

Abstract
Infant acute lymphoblastic leukemia (ALL) with MLL rearrangements (MLL-R) represents a distinct leukemia with a poor prognosis. To define its mutational landscape, we performed whole genome, exome, RNA and targeted DNA sequencing on 65 infants (47 MLL-R and 18 non-MLL-R) and 20 older children (MLL-R cases) with leukemia. Our data demonstrated infant MLL-R ALL to have one of the lowest frequencies of somatic mutations of any sequenced cancer, with the predominant leukemic clone carrying a mean of 1.3 non-silent mutations. Despite the paucity of mutations, activating mutations in kinase/PI3K/RAS signaling pathways were detected in 47%. Surprisingly, however, these mutations were often sub-clonal and frequently lost at relapse. In contrast to infant cases, MLL-R leukemia in older children had more somatic mutations (a mean of 6.5/case versus 1.3/case, P=7.15×10−5) and contained frequent mutations (45%) in epigenetic regulators, a category of genes that with the exception of MLL was rarely mutated in infant MLL-R ALL.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) arising in infants less than one year of age accounts for 2.5 to 5% of all childhood ALL. Up to 80% of the infant ALL cases are characterized by rearrangements of the Mixed Lineage Leukemia (MLL) gene at 11q231,2. Although current event free survival rates (EFS) for childhood ALL have reached greater than 85%3, the outcome for infant ALL with MLL rearrangements (MLL-R) remains poor with an EFS of only 28–36%1–3. Thus new therapeutic approaches are needed to improve cure rates for these patients. Studies on this leukemia subtype have demonstrated that the MLL translocation often occurs in utero, and that clinically overt leukemia develops with a very short latency, with rare cases presenting at birth4,5. These observations suggest that MLL-R ALL requires few additional mutations to induce full transformation. Consistent with this, genome-wide studies on MLL-R ALL using single nucleotide polymorphism (SNP) arrays have shown that this leukemia subtype contains on average only one copy number alteration (CNA) per case6,7. Nevertheless, murine models of MLL-R leukemia suggest that expression of the MLL chimeric gene alone is insufficient for full transformation8. To gain a better understanding of the complete landscape of somatic mutations in infant MLL-R ALL, we performed a genome-wide analysis on this leukemia subtype as part of the St. Jude Children’s Research Hospital – Washington University Pediatric Cancer Genome Project (PCGP)9.
RESULTS
Infant MLL-R ALL has an exceedingly low mutation frequency
Paired-end whole genome sequencing (WGS) was performed on diagnostic leukemia cells and matched remission bone marrow or peripheral blood cells from a discovery cohort of 22 infants with MLL-R ALL (Supplementary Tables 1 and 2 and Supplementary Fig. 1). The leukemic genomes had an average haploid coverage of 39X. Somatic alterations including single nucleotide variations (SNVs), insertions/deletions (indels), structural variations (SV) and CNAs were detected using multiple analytical pipelines10,11 and validated using orthogonal DNA sequencing approaches (Online Methods).
WGS revealed that the infant MLL-R ALL genomes contained an average of 111 somatic sequence mutations (range 29–229) and 10 CNAs/SVs (range 2–26), per case (Supplementary Fig. 2 and Supplementary Tables 3 and 4), yielding a median somatic coding mutation rate of 7.30×10−8 per base (range 0–2.29×10−7). With the exception of pediatric low-grade glioma12, this is 2–180 fold lower than those reported in adult and pediatric cancers (Supplementary Table 5). The mutation spectrum across the cohort did not suggest any specific mutational mechanisms with the most common nucleotide changes being C>T/G>A transition (Supplementary Fig. 2). The number of somatic alterations affecting the coding region of annotated genes or regulatory RNAs averaged 8.2/case, including 2.2 non-silent SNVs (range 0–4) and 6.0 CNA/SVs (range 2–19) per case (Fig. 1a–c, Supplementary Tables 3, 4 and 6–8 and Supplementary Figs. 2–3). RNA sequencing (RNAseq) in 21/22 cases demonstrated 48% of the non-silent SNVs to be expressed (Supplementary Tables 9–11 and Supplementary Figs. 4 and 5). Despite the paucity of mutations, 81% of the expressed missense mutations were predicted to have a deleterious effect on protein function.
Figure 1.

Somatic mutations detected by whole genome sequencing of infant MLL-R ALL. (a–c) CIRCOS plots of somatic non-silent mutations in three infant MLL-R ALL cases. (a) INF006 contained a balanced t(4;11)(q21;q23) encoding the MLL-AFF1 and gain of chromosome X. (b) INF009 contained a balanced t(11;19)(q23;p13.3) with the break on chromosome 19 occurring 22.7kb 5` of MLLT1 resulting in a spliced in-frame MLL-MLLT1 chimeric gene. The translocation had an inverted intrachromosomal duplication spanning 0.3kb at the breakpoint on chromosome 19 that resulted in an out-of-frame RFX2-MLL fusion. In addition, an unrelated intrachromosomal deletion of 46bp was detected on chromosome 6. (c) INF004 contained a complex three-way translocation involving chromosomes 9, 10, and 11 that encoded the MLL-MLLT10 on the derivative chromosome 11, an out-of-frame MLLT10-CNTNAP3B chimeric gene on chromosome 9, and a truncated 3` MLL on chromosome 10. The case also contained a t(8;14)(q24;q11.2) that resulted in the juxtaposition of MYC to the T-cell antigen receptor alpha (TRA) gene as denoted by the ∧. In addition, a non-silent SNV in COL13A1 and a deletion on chromosome 9 disrupted PAX5. (d) Structure of the INF004 complex translocation involving chromosomes 9p11 (blue), 10p12 (yellow), and 11q14–23 (burgundy). The genomic coordinates (hg18) are indicated above each genomic segment. The MLL-MLLT10 fusion gene is depicted by the solid arrow and all other rearrangements by dotted arrows. Copy number gains (red) and losses (blue) at the respective break points are shown. The final genomic products on chromosomes 9, 10 and 11 are shown in Supplementary Fig. 9. Ter, terminus; Cen, centromere.
Importantly, the majority of the CNAs/SVs were a direct consequence of the MLL rearrangement, with the break point of both MLL and its partner gene typically accompanied by the gain or loss of adjacent genetic material (Fig. 1d and see below). If the MLL-related CNAs/SVs are removed, the average CNAs/SVs per case decreases to 3.0 (Supplementary Table 12). Additionally, 40% of the identified somatic SNVs were detected at a mutant allele frequency (MAF) below 30% despite the high tumor purity (average 92%, range 74%–100%, Supplementary Tables 1, 4, 8 and 13). This suggests that these mutations reside in minor diagnostic sub-clones. Confirmation of this was achieved by performing custom capture and deep sequencing at an average of 437X coverage of all tier 1–3 somatic mutations identified in five cases. This analysis demonstrated the presence of significant intra-tumor heterogeneity (Supplementary Figs. 6 and 7). If we focus only on the mutations (including SNVs and indels) present in the dominant leukemia clone, the average number of non-silent mutations decreases to 1.3/case (Supplementary Table 13), with only 0.6 expressed at the RNA level (Supplementary Table 11 and Supplementary Fig. 5). A direct comparison of these mutation frequencies to those observed across 29 human cancers revealed infant ALL to have one of the lowest number of somatic mutations (Supplementary Fig. 8)13–15.
The majority of the MLL rearrangements are unbalanced
WGS detected a total of 133 SVs and CNAs across the infant ALL cohort that affected annotated genes (Supplementary Tables 3, 7, 12 and 14 and Supplementary Note). Approximately half (n=67) of the 133 SVs/CNAs, were a direct result of the MLL rearrangement. Within the remaining half (n=66) of the 133 SVs/CNAs, the most frequently affected genes were PAX5 (5 cases), CDKN2A/CDKN2B (3 cases) and the non-coding RNA genes DLEU1/2 (3 cases). The SVs/CNAs affecting these genes would be predicted to result in loss of function of a single allele.
Consistent with previous studies16–19, more than half of the MLL rearrangements were complex, involving three or more chromosomes, and/or accompanied by large insertions, deletions, and/or inversions of sequences adjacent to the breakpoints (Fig. 1d, Supplementary Figs. 3 and 9–16 and Supplementary Table 8). Moreover, even “so-called” simple cytogenetically balanced MLL translocations that involved only two chromosomes were found at the base pair level to have focal deletions and/or insertions of sequences at the breakpoints (Supplementary Table 15). As a result, although each MLL rearrangement would be predicted to encode an in-frame MLL-partner gene fusion protein, only 10/22 of the analyzed cases would be predicted to encode an in-frame reciprocal partner gene-MLL fusion protein. RNAseq on available samples demonstrated that 6 of 9 cases with a predicted reciprocal fusion expressed the reciprocal product, consistent with previous reports (Supplementary Table 16 and 17)20. Two of the predicted in-frame reciprocal fusion proteins involved genes with a known role in cancer: KRAS-MLL and AFF1-RAD51B-MLL (Supplementary Table 17 and Supplementary Figs. 11 and 12). Some of the complex MLL rearrangements also resulted in alterations of genes adjacent to MLL and/or the MLL fusion partner gene (Supplementary Table 16). An analysis of the sequence surrounding the breakpoints of MLL and its partner genes suggests that the predominant mechanism of rearrangement involved non-homologous end joining21.
RNAseq was performed on 12 diagnostic MLL-R cases from a validation cohort of MLL-R infant ALL (see below) identified a novel fusion gene between MLL and ubiquitin specific peptidase 2 (USP2) located at 11q23.3 approximately 1Mb from MLL on the reverse strand (Supplementary Table 17 and Supplementary Fig. 17). RNAseq also identified two novel non-MLL in-frame fusion genes in INF016: CABIN1-TRAPPC10 and PAX5-KANK1 (Supplementary Table 17), and an out-of-frame DDTL-CABIN1 fusion. Upon manual review, CABIN1-TRAPPC10 and DDTL-CABIN1 were identified in very few WGS reads, whereas the PAX5-KANK1 fusion lacked any WGS reads, suggesting that both fusions were present in minor sub-clones.
Mutations in the tyrosine kinase/PI3K/RAS signaling pathway
Despite the paucity of somatic mutations in the discovery cohort, activating mutations in tyrosine kinase/PI3K/RAS pathways were observed, with recurrent mutations in KRAS (n=4), NRAS (n=2), and non-recurrent mutations in FLT3, NF1, PTPN11, and PIK3R1 (Supplementary Tables 6 and 8). In contrast to the non-silent SNVs where only 48% of the mutant alleles were expressed, 100% of the activating kinase/PI3K/RAS pathway mutant alleles were expressed (Supplementary Table 11 and Supplementary Fig. 5). To extend these results, we sequenced the exons of 232 genes that included all mutated genes identified in the discovery cohort, as well as other genes in the kinase/PI3K/RAS signaling pathways, in a validation cohort (for a list of sequenced genes see Supplementary Notes) consisting of an additional 43 infant ALL cases, of which 25 harbored an MLL-R. Each sample was also analyzed for CNAs by SNP arrays (Online Methods and Supplementary Tables 18–23). Recurrent mutations were identified in 21 genes/gene loci across the combined infant MLL-R ALL cohorts (Fig. 2a and Supplementary Table 24). Importantly, activating mutations were identified in tyrosine kinase/PI3K/RAS pathways in 22/47 (47%) of the infant MLL-R cases (Fig. 2a,b, Supplementary Table 25 and Supplementary Figs. 18 and 19). The tyrosine kinase/PI3K/RAS mutations were observed in association with each of the different types of MLL rearrangements identified in infant ALL (Fig. 2b). In every case analyzed by RNAseq, the activating mutant alleles were expressed irrespective of their MAF (Fig. 2b, Supplementary Table 11). Furthermore, gene set enrichment analysis within the MLL-AFF1 cohort revealed the presence of expression signatures consistent with RAS pathway activation (Supplementary Table 26 and Supplementary Figs. 20–23).
Figure 2.
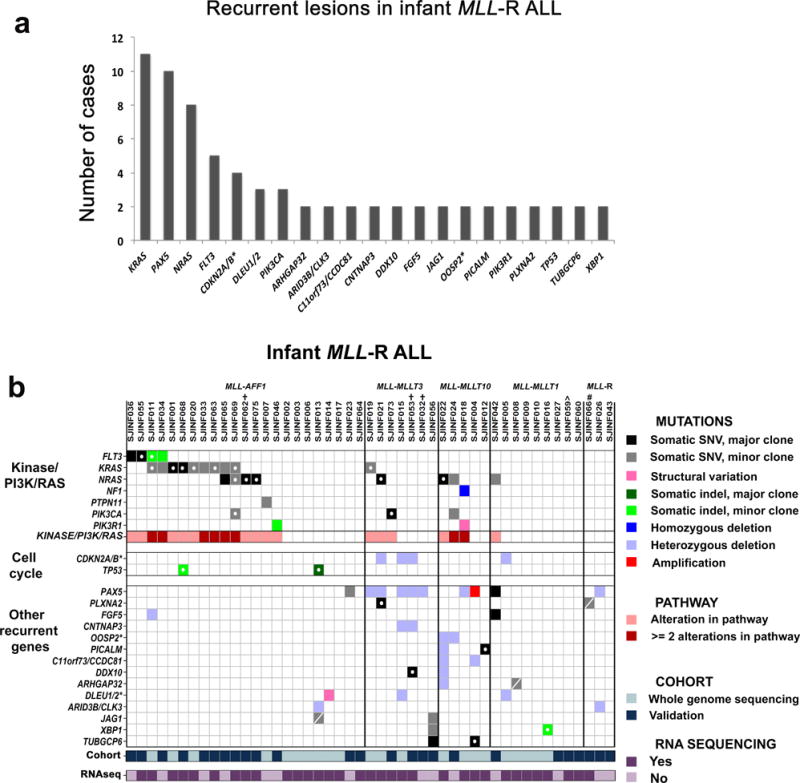
Recurrently mutated genes detected in 47 cases of infant MLL-R ALL. WGS was performed on 22 leukemic samples (discovery cohort) and targeted capture sequencing of the coding exons of 232 genes on an additional 25 infant MLL-R ALL samples (validation cohort). (a) Recurrent mutations were identified in 21 genes/loci in the combined infant MLL-R ALL cohorts. The * indicates that more than two genes were targeted in this locus (see Supplementary Tables 7, 8, 14, 19 and 24). (b) Distribution of mutated genes across the 47 infant MLL-R cases. For mutations in kinases or in genes in the PI3K/RAS pathway, only those known to confer activation of the pathway are shown. * indicates that more than two genes were targeted within the locus in one or more of the cases with only the first gene in the locus listed. † indicates that the sample lacked a matched non-leukemic sample. ∧ indicates that this sample is an identical twin to 060; the matched non-leukemic sample from 060 was used. # indicates that this sample has a novel MLL-USP2 fusion identified through RNA sequencing. A SNV marked with an open circle designates that the mutant allele is expressed as determined by RNA sequencing and a dash indicate that it is not expressed at the level of detection for our analysis.
The majority of the identified tyrosine kinase/PI3K/RAS mutations have been previously shown to be activating mutations (or inactivating mutations in the case of the homozygous deletion of NF1 in INF018)22–29. However, two cases contained a N676K FLT3 mutation that was previously reported in an acute myeloid leukemia (AML) patient following the development of resistance to the kinase inhibitor PKC41230,31, and as a recurrent mutation in core binding factor leukemia32 (Supplementary Fig. 24 and Supplementary Note). Similarly, we identified two cases that contained novel PIK3R1 mutations, including an internal tandem duplication (ITD) in the inter-SH2 domain (Supplementary Figs. 25–27 and Supplementary Note). We functionally demonstrated that the N676K FLT3 and PIK3R1 mutations were activating, resulting in factor independent growth of the IL-3-dependent murine leukemia cell line BaF3 (Supplementary Figs. 28 and 29).
A surprising observation was that 65% (20/31, observed in 22 cases) of the activating tyrosine kinase/PI3K/RAS mutations had MAFs <30%, suggesting that they were present in minor sub-clones (Supplementary Note, Supplementary Table 25 and 27). Moreover, although eight cases contained two or more mutations in this pathway, the MAF for each gene was below 30% in six cases. To further explore the importance of the identified tyrosine kinase/PI3K/RAS pathway mutations, we analyzed seven matched diagnostic and relapse infant MLL-R samples, including five with a mutation in this pathway at diagnosis. Although the activating mutation was maintained or increased at relapse in two cases (INF001 and INF65), they were lost in two (INF033 and INF042), while in another case (INF073) the PIK3CA MAF decreased from 39% to 15% (Supplementary Table 28). These data suggest that the tyrosine kinase/PI3K/RAS pathway activating mutations may not be necessary for maintenance of the leukemic cells.
Although a trend toward poorer EFS and Overall Survival (OS), and an increased cumulative incidence of relapse was observed in patients containing an activating pathway mutation, this did not reach statistical significance (Supplementary Fig. 30). There was no difference in the age at diagnosis of infant MLL-R patients with or without an activating mutation (Supplementary Fig. 31a,b). However, within the MLL-AFF1 infant cohort (N=23), those with an activating mutation were on average younger than those lacking a mutation (mean 4.01 versus 6.75 months, P=0.0473, Supplementary Fig. 32a). Furthermore, there was an age difference among patients with activating mutations in a major or minor clone, or those lacking an activating mutation (mean 2.77, 5.09 and 6.75 months respectively, P=0.0228, Supplementary Fig. 32b), suggesting that an activating mutation among MLL-AFF1+ patients may be associated with decreased disease latency.
Clonal evolution at relapse in infant MLL-R ALL
To further explore the relationship between diagnosis and relapse in infant MLL-R ALL, we compared all somatic mutations between the diagnostic and relapse samples from two infant patients with MLL-R ALL, each relapsing three years from diagnosis (INF001D/INF001R and INF002D/INF002R). Non-tumor as well as diagnostic and relapse samples were analyzed by WGS followed by custom capture enrichment and deep sequencing allowing us to accurately calculate the MAF for all SNVs for each leukemia. In both cases, similar to non-MLL-R leukemia, relapse was associated with a marked increase in the total number of SNVs and CNAs/SVs (Fig. 3a,b, Supplementary Figs. 33–35 and Supplementary Tables 3, and 29–30).
Figure 3.
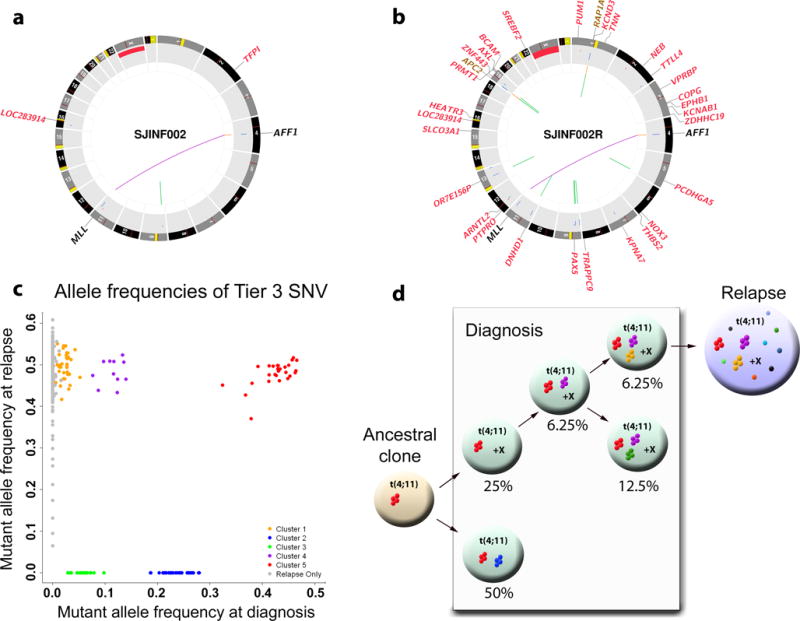
Clonal evolution from diagnosis to relapse in infant INF002. (a,b) CIRCOS plots showing the non-silent SNVs and SVs detected by WGS at (a) diagnosis and (b) relapse. (c) Five mutation clusters identified at diagnosis with MAF at 0.426 (red), 0.236 (blue), 0.114 (purple), 0.058 (green), and 0.024 (orange), respectively. The diagnostic mutations with a MAF of 0.426 are consistent with heterozygous mutations present in every leukemic cell, whereas clusters with lower MAF represent mutations present in minor leukemic sub-clones. (d) Evolutionary trajectory from diagnosis to relapse. Five clones were determined from diagnosis. Only a subclone with estimated population frequency of 6.25% survived therapy and persisted to relapse. It contained 3 mutation clusters (red, purple orange) from diagnosis but also acquired additional mutations at relapse. The relapsed tumor is likely to contain only a single clone as there is only 1 major MAF cluster (Supplementary Fig. 6b). “X” indicates that these clones also have an extra copy of “X”.
The SNVs occurring in unique sequences outside of genes or gene regulatory regions (ie., tier 3 SNVs) were analyzed at diagnosis and relapse for each pair by assigning them to statistically significant clusters based on their MAFs (Online Methods). Five mutation clusters were identified in INF002 at diagnosis with MAFs of 0.426, 0.236, 0.114, 0.058, and 0.024, respectively (Fig. 3c,d). The diagnostic mutations with a MAF of 0.426 are consistent with heterozygous mutations present in every leukemic cell, whereas clusters with lower MAF represent mutations present in minor sub-clones. After adjusting for tumor purity, the diagnostic sample was predicted to contain five related clones with 6.25%–50% population frequencies. At relapse, the majority of tier 3 mutations occurred at a MAF of 0.5 suggesting the presence of a single clone descending from a minor clone present as 6.25% of the diagnosis sample. INF001 followed the same trend (Supplementary Fig. 33). In both cases, the founder clone at relapse acquired additional mutations following treatment although the mutation spectrum did not significantly differ between the diagnostic and relapse samples for these two patients (Supplementary Fig. 36)
Genomic landscape of infant non-MLL-R ALL
Targeted gene resequencing of the 232 genes and SNP arrays were also performed on 18 infant non-MLL-R ALLs (Online Methods and Supplementary Tables 18, 21–23). There was no significant difference in the average number of SNVs in these 232 genes or frequency of tyrosine kinase/RAS/PI3K pathway mutations between infant MLL-R (22/47, 47%) and infant non-MLL-R ALL (7/18, 39%, P=0.59); again, all activating mutations were expressed in the six cases subjected to RNAseq. In contrast to SNVs, infants lacking MLL-R had significantly more CNAs than infants with MLL-R (average of 2.2/case versus 1.0/case); however, this increased frequency was associated with age and was independent of the presence or absence of MLL-R (P=0.005, Supplementary Figs. 37–39). CDKN2A/B deletions were more frequent in the infant non-MLL-R cases, approaching that seen in standard non-Philadelphia positive childhood ALL (5/18, 28% versus 4/47, 8.5%, P=0.058)33.
Six of the 18 non-MLL-R infant cases had RNA available for sequencing; four of which carried either novel fusion genes, and/or altered genes that would be predicted to encode truncated proteins (Supplementary Table 17). Specifically, we identified two novel fusions containing NUTM1: BRD9-NUTM1 (INF049) and ACIN1-NUTM1 (INF074). The BRD9-NUTM1 fusion product created by the t(5;15)(p15;q14), consists of the first 14 exons of BRD9 fused to exons 3–8 of NUTM1. This novel fusion is analogous to the BRD3-NUTM1 and BRD4-NUTM1 found in midline carcinomas34. Our patient was described as a t(5;15)(p15;q12), which has been a previously reported cytogenetic finding in infant non-MLL-R ALL, and thus the BRD9-NUTM1 fusion may be recurrent in infants with t(5;15)(p15;q11–13)35,36. The ACIN1-NUTM1 fusion involves the same exons of NUTM1, but this time fused to the first 4 exons of ACIN1 at 14q11. Our patient had an ins(15;14)(q22;q11.2q32.1), indicating that NUTM1 may be a candidate gene for cytogenetic rearrangements involving 15q37. INF061 had both a BICD2-JAK2 and an ARHGEP32-CAPRINI fusion, as well as a truncation of the cohesin gene STAG2 (Supplementary Table 17). Finally, INF070 carried a truncation in ARID1B, a member of the SWI/SNF transcriptional complex; truncations of this gene have been described in 7% of pediatric neuroblastoma38. Overall, the SV and CNAs differ significantly between MLL-R and non-MLL-R infant ALL; underscoring the difference in biology and clinical outcome1.
Epigenetic mutations in non-infant MLL-R leukemia
Although MLL-R occur at a high frequency in infant ALL, this genetic lesion is also seen in older children with ALL or AML39. To compare the MLL-R mutational profile between infants and older children, we performed whole exome sequencing (WES) and SNP array analysis on 20 non-infant MLL-R patients (7–19 years of age, 9 ALLs, 10 AMLs, and 1 case of acute undifferentiated leukemia), as well as RNAseq on 18/20 cases (Supplementary Tables 9–11, 17, 31–32, and Supplementary Figs. 4 and 40). This analysis revealed that the major clone in non-infant MLL-R leukemia harbor a significantly higher number of non-silent somatic SNVs/indels than infant MLL-R ALL (mean 6.5/case versus 1.3/case, P=7.15×10−5, and for expressed genes: mean 3.2/case versus 0.6/case, P=1.6×10−3, Fig. 4a and Supplementary Tables 33 and 34, see Supplementary Table 35 for a mutation summary of all three cohorts). Although there was a trend towards a lower basal mutation rate in infants compared to non-infants (P=0.15), multiple linear regression analysis demonstrated that the significantly higher number of mutations in older children could not be solely attributed to the difference in the basal mutation rates (Supplementary Notes, Supplementary Figs. 41 and 42). This suggests that overt leukemia in older children with MLL-R may require more cooperating mutations. Similar to infants with MLL-R, activating mutations in tyrosine kinase/PI3K/RAS pathways were identified in 50% of the non-infant leukemias, with recurrent mutations in FLT3 (n=3), KRAS (n=3), NRAS (n=3), and non-recurrent mutations in CBL, PIK3CD, PTPN11, and PPM1J; all of which were expressed at the RNA level (Supplementary Tables 11, 36 and Supplementary Figs. 43 and 44). In contrast to infant MLL-R ALL cases, the majority of the tyrosine kinase/PI3K/RAS pathway mutations in the non-infant MLL-R leukemias were present in the major clone (Supplementary Table 25 and Supplementary Fig. 44). This observation extended to all identified somatic exonic mutations, whereas in infant MLL-R ALLs these mutations are more commonly seen in minor clones (P<0.0001, Supplementary Fig. 45). Non-infant MLL-R leukemias had a significantly higher number of CNAs as compared to infant MLL-R ALL (average 2.6/case versus 1.0/case, P=0.0234) (Supplementary Fig. 46). Deletions in CDKN2A/B were noted in 3/9 ALL cases (33%), but in none of the AML cases. None of the 20 non-infant MLL-R leukemias harbored a focal PAX5 lesion, which is in contrast to MLL-R infant ALL where PAX5 alterations were present in 5/22 cases (23%) (Supplementary Figs. 39, 44 and 47 and Supplementary Tables 37–39). RNAseq identified two novel in-frame non-MLL fusions, SETD2-CCDC12 (SJMLL009) and PABPC1L-YWHAB (SJMLL019). In addition, eight events identified in six cases resulted in out-of-frame fusions, one of which included MLL and four of which included MLL-partner genes (Supplementary Table 17). At the RNA level, a unique gene expression signature was noted in older children with MLL-AFF1 B-lineage disease (Supplementary Fig. 48 and Supplementary Tables 40 and 41).
Figure 4.
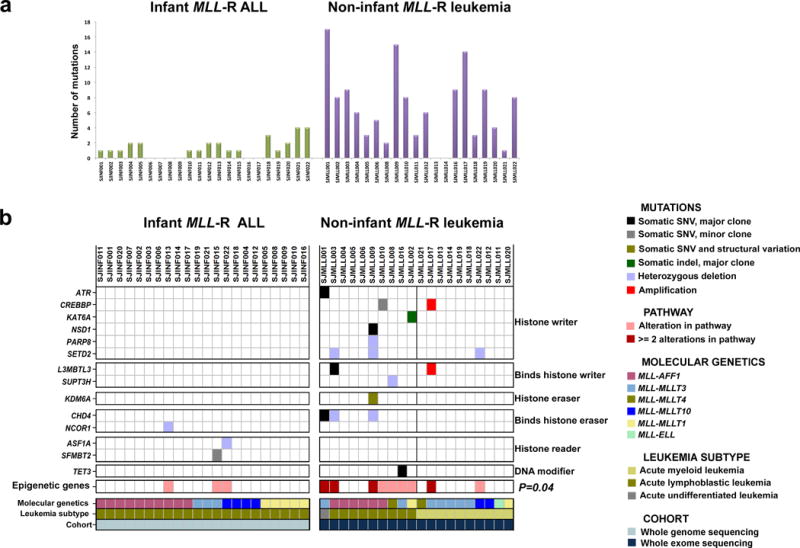
Mutational profiles of infant and non-infant MLL-R leukemia. (a) The number of non-silent SNVs and indels in the dominant leukemia clone affecting annotated genes in infant MLL-R ALL and non-infant MLL-R leukemia. (b) Distribution of somatic SNVs, indels, and CNAs in epigenetic regulatory genes in infant MLL-R ALL and non-infant MLL-R leukemias showing that these genes are significantly more often mutated in non-infant MLL-R leukemia (two-sided Fishers exact test, P=0.04). The black line between SJMLL002 and SJMLL021 indicate the separation between the lymphoblastic and myeloid non-infant leukemias. The only gene mutation that was found not to be expressed at the detection limit of our RNAseq analysis was the one in L3MBTL3.
An interesting observation in non-infant MLL-R leukemias was the presence of somatic mutations in genes whose products play a direct role in epigenetic regulation. The dominant epigenetic regulatory proteins are encoded by 633 genes (Supplementary Table 42)40. If we exclude MLL, which is altered in every case, somatic mutations were identified in 11 epigenetic regulatory genes (CHD4, SETD2, CREBBP, L3MBTL3, ATR, KAT6A, KDM6A, NSD1, PARP8, SUPT3H, and TET3) in 9/20 (45%) of the non-infant MLL-R leukemias (Fig. 4b and Supplementary Fig. 44). By contrast, only 3/22 (14%) of infant MLL-R ALLs harbored somatic mutations in epigenetic regulatory genes (P=0.04) (Fig. 4b).
DISCUSSION
In this analysis of the genetic landscape of infant MLL-R ALL, we demonstrate that this highly aggressive leukemia contains remarkably few somatic mutations, having one of the lowest somatic coding mutation rates observed in a human cancer to date41,42. The only sequenced cancer that has a lower number of somatic coding mutations is pediatric low-grade glioma, a clinically indolent tumor12. The observed low mutational burden is consistent with the known oncogenic potency of MLL fusion proteins and the very short disease latency seen in infant MLL-R ALL, with some patients presenting with overt leukemia at birth. However, despite the low overall number of cooperating mutations, tyrosine kinase/PI3K/RAS signaling pathways was activated in almost half of infant MLL-R ALL cases. While mutations in RAS have been previously described in MLL-R ALL43–45, we demonstrate for the first time recurrent activating mutations targeting the PI3K complex. Specifically, we found activating mutations in PIK3CA and PIK3R1 in 11% of the cases. The high frequency of activating mutations in tyrosine kinase/PI3K/RAS pathway genes in MLL-R ALLs underscores the biologic cooperativity between MLL fusion proteins and enhanced signaling through these pathways46–48.
Although the allele frequency of the MLL-chimeric genes indicated that it is present in every leukemic cell, our detailed analysis of the mutant allele frequencies of the other identified somatic mutations suggest that they are often present in minor clones. For example, 65% of the activating tyrosine kinase/PI3K/RAS mutations had MAFs <30%, suggesting that they were present in minor diagnostic clones. In addition, although eight infant MLL-R ALL cases contained two or more mutations in genes in this pathway, the MAF for each mutation was below 30% in six of these cases. Importantly, all of the activating tyrosine kinase/PI3K/RAS mutations were expressed, which was in contrast to other non-silent mutations in the discovery cohort, where only 38% were expressed. Even more surprising, in our analysis of seven matched diagnostic and relapse infant MLL-R samples, five of which contained a diagnostic mutation in the tyrosine kinase/PI3K/RAS pathway, we found that these mutant genes were maintained at relapse in two cases, decreased in one, and lost in two. Consistent with these observations, a recent report describing three paired MLL-AFF1 cases, showed that two lost the clone carrying the RAS mutation at relapse45.
The presence of RAS activating mutations in infant MLL-R leukemia has been previously suggested to be an independent predictor of a poor prognosis43. Although our cohort is small, our analysis suggested a trend toward a lower EFS and OS in cases with tyrosine kinase/PI3K/RAS pathway mutations, especially when the mutation was present at a MAF >30%. In contrast to the Interfant study43, within the MLL-AFF1 cohort, those with an activating mutation were on average younger than those lacking a mutation. This age difference was even more pronounced when considering the MLL-AFF1 cases with an activating mutation in the major clone, indicating that the presence of an activating mutation in this pathway may decrease the time required for the development of clinically overt disease. These data suggests that the co-existence of the MLL fusion protein with an activating mutation in tyrosine kinase/PI3K/RAS signaling pathways is not essential for either the establishment or maintenance of the leukemia; however its presence likely confers a growth advantage. Our data also raises the possibility that the high degree of clonal heterogeneity observed in infant MLL-R ALL49 may directly contribute to its poor prognosis, in that conventional ALL treatment regimens may not fully eliminate all clones, allowing the emergence of relapsed disease from a minor clone present at the time of diagnosis.
Interestingly, in non-infant MLL-R leukemias most somatic mutations were found to reside within a single dominant clone. In addition, these latter cases harbored significantly more non-silent mutations than infant MLL-R leukemias, suggesting that in the older patients, MLL fusion genes appear to require an increased number of cooperating mutations to generate overt disease. This raises the possibility that there may be a fundamental difference in the target cell of transformation, the microenvironment, or both between infants and older patients. This is further supported by the finding that non-infant MLL-R leukemias carry significantly more mutations in genes encoding epigenetic regulators than infant cases, suggesting that the target cell in infants may already have a chromatin state that is more permissive to transformation by the MLL gene rearrangement.
These data provide a unique look into the underlying molecular pathology of this highly aggressive pediatric leukemia. The data suggest that in infants, the leukemia is initiated in a hematopoietic progenitor cell (HPC) that may have a different chromatic state than more mature HPCs, and that within this context, the MLL chimeric gene plays a dominant role in establishing overt leukemic cells. These cells gain few additional mutations during their proliferative expansion, although the frequent targeting of genes within tyrosine kinase/PI3K/RAS signaling pathways suggest that activation of this pathway can cooperate in leukemogenesis, although not to a level that results in the establishment of a dominant clone. Moreover, the loss of these latter mutations at relapse in a number of patients suggests that specifically targeting these activating lesions will likely provide little therapeutic benefit. On the other hand, the clear implication that the MLL fusion protein is a potent driver gene in this aggressive leukemia, and that the leukemic cells lack a high mutational rate raises the possibility that therapy directed toward the MLL fusion protein, or proteins required for its biological actions, may be a fruitful approach for targeted therapy in this aggressive leukemia.
ONLINE METHODS
Patients
Paired-end whole genome sequencing on diagnostic leukemia blasts and matched germ line samples was performed on a discovery cohort of 22 infants with MLL rearranged (MLL-R) acute lymphoblastic leukemia (ALL) and 2 matched relapse samples (INF001R and INF002R) using the Illumina sequencing platform (Illumina Inc., San Diego, CA). The infants were treated at St Jude Children’s Research Hospital, Memphis, Tennessee and diagnosed during a period of 1992–2008. The cohort consisted of 10 cases with the t(4;11)[MLL-AFF1(AF4)], 5 cases with the t(11;19)[MLL-MLLT1(ENL)], 4 cases with the t(10;11)[MLL-MLLT10(AF10)], and 3 cases with the t(9;11)[MLL-MLLT3(AF9)]. 11q23/MLL rearrangements were evaluated by florescence in situ hybridization (FISH) to confirm MLL gene rearrangements and/or reverse-transcription PCR for MLL-AFF1(AF4), MLL-MLLT3(AF9), MLL-MLLT10(AF10), MLL-MLLT1(ENL) and MLL-ELL. Samples were cytogenetically analyzed and screened by RT-PCR for the presence of ETV6-RUNX1, TCF3-PBX1, and BCR/ABL1 as part of routine clinical diagnostics. See Supplementary Table 1 for the clinical and genetic characteristics of the 22 infant MLL-R ALL cases.
The validation cohort consisted of 43 additional infant leukemia cases, 25 of which contained a MLL-R (Supplementary Table 18). In 38 of the cases, non-leukemic normal (germline) sample was available. INF059 and INF060 were identical twins and a matched non-leukemic sample was only available for INF060; thus this sample was used as a germline sample also for INF059.
In addition, five matched relapse infant MLL-R leukemia cases were investigated using either exome sequencing (SJINF033, SJINF065, SJINF073) or targeted capture sequencing (SJINF042, SJINF060). Two matched relapse samples from non-MLL-R infant cases were also whole exome sequenced (SJINF039 and SJINF061).
Whole exome sequencing was performed on 20 cases of non-infant MLL-R leukemia [1 Acute undifferentiated leukemia (AUL), 9 ALL cases, and 10 acute myeloid leukemia (AML)] (See Supplementary Table 31 for their clinical and genetic characteristics) and their matched normal non-leukemic DNA sample. All patients were treated at St. Jude Children’s Research Hospital. At diagnosis, the 11q23 rearrangements were detected as described above.
Tumor and germline samples were obtained with informed consent using a protocol approved by the St. Jude Children’s Research Hospital institutional review board. The study was approved by the Institutional Review Boards of St. Jude Children’s Research Hospital and Washington University.
Copy number analyses using Affymetrix SNP arrays
Of the 22 infant MLL-R ALL cases from the discovery cohort, 10 had copy number analyses performed using Affymetrix 500k SNP array (Affymetrix) as part of other studies and 12 had copy number data from SNP 6.0 array data (Affymetrix). In this study, we performed copy number analysis using the Affymetrix 6.0 SNP array for all samples in the validation cohort and for the non-infant MLL-R cases.
Illumina library construction for whole genome sequencing
All methods in the library construction and whole genome DNA sequencing have been described previously50,51. Detailed information regarding the coverage is included in Supplementary Table 2 and Supplementary Fig. 1.
Agilent liquid capture and library construction for whole exome sequencing
Exon sequence was captured from 3ug of genomic DNA using the Agilent SureSelect Human All Exon 50MB Kit (Agilent Technologies) following the manufacturer’s instructions. After elution, the cDNA libraries were sequenced (paired end 2×101 cycles) on Illumina sequencers (Illumina).
RNA Sequencing
RNA sequencing was performed on 21 diagnostic and 2 paired relapse cases from the discovery cohort (not INF007), 12 diagnostic and 4 relapse MLL-R infant ALLs and 6 non–MLL-R infants from the validation cohort, and on 18/20 cases from the non-infant MLL-R cohort (Supplementary Tables 9–11). Total RNA was extracted with Trizol (Ambion) per manufacturer’s protocol. One microgram of total RNA was DNase I (Ambion) treated at room temperature for 15 minutes followed by phenol/chloroform/isoamyl extraction and ethanol precipitation. The integrity of the DNase I treated RNA was analyzed on the Agilent 2100 Bioanalyzer (Agilent Technologies,) prior to poly-A mRNA selection. Poly-A mRNA selection and subsequent cDNA synthesis was done using Illumina TruSeq RNA sample preparation kit according to manufacturer’s protocol. The cDNA was fragmented (200bp peak) with a Covaris E210 ultrasonicator prior to library preparation utilizing Illumina TruSeq RNA sample preparation kit according to manufacturer’s protocol. The adapter ligated fragments were PCR amplified for 10 cycles. The quality and size of the final library preparation was analyzed on the Agilent 2100 Bioanalyzer. Sequencing was performed on the Illumina HiSeq2500 in rapid mode to generate paired end 100 cycle reads. Putative in-frame fusions were validated by RT-PCR followed by Sanger sequencing. Primer sequences are available upon request.
Analysis of whole genome sequencing data
Methods employed for WGS mapping, coverage and quality assessment, single nucleotide variations (SNV) / indel detection, tier annotation for sequence mutations, prediction of deleterious effects of missense mutations, and identification of loss-of-heterozygosity have been described previously11. Briefly, transcripts from Ensembl52 build (54_36) and Genbank53,54 (build download May 21, 2009) were used for annotation. Sequence variants were classified into the following four tiers: 1) Tier 1: Coding synonymous, nonsynonymous, splice site, and non-coding RNA variants; 2) Tier 2: Conserved variants (cutoff: conservation score greater than or equal to 500 based on either the phastConsElements28way table or the phastConsElements17way table from the UCSC genome browser, and variants in regulatory regions annotated by UCSC annotation (Regulatory annotations included are targetScanS, ORegAnno, tfbsConsSites, vistaEnhancers, eponine, firstEF, L1 TAF1 Valid, Poly(A), switchDbTss, encodeUViennaRnaz, laminB1, cpgIslandExt); 3) Tier 3: Variants in non-repeat masked regions; and 4) Tier 4: the remaining SNVs.
Structural variations including inter-chromosomal translocations (CTX), intra-chromosomal translocations (ITX), inversions (INV), deletions (DEL), and insertions (INS) were analyzed by CREST (Clipping REveals STructure)10 and annotated as previously described11. In addition, BreakDancer55 was run using the default parameters for INF013. Paired tumor/normal bam files were used to identify putative somatic SVs. All predicted transcripts were analyzed and transcripts that would lack a CDS start or stop site were filtered out. The CDS length were then computed by taking the sum of the lengths of the coding portions of the exons containing the CDS start and stop sites with the lengths of all intermediate exons. In cases where both breakpoints of an event occurred inside exons, we reduced both exons to a single fused exon that was built by assembling the reads at the SV breakpoints and aligning the assembled contig back to the reference. If the fused exon lay between the CDS start and stop sites, the fused exon’s length were included in the CDS length calculation. In addition, if the predicted CDS length was a multiple of three, the fusion transcript was classified as “in-frame”. If the transcript was altered from the annotated CDS of one of the genes, it was classified as “modified in-frame”. In cases where the fusion occurred in the UTR, there could be modified transcripts with unmodified CDSes, so we explicitly checked for and flagged transcripts predicted to have modified in-frame CDSes.
Copy number variations (CNVs) were identified by evaluating the difference of read depth for each tumor and its matching normal DNA using the novel algorithm CONSERTING (COpy Number SEgmentation by Regression Tree In Next-Gen sequencing)11. Confidence for a CNV segment boundary was determined using a series of criteria, including: length of flanking segments, difference of CNV between neighboring segments, presence of sequence gaps on the reference genome, presence of structural variation (SV) breakpoints, and any CNV in the matching germline sample. All CNAs were manually reviewed and compared with Affymetrix SNP 6.0 or 500K CNA results. Regions of LOH were identified from the high quality SNVs. First, heterozygous SNVs with mutant allele frequency between 40–60% in the germline sample were used to estimate the LOH signal. For each heterozygous SNV, the LOH signal was calculated as the absolute mutant allele frequency difference between the tumor and germline samples. Second, chromosomes were segmented and segments were merged on the LOH signal.
Analysis of RNA sequencing data
For RNA-Seq, paired-end sequencing was performed using the HiSeq platform with 100bp read length. Paired-end reads from RNA-seq were aligned to the following 4 database files using BWA (0.5.10) aligner: (1) the human GRCh37-lite reference sequence, (2) RefSeq, (3) a sequence file representing all possible combinations of non-sequential pairs in RefSeq exons, (4) AceView database flat file downloaded from UCSC representing transcripts constructed from human ESTs. The mapping results from (2) to (4) were translated to human reference genome coordinates. In addition, they were aligned using STAR 2.3.0 to the human GRCh37-lite reference sequence without annotations. A BAM file was constructed by selecting the best alignment among the five mappings. Poor quality mappings were improved using SIM4 when possible to generate the final BAM. The coverage was calculated using an in-house pipeline. SV detection was carried out using CICERO, a novel algorithm that uses de novo assembly to identify structural variation in RNASeq (Li et al, manuscript in preparation). Putative fusions were validated by reverse transcription and polymerase chain reaction.
Digital gene expression profiling
The transcript expression levels were estimated as Fragments Per Kilobase of transcript per Million mapped reads (FPKM); gene FPKMs were computed by summing the transcript FPKMs for each gene using Cuffdiff256,57. A gene was considered “expressed” if the FPKM value >= 0.5 based on the distribution of FPKM gene expression levels. Genes that were not expressed in any sample were excluded from the final data matrix for downstream analysis. RNA expression profiles were analyzed using Qlucore Omics (Qlucore AB).
Targeted sequencing in the infant MLL-R and non-MLL-R validation cohorts
To determine the frequency of identified mutations in a larger infant cohort, additional sequencing was performed for a total of 232 genes by Agilent Targeted Capture (216 genes, see below) followed by Illumina Sequencing or by PCR followed by Sanger sequencing (16 genes, see below) in a validation cohort consisting of an additional 43 infant ALL cases, 25 of which harbored an MLL-R. The 232 genes included in the targeted sequencing were selected based on the following criteria a) they were targeted by a genetic lesion (SNVs, SVs or focal CNA (≦ 5 genes)) in the infant MLL-R discovery cohort as determined by whole genome sequencing (157/232 genes); or b) annotated within a recurrently mutated pathway or if they had been described to be of importance for cancer (59 genes). This set of genes (216) were analyzed by targeted capture. In addition, the coding exons from 16 genes were sequenced by Sanger sequencing (ABL1, AKT3, ALK, FOXO1, FOXO3, FOXO4, FOXO6, INPP5D, JAK2, MET, PDGFRA, PDGFRB, PTEN, RAF1, RET, SYK) and putative SNVs and indel variants were detected by SNPdetector58. Non-silent sequence mutations were selected for validation by Sanger sequencing of both the tumor and matching normal samples (where available).
Experimental validation of somatic mutations and structural variants
Validation of identified somatic Tier 1 SNVs was performed using PCR amplification of the leukemia and germ line DNA followed by Sanger (SJINF010 and SJINF019) or 454-based sequencing (SJINF006-009, SJINF011-018, SJINF020-022). We validated the tier 1–4 mutations by array-based capture followed by Illumina-based sequencing for INF001-005. Structural variations were validated by PCR and subsequent Sanger sequencing on whole genome amplified DNA or by capture based methods.
The 454-based sequencing was performed as previously described51. Briefly, the PCR products were subjected to library construction followed by 454 Titanium sequencing. Read sequences and quality scores were extracted with sffinfo (454 proprietary software), and aligned to NCBI Build 36 or 37 using SSAHA2 with the SAM output option. Alignments were imported to BAM format using SAMtools. A SAMtools pileup file was generated, and the read counts were determined by VarScan59. In the analysis of the 454 reads, a minimum base quality of 15 was required, with at least 20 reads aligned, to report the allele frequencies.
Oligonucleotide primers for genomic PCRs were designed using the flanking sequences of each SV or SNV using Primer 360. For SJINF001-005 and 001R and 002R, the SVs were also validated using targeted capture followed by Illumina sequencing. 77% of the SVs reported herein have been experimentally validated.
Experimental validation of predicted in-frame fusion transcripts
Predicted in-frame predictions of fusion transcripts were validated by RT-PCR followed by Sanger sequencing of the purified PCR products. Primer sequences are available upon request.
Clonal evolution analysis
In order to decipher the potential clonal evolutionary path between the diagnostic and relapse tumor, we compared mutant allele frequency (MAF) of somatic SNVs detected at diagnosis and relapse. To have an accurate MAF readout, we performed deep sequencing using custom capture followed by Illumina sequencing on all tiers 1–3 mutations detected by WGS in the trio of diagnostic, relapse, and germline samples for case SJINF001 and SJINF002. The median coverage of targets in the primary tumors SJINF001_D, SJINF002_D and in the relapse tumors SJINF001_R, SJINF002_R, is 1534X, 894X, 769X, and 882X, respectively. The following criteria were used when assessing SNVs in the diagnostic tumor: 1) the mutant is missing from the matched germline sample and 2) the mutant allele frequency is significantly different between the germline and diagnostic sample (Fisher’s Exact test P≤ 0.05). SNVs located on chrX/Y or chromosomal segments with CNAs were further removed. The remaining Tier3 SNVs were used to perform normal mixture modeling using the mclust package (version 3.4.10) in R. The optimal model is determined by Bayesian information criterion (BIC).
FLT3 and PIK3 modeling
The structure of auto inhibited, inactive FLT3 kinase domain was used to model the Asn676Lys (N676K) mutation (PDBID: 1RJB). The structures of phosphorylated (PDBID: 2PSQ) and unphosphorylated (PDBID: 2PVF) FGFR2 were used to model the mechanism of kinase activation61,62. The N676K was introduced into the FLT3 kinase domain structure and energy minimized with default parameters in FOLDX63. Images and structure alignments were performed in Pymol64. The structure of PIK3R1/PIK3CA was used to interpret the insertion and deletion mutation sites (PDB: 3HIZ)65; this structure represents the inhibited state of the PIK3CA kinase domain. Prediction of disordered residues was performed by DISORED266 and were then visualized in Pymol64.
Clinical correlations
The association between the presence of an activating mutation or KRAS/NRAS mutations to clinical variables was tested using the exact Chi-square test. Event-free survival and overall survival distributions in different genetic groups were compared using the Log-Rank test. The cumulative incidence of any relapse was calculated according to the method of Kalbfleisch and Prentice and compared across genetic groups by using Gray’s test67,68. We have limited the outcome analyses to MLL-R infant patients treated at St. Jude Children’s Research Hospital (N=33 for 10-year overall survival (OS) and 10-year event free survival (EFS), and N=31 for the risk of relapse).
Pathway analysis
A genomic random interval (GRIN)69 model was used as a null model to evaluate the statistical significance of the pattern of overlap of genomic lesions with the loci of individual genes, predefined sets of genes, and each base-pair locus in the genome. The null model represents each lesion as an interval of fixed length and random location that may occur at any location along the chromosome with equal probability.
Other statistical analysis
Unless otherwise specified, all the statistical tests were done using R version 3.1.170. Two-sided Fisher’s exact test was used to compare mutation frequencies in different patient groups. Two-sided Wilcoxon rank sum test available from the R coin package (dealing with ties and reporting asymptotic P values) was used to compare number of mutation, BMR and number of CNAs between infant MLL-R ALL and non-infant MLL-R leukemias and to compare age between two patient groups. Kruskal-Wallis test was used to compare age among more than two patient groups. Two-sided tests for association using Spearman’s rho between mutation and age was done using the R cor.test function.
DNA constructs and retrovirus production
Full length wild type FLT3, PIK3CA, and PIK3R1 were amplified from human cDNA and sequence variants (FLT3-D835Y (Ctrl), FLT3-D600H, FLT3-N676K, FLT3-D839G, FLT3-P934L, PIK3CA-E542K (Ctrl), PIK3CA-H1047R (Ctrl), and PIK3R1-Q572* (Ctrl)) were introduced using the QuickChange II XL Site Directed Mutagenesis Kit (Stratagene). The FLT3-ITD, PIK3R1-Dup (INF018), PIK3R1-R1a* (INF070), PIK3R1-R1b (INF070), were cloned from primary patient material. All mutations were verified by sequencing the complete open reading frame of FLT3, PIK3CA and PIK3R1. The mutant cDNAs were cloned into the defective mouse stem cell virus (MSCV) co-expressing our mutations (FLT3-WT, FLT3-ITD, FLT3-D835Y, FLT3-D600H, FLT3-N676K, FLT3-D839G, FLT3-P934L, PIK3R1-WT, PIK3R1-Dup, PIK3R1-R1a*, PIK3R1-R1b, PIK3R1-Q572*) and green fluorescent protein (GFP) or a MSCV virus co-expressing the mutant cDNA (PIK3CA-WT, PIK3CA-E542K, PIK3CA-H1047R) and Cherry. Full-length protein expression was verified by western blotting. Retroviral supernatants were produced using the ecotropic Phoenix packaging cell line (G.P. Nolan, Stanford University) and used to transduce BaF3 cells.
BaF3 cell culture and cytokine independence assay
The BaF3 cells were cultured in RPM1-1640 supplemented with 10% fetal calf serum (FSC) and 10ng/ml IL-3 (PeproTech). To analyze cytokine independence, cells were transduced with the retroviral constructs, flow sorted 48h after transduction to obtain GFP or GFP/Cherry double positive cells, and then seeded in IL-3 free media. Cells were seeded at a density of 0.2 million/ml in 5 ml in 6 well plates and counted in a Vicell cell counter (Beckman Coulter) every day for 7 days by tryptan blue exclusion. Cells were split when cell numbers reached 2 millions/ml to 0.2 million/ml. Each experiment was performed in triplicate.
Western blot
IL-3 (Pepro Tech) was withdrawn from the BaF3 cells transduced with the FLT3 or PIK3 constructs overnight, followed by 6h serum starvation in 0.3% fetal calf serum (Hyclone). For FLT3, cells were resuspended in 1 ml of media with or without 100ng/ml FLT3 ligand (GenScript) for 5 minutes, washed once in ice cold PBS and lysed with 1X RIPA (Cell Signaling Technology) containing Protease Inhibitor Cocktail (Sigma-Aldrich), PhosSTOP (Roche) and PMSF (Sigma-Aldrich). Western Blot was performed according to standard protocols. In brief, 50μg of protein were separated on 10% Bis-Tris Gels (Life Technologies) and transferred to PVDF membranes (Life Technologies). The following antibodies were used: anti-FLT3 (clone 8F2), anti-phospho-FLT3 (Y591, clone 54H10), anti-STAT5, anti-phospho-STAT5 (Y694, clone C11C5), anti-ERK1/2, anti-phospho-ERK1/2 (T202/Y204, clone 20G11), anti-GAPDH (Santa Cruz Biotechnology), anti-PIK3CA, anti-PIK3R1, anti-AKT (clone 40D4), anti-phospho-AKT (S473, clone D9E), anti-pS6 (clone 5G10), anti-phospho-pS6 (S240/244). All antibodies, except GAPDH, were purchased from Cell Signaling Technology. The phospho antibodies were used at a 1:500 dilution. The total antibodies were used at a 1:2000 dilution and the control antibody was used at a 1:10000 dilution.
Supplementary Material
Acknowledgments
We thank all the patients and their parents from the St. Jude Children’s Research Hospital, USA, Department of Pediatrics at Lund University Hospital, Sweden, and children’s hospitals associated with the Australian and New Zealand Children’s Haematology and Oncology Group. We thank Bill Pappas and Scott Malone for information technology infrastructure. We thank the Tissue Resources Laboratory, the Flow Cytometry and Cell Sorting Core, and the Clinical Applications of Core Technology Laboratories of the Hartwell Center for Bioinformatics and Biotechnology of St. Jude Children’s Research Hospital. This work was funded by The St. Jude Children’s Research Hospital – Washington University Pediatric Cancer Genome Project, the American Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital, and supported by a grant from the National Institutes of Health (P30 CA021765). A.K.A was supported by the Swedish Childhood Cancer Society, the Swedish Research Council, the Swedish Cancer Society, BioCARE, and Gunnar Nilsson Cancer Foundation. C.G.M. is a Pew Scholar in the Biomedical Sciences and a St Baldrick’s Scholar.
Footnotes
AUTHOR CONTRIBUTIONS A.K.A., J.R.D., T.A.G., J.Z., J.M. and R.K.W. designed all experiments. J.Z. and J.M. led the sequencing analysis. J.Z., J.M., J.W., X.C., M.P., M.R., G.W., Y.I., J.B., P.G., M.E., P.N., L.W., C.L., L.D. and E.R.M. performed the computational data analyses. G.S. provided bioinformatics support. A.K.A., L.H. and C.G.M. analysed SNP array data. R.H. and R.K. performed structural modelling. A.L.G. and J.D. performed functional work on the FLT3 and PI3K mutations. J.N., J.E., M.P., B.V., D.Y. performed validation experiments. K.B. performed the RNA sequencing. J.E. and J.M, performed exome sequencing. C.G.M., H.M. and D.P.T. prepared samples. S.P., K.G., L.S., C.C. and D.P. performed statistical analysis. R.S., N.C.V., A.C., A.R., D.C., JH., T.F. and C.H.P, provided annotated patient samples. S.R. and S.S. provided molecular genetics, cytogenetics and FISH data. J.M. and T.A.G. preformed critical reading and contributed to the writing of the manuscript. A.K.A. and J.R.D. wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
The sequence and single nucleotide polymorphism microarray data have been deposited at https://www.ebi.ac.uk/ega/datasets/EGAS00001000246.
References
- 1.Pieters R, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–250. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 2.Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeutic aspects of infant leukemia. Blood. 2000;96:24–33. [PubMed] [Google Scholar]
- 3.Pui CH, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford AM, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 5.Gale KB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94:13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 7.Bardini M, et al. Implementation of array based whole-genome high-resolution technologies confirms the absence of secondary copy-number alterations in MLL-AF4-positive infant ALL patients. Leukemia. 2011;25:175–178. doi: 10.1038/leu.2010.232. [DOI] [PubMed] [Google Scholar]
- 8.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 9.Downing JR, et al. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–622. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillert E, et al. A DNA damage repair mechanism is involved in the origin of chromosomal translocations t(4;11) in primary leukemic cells. Oncogene. 1999;18:4663–4671. doi: 10.1038/sj.onc.1202842. [DOI] [PubMed] [Google Scholar]
- 17.Reichel M, et al. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): the DNA damage-repair model of translocation. Oncogene. 1998;17:3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- 18.Super HG, et al. Identification of complex genomic breakpoint junctions in the t(9;11) MLL-AF9 fusion gene in acute leukemia. Genes Chromosomes & Cancer. 1997;20:185–195. [PubMed] [Google Scholar]
- 19.Meyer C, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27:2165–2176. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Downing JR, et al. The der(11)-encoded MLL/AF-4 fusion transcript is consistently detected in t(4;11)(q21;q23)-containing acute lymphoblastic leukemia. Blood. 1994;83:330–335. [PubMed] [Google Scholar]
- 21.Zhang Y, Rowley JD. Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) 2006;5:1282–1297. doi: 10.1016/j.dnarep.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 23.Tyner JW, et al. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith G, et al. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours – implications for personalised cancer medicine. Br J Cancer. 2010;102:693–703. doi: 10.1038/sj.bjc.6605534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janakiraman M, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–5911. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith CC, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121:3165–3171. doi: 10.1182/blood-2012-07-442871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bentires-Alj M, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 28.Loh ML, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 29.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA) Proc Natl Acad Sci U S A. 2012;109:15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heidel F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 31.von Bubnoff N, et al. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69:3032–3041. doi: 10.1158/0008-5472.CAN-08-2923. [DOI] [PubMed] [Google Scholar]
- 32.Opatz S, et al. Exome sequencing identifies recurring FLT3 N676K mutations in core-binding factor leukemia. Blood. 2013;122:1761–1769. doi: 10.1182/blood-2013-01-476473. [DOI] [PubMed] [Google Scholar]
- 33.Mullighan CG, Williams RT, Downing JR, Sherr CJ. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008;22:1411–1415. doi: 10.1101/gad.1673908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 35.Heerema NA, et al. Cytogenetic features of infants less than 12 months of age at diagnosis of acute lymphoblastic leukemia: impact of the 11q23 breakpoint on outcome: a report of the Childrens Cancer Group. Blood. 1994;83:2274–2284. [PubMed] [Google Scholar]
- 36.De Lorenzo P, et al. Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia. 2014;28:428–430. doi: 10.1038/leu.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heerema NA, et al. Abnormalities of chromosome bands 15q13–15 in childhood acute lymphoblastic leukemia. Cancer. 2002;94:1102–1110. [PubMed] [Google Scholar]
- 38.Sausen M, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45:12–17. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szczepanski T, Harrison CJ, van Dongen JJ. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol. 2010;11:880–889. doi: 10.1016/S1470-2045(09)70369-9. [DOI] [PubMed] [Google Scholar]
- 40.Huether R, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobbins SE, et al. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2013;52:954–960. doi: 10.1002/gcc.22090. [DOI] [PubMed] [Google Scholar]
- 42.Chang VY, Basso G, Sakamoto KM, Nelson SF. Identification of somatic and germline mutations using whole exome sequencing of congenital acute lymphoblastic leukemia. BMC Cancer. 2013;13:55. doi: 10.1186/1471-2407-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Driessen EM, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013;98:937–944. doi: 10.3324/haematol.2012.067983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang DC, et al. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006;106:950–956. doi: 10.1002/cncr.21687. [DOI] [PubMed] [Google Scholar]
- 45.Prelle C, Bursen A, Dingermann T, Marschalek R. Secondary mutations in t(4;11) leukemia patients. Leukemia. 2013;27:1425–1427. doi: 10.1038/leu.2012.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim WI, Matise I, Diers MD, Largaespada DA. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood. 2009;113:1086–1096. doi: 10.1182/blood-2008-01-132316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ono R, et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia. 2009;23:2197–2209. doi: 10.1038/leu.2009.177. [DOI] [PubMed] [Google Scholar]
- 48.Tamai H, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia. 2011;25:888–891. doi: 10.1038/leu.2011.15. [DOI] [PubMed] [Google Scholar]
- 49.Bardini M, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia. 2014 doi: 10.1038/leu.2014.154. [DOI] [PubMed] [Google Scholar]
ONLINE METHODS REFERENCES
- 50.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flicek P, et al. Ensembl 2011. Nucleic Acids Res. 2011;39:D800–806. doi: 10.1093/nar/gkq1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009;37:D26–31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, et al. SNPdetector: a software tool for sensitive and accurate SNP detection. PLoS Comput Biol. 2005;1:e53. doi: 10.1371/journal.pcbi.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 61.Berman HM. The Protein Data Bank: a historical perspective. Acta Crystallogr A. 2008;64:88–95. doi: 10.1107/S0108767307035623. [DOI] [PubMed] [Google Scholar]
- 62.Griffith J, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–178. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 63.Schymkowitz J, et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schrödinger L. The PyMOL Molecular Graphics System. 2011 v1.3. [Google Scholar]
- 65.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 67.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 68.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. Wiley, N.Y: 1980. [Google Scholar]
- 69.Pounds S, et al. A genomic random interval model for statistical analysis of genomic lesion data. Bioinformatics. 2013;29:2088–2095. doi: 10.1093/bioinformatics/btt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.R Core Team: R A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; http://www.R-project.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.