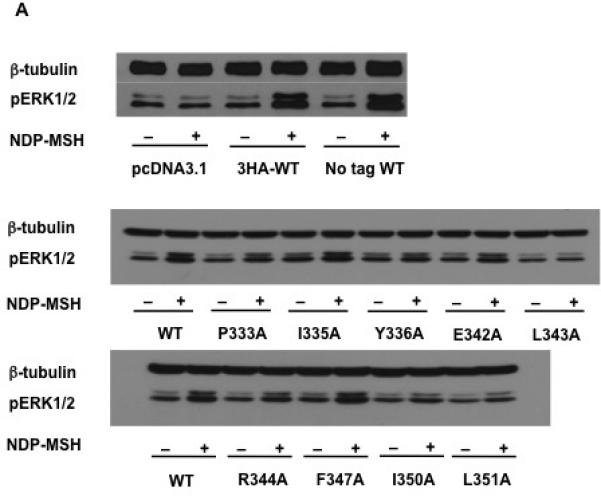
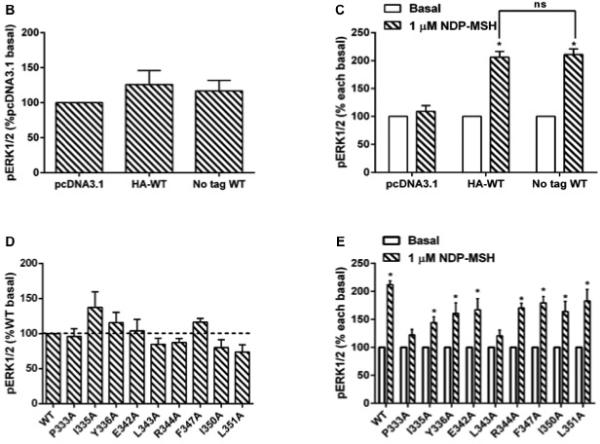
MAPK signaling of WT and mutant hMC3Rs. (A) HEK293T cells were transiently transfected with pcDNA3.1, WT (with or without HA tag) or mutant constructs, and 24h later cells were starved overnight and then harvested after treated with or without 1 μM NDP-MSH stimulation for 5 min. Western blot analysis was performed using antibody against pERK1/2 and β-tubulin as control. (B) Basal pERK1/2 levels of cells transfected with pcDNA3.1 or WT hMC3Rs (with or without HA tag); data are mean ± SEM of three independent experiments. (C) Stimulated pERK1/2 levels in cells transfected with pcDNA3.1 or WT hMC3R (with or without HA tag) after 1 μM NDP-MSH stimulation, expressed as percentage of each basal pERK1/2 activity from three independent experiments. Star (*) indicates significantly different from corresponding basal level (P < 0.05) and ns means no statistical difference between HA-tagged and non-tagged WT hMC3R (P > 0.05). (D) Data of the basal pERK1/2 of WT and mutant hMC3Rs are mean ± SEM of at least four independent experiments. (E) Data of the pERK1/2 levels of WT and mutant hMC3Rs after 1 μM NDP-MSH stimulation are expressed as percentage of each basal pERK1/2 activity from at least four independent experiments. Star (*) indicates significantly different from corresponding basal activity (P < 0.05).
The constitutive activities of WT and mutant hMC3Rs in activating ERK1/2 were studied. Our results showed that all the mutants, including the F347A that was shown to be constitutively active in cAMP pathway, had similar basal pERK1/2 levels as the WT hMC3R (Fig. 7D).
We then investigated the pERK1/2 levels of WT and mutant hMC3Rs after 1 μM NDP-MSH stimulation. As expected, the ERK1/2 phosphorylation was significantly enhanced in HEK 293T cells transfected with WT hMC3R. P333A and L343A did not respond to NDP-MSH stimulation with ERK1/2 activation while all other mutants had significantly increased pERK1/2 levels compared to the corresponding basal activity upon 1μM NDP-MSH stimulation (Fig. 7E).
Discussion
Although several physiological functions of the MC3R have now been described, it remains to be the most enigmatic member of the melanocortin receptor family. The recent identification of an additional Helix 8, linking the TM7 and the C-terminus of GPCRs, has also aroused great interest. Analysis of the role of Helix 8 in both family A and B GPCRs confirmed its importance in receptor expression, ligand binding, signal transduction and internalization (Ernst et al. 2000; Faussner et al. 2005; Kuwasako et al. 2011; Marin et al. 2000; Tetsuka et al. 2004). In present study, we performed detailed study of the 20 residues in Helix 8 and the DPLIY motif of hMC3R using the classical method of site-directed alanine-scanning mutagenesis (Cunningham and Wells 1989).
The flow cytometric analysis demonstrated that all mutants were expressed normally on cell surface. Receptor retention or mislocalization, which has previously been described as the major defect for human disease caused by GPCR mutations (Tao 2006; Tao and Conn 2014), was not observed in present study. Similarly, in our studies on the second and third intracellular loops, we also showed that the alanine mutants are expressed normally at the cell surface (Huang and Tao 2014; Wang and Tao 2013).
Ligand binding studies revealed that 11 residues (P333, L334, Y336, S340, E342, L343, R344, F347, R348, I350 and L351) were important for NDP-MSH binding. Although it is uncommon that residues important for ligand binding are located in the cytoplasmic side of receptor, observations have been reported previously in MC3R (Huang and Tao 2014) as well as other GPCRs, such as gonadotropin-releasing hormone receptor (Lu et al. 2005), angiotensin II type 2 receptor (Moore et al. 2002), and V2 vasopressin receptor (Pan et al. 1994). These residues are not expected to directly participate in ligand binding; rather they might indirectly participate in ligand–receptor interaction through intramolecular interactions and conformational changes (Kobilka and Deupi 2007; Rovati et al. 2007).
The highly conserved DPLIY motif is one of the two crucial motifs (the other one is D/ERY motif in TM3) for receptor stabilization and activation (Park et al. 2008). P7.50 (the superscript number represents Ballesteros-Weinstein numbering (Ballesteros and Weinstein 1995)) has been regarded to be critical in inducing structurally important helical break due to its distinctive cyclic structure of side chain. Although L7.51 was not studied in detail before, its adjacent residue I7.52 has been recognized to be important in multiple aspects of MC3R function (Tao 2007). L7.51 might contribute to the supposed interaction between the I7.52 and hydrophobic residues in Helix 8 to maintain the receptor conformation necessary for ligand binding. Y7.53 has been shown to be critical for receptor activation and signal transduction in many GPCRs, such as MC4R, gonadotropin-releasing receptor, and 5HT2C receptor (Arora et al. 1996; Prioleau et al. 2002; Roth et al. 2009). The conformational change of tyrosine enables it to insert into the space previously occupied by TM6 and hinder its inward tilt to stabilize the active state (Scheerer et al. 2008). Mutations of the tyrosine residue in some other receptors result in either no ligand binding or defective signaling (Feng and Song 2001). Our result here highlighted the importance of Y7.53 in hMC3R.
The D/E(x)7LL motif, composed of highly conserved di-leucine sequence with an upstream acidic residue, has been shown to be present in the C-terminus of several GPCRs such as α1-, α2-adrenergic receptors and dopamine receptor (Schulein et al. 1998). The corresponding residue of glutamate (E3357.63) in V2 vasopressin receptor was reported to be crucial for establishing a transport-competent folding state to support the escape of the receptor from the ER (Schuleinet al. 1998). The di-leucine motif, which should be strictly defined as dihydrophobic pair due to its consisting of mainly leucine, isoleucine or valine, was shown to play an important role in cell surface targeting, receptor internalization and protein trafficking (Gabilondo et al. 1997; Ho and MacKenzie 1999). The alanine mutation of di-leucine pair was reported to have no effect on ligand affinity in β2-adrenergic receptor (Gabilondo et al. 1997), whereas the same mutation of di-isoleucine was shown to impair NDP-MSH binding to MC4R (VanLeeuwen et al. 2003). Our data demonstrated that the maximal binding of E3427.59A, I3507.67A, and L3517.68A had decreased by 90%, 87% and 83%, respectively, compared with WT hMC3R, although all the mutant receptors could be normally expressed on cell surface.
The interaction between two highly conserved aromatic residues, tyrosine in the D/NPxxY motif and phenylalanine in Helix 8 has been highlighted in several studies. However, the conserved phenylalanine in melanocortin receptor family is substituted by a leucine residue (L3437.60 in hMC3R) and the function of replaced phenylalanine might be taken by another aromatic residue, F3477.64. As discussed above, the I7.52 in DPLIY motif of MC3R might interact with hydrophobic residues in Helix 8 to maintain the receptor conformation. L3437.60 is a preferable choice for taking part in the hydrophobic interaction since the mutant L3437.60A did not exhibit any binding or signaling in response to NDP-MSH stimulation in our study.
One residue, R3397.56, was demonstrated to be crucial for normal receptor signaling. Alanine mutation of this arginine resulted in normal ligand binding but severely decreased signaling potency. Missense mutations of the corresponding residue of arginine in hMC4R, including R3057.56S, R3057.56Q, and R3057.56W, have been identified from obese patient (Tao 2009). R3057.56S and R3057.56Q have been shown to have partial cAMP response to α-MSH stimulation (Calton et al. 2009; Stutzmann et al. 2008) and R3057.56W exhibits normal binding but severely impaired signaling in response to α-MSH stimulation (Roubert et al. 2010), which is in agreement with our results of R3397.56A hMC3R.
The MC3R has been reported to activate the ERK1/2 signaling pathway (see Introduction). Unbalanced cAMP and ERK1/2 signaling was reported in some human MC4R mutants (He and Tao 2014; Huang and Tao 2012; Mo et al. 2012) (reviewed in (Tao 2014)). In our recent study of the DRY motif and intracellular loop 2 of hMC3R, the presence of this biased signaling was also suggested (Huang and Tao 2014). To test it further, in the current study, the ERK1/2 phosphorylation levels of nine hMC3R mutants, including eight binding- or signaling-defective mutants (P333A, I335A, Y336A, E342A, L343A, R344A, I350A, and L351A) which led to less than 20% cAMP production compared with WT hMC3R, and one constitutively active mutant (F347A) with enhanced basal cAMP activity, were investigated using western blots.
Non-specific effects were excluded in the present study by using the empty vector pcDNA3.1 and non-tagged WT hMC3R. The results indicated that there was no endogenous receptor in HEK293T cells responding to NDP-MSH stimulation and HA tag did not have any effect on MC3R activation of ERK1/2 (Fig. 7A-C). All nine mutants had similar basal pERK1/2 levels as the WT MC3R. Notably, F347A, which had increased basal cAMP level, did not show any increase in basal pERK1/2 level compared with the WT hMC3R. Upon 1μM NDP-MSH stimulation, two binding-defective mutants, P333A and L343A, did not activate ERK1/2. However, all the other tested mutants, especially I335A, Y336A, E342A, and R344A that had almost no cAMP response, had shown significant ERK1/2 activation. Two mutants with no detectable binding, I335A (Tao 2007) and R344A (present study), responded to NDPMSH stimulation with increased pERK1/2 levels. Although it is uncommon, similar observations have been previously reported in other GPCRs, such as MC4R (Huang and Tao 2012) and gonadotropin-releasing hormone receptor (Bedecarrats et al. 2003). This might due to the faster dissociation of the ligand from the MC3R mutants compared with WT receptor making the binding difficult to detect, but ERK1/2 could still be activated by this transient binding (Huang and Tao 2014). NDP-MSH was reported to induce ERK1/2 phosphorylation in HEK293 cells transfected with WT MC3R in a very fast and potent pattern, peaking at only 5 min with an EC50 of 3.3 ± 1.5 nM (Chai et al. 2007). These data suggest these mutants were biased receptors, supporting two recent studies on biased signaling at the MC3R (Montero-Melendezet al. 2015; Yang et al. 2015).
In summary, for the systemic study of the DPLIY motif and Helix 8 of hMC3R, we have identified residues that were crucial for ligand binding and cAMP generation. One constitutively active mutant was identified and the existence of biased signaling was confirmed in MC3R mutants. The data obtained in our present research indicated that the highly conserved DPLIY motif and Helix 8 play important roles in MC3R activation and signaling transduction. Our result established a theoretical basis for the structure-functional relationship of MC3R and will be useful for further investigation of its physiological role in energy homeostasis.
Acknowledgements
This study was supported by grants from the National Institutes of Health R15DK077213 and Animal Health and Disease Research Program of Auburn University College of Veterinary Medicine. Zhao Yang was supported by a fellowship from China Scholarship Council of the People's Republic of China. Zhi-Li Huang was supported by a fellowship from Shenzhen Polytechnic. We thank Dr. Shuxiu Wang for generating the mutant constructs and performing some of the preliminary experiments.
Footnotes
Disclosure:
The authors have nothing to disclose.
References
- Arora KK, Cheng Z, Catt KJ. Dependence of agonist activation on an aromatic moiety in the DPLIY motif of the gonadotropin-releasing hormone receptor. Molecular Endocrinology. 1996;10:979–986. doi: 10.1210/mend.10.8.8843414. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- Barak LS, Menard L, Ferguson SS, Colapietro AM, Caron MG. The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the β2-adrenergic receptor. Biochemistry. 1995;34:15407–15414. doi: 10.1021/bi00047a003. [DOI] [PubMed] [Google Scholar]
- Bedecarrats GY, Linher KD, Janovick JA, Beranova M, Kada F, Seminara SB, Conn PM, Kaiser UB. Four naturally occurring mutations in the human GnRH receptor affect ligand binding and receptor function. Molecular and Cellular Endocrinology. 2003;205:51–64. doi: 10.1016/s0303-7207(03)00201-6. [DOI] [PubMed] [Google Scholar]
- Begriche K, Marston OJ, Rossi J, Burke LK, McDonald P, Heisler LK, Butler AA. Melanocortin-3 receptors are involved in adaptation to restricted feeding. Genes, Brain and Behavior. 2012;11:291–302. doi: 10.1111/j.1601-183X.2012.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- Calton MA, Ersoy BA, Zhang S, Kane JP, Malloy MJ, Pullinger CR, Bromberg Y, Pennacchio LA, Dent R, McPherson R, et al. Association of functionally significant Melanocortin-4 but not Melanocortin-3 receptor mutations with severe adult obesity in a large North American case-control study. Human Molecular Genetics. 2009;18:1140–1147. doi: 10.1093/hmg/ddn431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania A, Gatti S, Colombo G, Lipton JM. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacological Reviews. 2004;56:1–29. doi: 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- Chai B, Li JY, Zhang W, Ammori JB, Mulholland MW. Melanocortin-3 receptor activates MAP kinase via PI3 kinase. Regulatory Peptides. 2007;139:115–121. doi: 10.1016/j.regpep.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Chandramohan G, Durham N, Sinha S, Norris K, Vaziri ND. Role of γ melanocyte-stimulating hormone-renal melanocortin 3 receptor system in blood pressure regulation in salt-resistant and salt-sensitive rats. Metabolism. 2009;58:1424–1429. doi: 10.1016/j.metabol.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nature Genetics. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and Cellular Biology. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhajlani V. Distribution of cDNA for melanocortin receptor subtypes in human tissues. Biochemistry and Molecular Biology International. 1996;38:73–80. [PubMed] [Google Scholar]
- Cone RD. The central melanocortin system and energy homeostasis. Trends in Endocrinology and Metabolism. 1999;10:211–216. doi: 10.1016/s1043-2760(99)00153-8. [DOI] [PubMed] [Google Scholar]
- Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- Daniels D, Patten CS, Roth JD, Yee DK, Fluharty SJ. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Research. 2003;986:1–11. doi: 10.1016/s0006-8993(03)03162-7. [DOI] [PubMed] [Google Scholar]
- Delos Santos NM, Gardner LA, White SW, Bahouth SW. Characterization of the residues in helix 8 of the human β1-adrenergic receptor that are involved in coupling the receptor to G proteins. Journal of Biological Chemistry. 2006;281:12896–12907. doi: 10.1074/jbc.M508500200. [DOI] [PubMed] [Google Scholar]
- Ernst OP, Meyer CK, Marin EP, Henklein P, Fu WY, Sakmar TP, Hofmann KP. Mutation of the fourth cytoplasmic loop of rhodopsin affects binding of transducin and peptides derived from the carboxyl-terminal sequences of transducin 〈 and © subunits. Journal of Biological Chemistry. 2000;275:1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. New England Journal of Medicine. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Faussner A, Bauer A, Kalatskaya I, Schussler S, Seidl C, Proud D, Jochum M. The role of helix 8 and of the cytosolic C-termini in the internalization and signal transduction of B1 and B2 bradykinin receptors. FEBS Journal. 2005;272:129–140. doi: 10.1111/j.1432-1033.2004.04390.x. [DOI] [PubMed] [Google Scholar]
- Feng W, Song ZH. Functional roles of the tyrosine within the NP(X)(n)Y motif and the cysteines in the C-terminal juxtamembrane region of the CB2 cannabinoid receptor. FEBS Letters. 2001;501:166–170. doi: 10.1016/s0014-5793(01)02642-4. [DOI] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. PNAS. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabilondo AM, Hegler J, Krasel C, Boivin-Jahns V, Hein L, Lohse MJ. A dileucine motif in the C terminus of the β2-adrenergic receptor is involved in receptor internalization. PNAS. 1997;94:12285–12290. doi: 10.1073/pnas.94.23.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. Journal of Biological Chemistry. 1993;268:8246–8250. [PubMed] [Google Scholar]
- Getting SJ, Christian HC, Lam CW, Gavins FN, Flower RJ, Schioth HB, Perretti M. Redundancy of a functional melanocortin 1 receptor in the anti-inflammatory actions of melanocortin peptides: studies in the recessive yellow (e/e) mouse suggest an important role for melanocortin 3 receptor. Journal of Immunology. 2003;170:3323–3330. doi: 10.4049/jimmunol.170.6.3323. [DOI] [PubMed] [Google Scholar]
- Getting SJ, Lam CW, Chen AS, Grieco P, Perretti M. Melanocortin 3 receptors control crystal-induced inflammation. The FASEB Journal. 2006;20:2234–2241. doi: 10.1096/fj.06-6339com. [DOI] [PubMed] [Google Scholar]
- Getting SJ, Riffo-Vasquez Y, Pitchford S, Kaneva M, Grieco P, Page CP, Perretti M, Spina D. A role for MC3R in modulating lung inflammation. Pulmonary Pharmacology and Therapeutics. 2008;21:866–873. doi: 10.1016/j.pupt.2008.09.004. [DOI] [PubMed] [Google Scholar]
- He S, Tao YX. Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene. International Journal of Biological Sciences. 2014;10:1128–1137. doi: 10.7150/ijbs.10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Progress in Molecular Biology and Translational Science. 2013;114:147–191. doi: 10.1016/B978-0-12-386933-3.00005-4. [DOI] [PubMed] [Google Scholar]
- Ho G, MacKenzie RG. Functional characterization of mutations in melanocortin-4 receptor associated with human obesity. Journal of Biological Chemistry. 1999;274:35816–35822. doi: 10.1074/jbc.274.50.35816. [DOI] [PubMed] [Google Scholar]
- Huang H, Tao YX. Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor. Journal of Molecular Endocrinolgoy. 2012;49:237–248. doi: 10.1530/JME-12-0161. [DOI] [PubMed] [Google Scholar]
- Huang H, Tao YX. Functions of the DRY motif and intracellular loop 2 of human melanocortin 3 receptor. Journal of Molecular Endocrinolgoy. 2014;53:319–330. doi: 10.1530/JME-14-0184. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jegou S, Boutelet I, Vaudry H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. Journal of Neuroendocrinology. 2000;12:501–505. doi: 10.1046/j.1365-2826.2000.00477.x. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends in Pharmacological Sciences. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kuwasako K, Kitamura K, Nagata S, Hikosaka T, Kato J. Structure-function analysis of helix 8 of human calcitonin receptor-like receptor within the adrenomedullin 1 receptor. Peptides. 2011;32:144–149. doi: 10.1016/j.peptides.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Lee YS, Poh LK, Kek BL, Loke KY. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes. 2007;56:2622–2630. doi: 10.2337/db07-0225. [DOI] [PubMed] [Google Scholar]
- Lee YS, Poh LK, Loke KY. A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. Journal of Clinical Endocrinology and Metabolism. 2002;87:1423–1426. doi: 10.1210/jcem.87.3.8461. [DOI] [PubMed] [Google Scholar]
- Lu ZL, Gallagher R, Sellar R, Coetsee M, Millar RP. Mutations remote from the human gonadotropin-releasing hormone (GnRH) receptor-binding sites specifically increase binding affinity for GnRH II but not GnRH I: evidence for ligand-selective, receptor-active conformations. Journal of Biological Chemistry. 2005;280:29796–29803. doi: 10.1074/jbc.M413520200. [DOI] [PubMed] [Google Scholar]
- Marin EP, Krishna AG, Zvyaga TA, Isele J, Siebert F, Sakmar TP. The amino terminus of the fourth cytoplasmic loop of rhodopsin modulates rhodopsin-transducin interaction. Journal of Biological Chemistry. 2000;275:1930–1936. doi: 10.1074/jbc.275.3.1930. [DOI] [PubMed] [Google Scholar]
- Mencarelli M, Walker GE, Maestrini S, Alberti L, Verti B, Brunani A, Petroni ML, Tagliaferri M, Liuzzi A, Di Blasio AM. Sporadic mutations in melanocortin receptor 3 in morbid obese individuals. European Journal of Human Genetics. 2008;16:581–586. doi: 10.1038/sj.ejhg.5202005. [DOI] [PubMed] [Google Scholar]
- Mioni C, Giuliani D, Cainazzo MM, Leone S, Bazzani C, Grieco P, Novellino E, Tomasi A, Bertolini A, Guarini S. Further evidence that melanocortins prevent myocardial reperfusion injury by activating melanocortin MC3 receptors. European Journal of Pharmacology. 2003;477:227–234. doi: 10.1016/s0014-2999(03)02184-8. [DOI] [PubMed] [Google Scholar]
- Mo XL, Yang R, Tao YX. Functions of transmembrane domain 3 of human melanocortin-4 receptor. Journal of Molecular Endocrinolgoy. 2012;49:221–235. doi: 10.1530/JME-12-0162. [DOI] [PubMed] [Google Scholar]
- Montero-Melendez T, Gobbetti T, Cooray SN, Jonassen TE, Perretti M. Biased agonism as a novel strategy to harness the proresolving properties of melanocortin receptors without eliciting melanogenic effects. Journal of Immunology. 2015;194:3381–3388. doi: 10.4049/jimmunol.1402645. [DOI] [PubMed] [Google Scholar]
- Moore SA, Patel AS, Huang N, Lavin BC, Grammatopoulos TN, Andres RD, Weyhenmeyer JA. Effects of mutations in the highly conserved DRY motif on binding affinity, expression, and G-protein recruitment of the human angiotensin II type-2 receptor. Brain Research Molecular Brain Research. 2002;109:161–167. doi: 10.1016/s0169-328x(02)00552-1. [DOI] [PubMed] [Google Scholar]
- Mustafi D, Palczewski K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Molecular Pharmacology. 2009;75:1–12. doi: 10.1124/mol.108.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni XP, Butler AA, Cone RD, Humphreys MH. Central receptors mediating the cardiovascular actions of melanocyte stimulating hormones. Journal of Hypertension. 2006;24:2239–2246. doi: 10.1097/01.hjh.0000249702.49854.fa. [DOI] [PubMed] [Google Scholar]
- Nijenhuis WA, Oosterom J, Adan RA. AgRP(83-132) acts as an inverse agonist on the human melanocortin-4 receptor. Molecular Endocrinology. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- Pan Y, Wilson P, Gitschier J. The effect of eight V2 vasopressin receptor mutations on stimulation of adenylyl cyclase and binding to vasopressin. Journal of Biological Chemistry. 1994;269:31933–31937. [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Prioleau C, Visiers I, Ebersole BJ, Weinstein H, Sealfon SC. Conserved helix 7 tyrosine acts as a multistate conformational switch in the 5HT2C receptor. Identification of a novel “locked-on” phenotype and double revertant mutations. Journal of Biological Chemistry. 2002;277:36577–36584. doi: 10.1074/jbc.M206223200. [DOI] [PubMed] [Google Scholar]
- Rached M, Buronfosse A, Begeot M, Penhoat A. Inactivation and intracellular retention of the human I183N mutated melanocortin 3 receptor associated with obesity. Biochimica et Biophysica Acta. 2004;1689:229–234. doi: 10.1016/j.bbadis.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Renquist BJ, Lippert RN, Sebag JA, Ellacott KL, Cone RD. Physiological roles of the melanocortin MC3 receptor. European Journal of Pharmacology. 2011;660:13–20. doi: 10.1016/j.ejphar.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for © melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. PNAS. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth CL, Ludwig M, Woelfle J, Fan ZC, Brumm H, Biebermann H, Tao YX. A novel melanocortin-4 receptor gene mutation in a female patient with severe childhood obesity. Endocrine. 2009;36:52–59. doi: 10.1007/s12020-009-9156-4. [DOI] [PubMed] [Google Scholar]
- Roubert P, Dubern B, Plas P, Lubrano-Berthelier C, Alihi R, Auger F, Deoliveira DB, Dong JZ, Basdevant A, Thurieau C, et al. Novel pharmacological MC4R agonists can efficiently activate mutated MC4R from obese patient with impaired endogenous agonist response. Journal of Endocrinology. 2010;207:177–183. doi: 10.1677/JOE-09-0336. [DOI] [PubMed] [Google Scholar]
- Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Molecular Pharmacology. 2007;71:959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-〈-melanocyte-stimulating hormone: a highly potent 〈-melanotropin with ultralong biological activity. PNAS. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, Rosenthal W. A dileucine sequence and an upstream glutamate residue in the intracellular carboxyl terminus of the vasopressin V2 receptor are essential for cell surface transport in COS.M6 cells. Molecular Pharmacology. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- Stutzmann F, Tan K, Vatin V, Dina C, Jouret B, Tichet J, Balkau B, Potoczna N, Horber F, O'Rahilly S, et al. Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. 2008;57:2511–2518. doi: 10.2337/db08-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift S, Leger AJ, Talavera J, Zhang L, Bohm A, Kuliopulos A. Role of the PAR1 receptor 8th helix in signaling: the 7-8-1 receptor activation mechanism. Journal of Biological Chemistry. 2006;281:4109–4116. doi: 10.1074/jbc.M509525200. [DOI] [PubMed] [Google Scholar]
- Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacology and Therapeutics. 2006;111:949–973. doi: 10.1016/j.pharmthera.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Tao YX. Functional characterization of novel melanocortin-3 receptor mutations identified from obese subjects. Biochimica et Biophysica Acta. 2007;1772:1167–1174. doi: 10.1016/j.bbadis.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Tao YX. Mutations in melanocortin-4 receptor and human obesity. Progress in Molecular Biology and Translational Science. 2009;88:173–204. doi: 10.1016/S1877-1173(09)88006-X. [DOI] [PubMed] [Google Scholar]
- Tao YX. The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocrine Reviews. 2010a;31:506–543. doi: 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX. Mutations in the melanocortin-3 receptor (MC3R) gene: Impact on human obesity or adiposity. Current Opinion in Investigational Drugs. 2010b;11:1092–1096. [PubMed] [Google Scholar]
- Tao YX. Constitutive activity in melanocortin-4 receptor: Biased signaling of inverse agonists. Advances in Pharmacology. 2014;70:135–154. doi: 10.1016/B978-0-12-417197-8.00005-5. [DOI] [PubMed] [Google Scholar]
- Tao YX, Conn PM. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocrine Reviews. 2014;35:602–647. doi: 10.1210/er.2013-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX, Huang H, Wang ZQ, Yang F, Williams JN, Nikiforovich GV. Constitutive activity of neural melanocortin receptors. Methods in Enzymology. 2010;484:267–279. doi: 10.1016/B978-0-12-381298-8.00014-9. [DOI] [PubMed] [Google Scholar]
- Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–4551. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- Tao YX, Segaloff DL. Functional characterization of melanocortin-3 receptor variants identify a loss-of-function mutation involving an amino acid critical for G protein-coupled receptor activation. Journal of Clinical Endocrinology and Metabolism. 2004;89:3936–3942. doi: 10.1210/jc.2004-0367. [DOI] [PubMed] [Google Scholar]
- Tetsuka M, Saito Y, Imai K, Doi H, Maruyama K. The basic residues in the membrane-proximal C-terminal tail of the rat melanin-concentrating hormone receptor 1 are required for receptor function. Endocrinology. 2004;145:3712–3723. doi: 10.1210/en.2003-1638. [DOI] [PubMed] [Google Scholar]
- VanLeeuwen D, Steffey ME, Donahue C, Ho G, MacKenzie RG. Cell surface expression of the melanocortin-4 receptor is dependent on a C-terminal di-isoleucine sequence at codons 316/317. Journal of Biological Chemistry. 2003;278:15935–15940. doi: 10.1074/jbc.M211546200. [DOI] [PubMed] [Google Scholar]
- Versteeg DH, Van Bergen P, Adan RA, De Wildt DJ. Melanocortins and cardiovascular regulation. European Journal of Pharmacology. 1998;360:1–14. doi: 10.1016/s0014-2999(98)00615-3. [DOI] [PubMed] [Google Scholar]
- Wang SX, Fan ZC, Tao YX. Functions of acidic transmembrane residues in human melanocortin-3 receptor binding and activation. Biochemical Pharmacology. 2008;76:520–530. doi: 10.1016/j.bcp.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Tao YX. Functions of the third intracellular loop of the human melanocortin-3 receptor. Current Pharmaceutical Design. 2013;19:4831–4838. doi: 10.2174/1381612811319270005. [DOI] [PubMed] [Google Scholar]
- Wess J, Nanavati S, Vogel Z, Maggio R. Functional role of proline and tryptophan residues highly conserved among G protein-coupled receptors studied by mutational analysis of the m3 muscarinic receptor. EMBO Journal. 1993;12:331–338. doi: 10.1002/j.1460-2075.1993.tb05661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Huang H, Tao YX. Biased signaling in naturally occurring mutations in human melanocortin-3 receptor gene. International Journal of Biological Sciences. 2015;11:423–433. doi: 10.7150/ijbs.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Tao YX. Functional characterization of nine novel naturally occurring human melanocortin-3 receptor mutations. Biochimica et Biophysica Acta. 2012;1822:1752–1761. doi: 10.1016/j.bbadis.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. Two disparate ligand-binding sites in the human P2Y receptor. Nature. 2015;520:317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]