

Abstract
Recent studies have identified a cholestatic variant of nonalcoholic fatty liver disease (NAFLD) with portal inflammation and ductular reaction. Based on reports of biliary damage, as well as increased circulating free fatty acids (FFAs) in NAFLD, we hypothesized the involvement of cholangiocyte lipoapoptosis as a mechanism of cellular injury. Here, we demonstrate that the saturated FFAs palmitate and stearate induced robust and rapid cell death in cholangiocytes. Palmitate and stearate induced cholangiocyte lipoapoptosis in a concentration-dependent manner in multiple cholangiocyte-derived cell lines. The mechanism of lipoapoptosis relied on the activation of caspase 3/7 activity. There was also a significant up-regulation of the proapoptotic BH3-containing protein, PUMA. In addition, palmitate-induced cholangiocyte lipoapoptosis involved a time-dependent increase in the nuclear localization of forkhead family of transcription factor 3 (FoxO3). We show evidence for posttranslational modification of FoxO3, including early (6 hours) deacetylation and dephosphorylation that coincide with localization of FoxO3 in the nuclear compartment. By 16 hours, nuclear FoxO3 is both phosphorylated and acetylated. Knockdown studies confirmed that FoxO3 and its downstream target, PUMA, were critical for palmitate- and stearate-induced cholangiocyte lipoapoptosis. Interestingly, cultured cholangiocyte-derived cells did not accumulate appreciable amounts of neutral lipid upon FFA treatment.
Conclusion
Our data show that the saturated FFAs palmitate and stearate induced cholangiocyte lipoapoptosis by way of caspase activation, nuclear translocation of FoxO3, and increased proapoptotic PUMA expression. These results suggest that cholangiocyte injury may occur through lipoapoptosis in NAFLD and nonalcoholic steatohepatitis patients.
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome.1 NAFLD is a spectrum of liver diseases including simple steatosis, nonalcoholic steatohepatitis (NASH), advanced hepatic fibrosis, liver cirrhosis, and hepatocellular carcinoma.1 NAFLD is the most common liver disease in Western countries and it is highly associated with obesity, diabetes, dyslipidemia, and hypertension.1 Recently, a cholestatic presentation of NAFLD with ductular inflammation, bile duct loss and swelling, and bile duct proliferation was reported.2 They also showed that bridging fibrosis or cirrhosis was more common in patients with biliary injury.2 This suggests the involvement of biliary epithelial cell injury as a possible contributor to the severity of NAFLD or NASH.2,3 Bile duct epithelial cell expansion, termed the ductular reaction, is a response to injury and has been observed in NAFLD.3
NAFLD patients have elevated concentrations of circulating saturated free fatty acids (FFAs). FFA-induced hepatocyte lipoapoptosis is a recognized hallmark of NAFLD and includes the activation of p38-mitogen activated protein kinase (p38-MAPK), extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK).4 These stress-activated kinases have been well known to translocate into the nucleus upon activation and can phosphorylate several transcription factors including forkhead family transcription factors (FoxO) to regulate gene expression.5 Further, FoxO1 has been shown to be phosphorylated by p38 and ERK but not by JNK.5 Phosphorylation of FoxO3, at residue Ser7 by p38, has been shown before to promote its nuclear localization and apoptosis in response to doxorubicin treatment.6 FoxO3 nuclear translocation increases the expression of proapoptotic proteins such as p53-up-regulated modulator of apoptosis (PUMA), Bim, p27, and TNF-related apoptosis inducing ligand (TRAIL).7,8 Additionally, FoxO3 has been confirmed to have direct transcriptional activity on the PUMA promoter and induces the expression of PUMA protein.8 It has been well established that hepatocyte lipoapoptosis is dependent on the activation of JNK but the involvement of p38-MAPK and ERK has been ruled out.4,9 In the present study we tested the activation of all three stress-dependent kinases and their role in cholangiocyte lipoapoptosis.
While hepatocyte lipoapoptosis due to FFAs has been established and implicated in the pathogenesis of NAFLD or NASH,4,9 the occurrence of cholangiocyte lipoapoptosis due to FFAs has not been thoroughly tested. The present study explores cholangiocyte lipoapoptosis using cholangiocyte cell culture models. The data are consistent with saturated FFA-induced cholangiocyte lipoapoptosis by way of activation of FoxO3 and up-regulation of the proapoptotic BH3-containing protein PUMA.
Materials and Methods
Materials
Palmitic acid (#P5585), stearic acid (#S4751), oleic acid (#O1008), and fatty acid-free bovine serum albumin (BSA; #A3803) were obtained from Sigma-Aldrich. The pan-caspase inhibitor Z-VAD-fmk and ERK inhibitor FR180204 were from Santa Cruz Biotechnology and the JNK inhibitor SP600125 was from Calbiochem. Magnetic protein G beads were purchased from New England Bio Labs.
Antibodies
Rabbit antisera against phospho-FoxO3 (Thr32) (#9464), acetylated-lysine (#9441), FoxO3 (#2497), FoxO1 (#2880), phospho-p38-MAPK (#9211), MAPK (#9212), phospho-ERK1/2 (#9101), ERK1/2, phospho-JNK (#9251), JNK (#9252), and p83-MAPK inhibitor SB204580 were from Cell Signaling. Rabbit antiserum against PUMA (#ab54288) was from Abcam. Goat anti-Lamin B (#sc-6216) and rabbit anti-actin (#sc-1615) were purchased from Santa Cruz Biotechnology. Peroxidase-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Cell Lines and Treatment
H69, a human immortalized cholangiocyte cell line and KMCH, Mz-ChA-1, and HuCCT-1, human cholangiocarcinoma cell lines were grown as described.11 BDEneu cells were a kind gift from Dr. Alphonse Sirica (Virginia Common-wealth University) and were grown as described.12 Cells were treated with the indicated concentrations of FFAs (400-800 μM) dissolved in isopropanol and added to media containing 1% fatty acid-free BSA for 24 hours. Vehicle treatment was isopropanol with a final concentration of <1% in the medium.
Measurement of Apoptosis
Percent apoptosis was quantified by characteristic nuclear morphology and visualized by treatment with the fluorescent DNA-binding dye, DAPI (4′, 6-diamidine-2′-phenylindole dihydrochloride) as described before.9 Briefly, cells were stained with 5 μg/mL of DAPI for 20-30 minutes at 37°C. Apoptotic nuclei (condensed, fragmented) were counted and presented as a percent of total nuclei. At least 100 cells were counted per well and experiments were performed in triplicate. Caspase 3/7 activity was measured by enzymatic fluorophore release (Apo-One) according to the manufacturer's instructions (Promega) with experiments performed in quadruplicate.
Oil Red O Staining
Intracellular lipid droplets were stained using oil red O dye. Cells were treated with 600 μM of the indicated FFAs for 24 hours. Cells were washed with phosphate-buffered saline (PBS) and fixed with 10% buffered formalin for 10 minutes. Oil red O (2 mg/mL) staining solution was added for 15 minutes and washed with water. Cells were mounted using Fluoromount G (Electron Microscopy Services). Images were obtained by Olympus 1X71 fluorescent microscopy using the rhodamine channel.
Nuclear Isolation, Immunoblot, and Immunofluorescence
Nuclear extracts were prepared as described.13 Cell lysates containing 30 μg of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a nitrocellulose membrane and visualized by immunoblotting. Mz-ChA-1 cells were labeled with anti-FoxO3 antibody and visualized by Alexafluor 488 secondary staining.
Lentiviral shRNA Transduction
Short hairpin RNA (shRNA) silencing lentiviral pLKO.1-puro vector targeting FoxO3 and PUMA were obtained from Sigma. FoxO3 shRNA#1 and #2 targets the nucleotide sequences 2185-2205 and 1626-1648 of FoxO3 mRNA (NM_001455.1), respectively. PUMA shRNA targets the nucleotide sequence 783-805 of PUMA mRNA (NM_014417.2). Stable transfections were carried out as described.14
Statistics
Statistical analysis was performed using one-way analysis of variance (ANOVA) with Bonferroni posthoc correction, unless indicated otherwise.
Results
FFAs Induced Cholangiocyte Lipoapoptosis
Biochemical characteristics of apoptosis include nuclear morphology changes and activation of the caspase cascade. We tested the effect of 400, 600, and 800 μM FFAs on cholangiocyte lipoapoptosis. These concentrations were chosen based on circulating levels of total FFAs found in NAFLD or NASH patients15 and previous studies.4,9,14 The saturated FFAs palmitate (PA) and stearate (SA) induced cholangiocytes to undergo lipoapoptosis in a concentration-dependent manner (Fig. 1). Both PA and SA resulted in 25%-60% apoptosis in H69 cells (Fig. 1A). The monounsaturated fatty acid, oleate (OA), showed no apoptosis at 400 μM and 600 μM. OA at 800 μM induced minimal apoptosis in H69 cells compared with 800 μM of PA- or SA-treated cells. To confirm the apoptotic nuclear changes, we tested the activation of caspase 3/7 in FFA-treated cells. PA and SA treatment of cholangiocytes resulted in significantly increased caspase 3/7 activity in a concentration-dependent manner. Indeed, 600 μM and 800 μM PA resulted in 4- and 6-fold increased caspase 3/7 activity in H69 cells, whereas 600 μM and 800 μM SA resulted in 5- and 8-fold increased caspase 3/7 activity, respectively (Fig. 1A). H69 cells, upon OA treatment, again showed a significant increase in caspase activity only at 800 μM OA (Fig. 1A). FFA-induced cholangiocyte lipoapoptosis was then tested in additional cholangiocyte-derived cell lines. PA induced an increase in the number of apoptotic nuclei in KMCH cells at 600 μM and 800 μM, whereas SA induced increased lipoapoptosis starting from 400 μM to 800 μM. Increased caspase activity was observed with increasing concentrations of both PA and SA in KMCH cells (Fig. 1B). Treatment of KMCH cells with OA did not induce apoptosis (Fig. 1B). Both Mz-ChA-1 and HuCCT cell lines showed similar FFA-induced apoptosis with increasing concentrations of PA and SA, but not with OA (Fig. 1C,D). Together, these results suggest that saturated FFAs SA and PA induced cholangiocyte lipoapoptosis.
Fig. 1.

FFAs induce cholangiocyte lipoapoptosis. (A) H69 cells were treated with 400, 600, or 800 μM palmitate (PA), stearate (SA), or oleate (OA) for 24 hours. Apoptotic nuclei were counted and expressed as a percent of total nuclei (left panel). H69 cells were treated in parallel as above for 24 hours, followed by quantitation of caspase 3/7 activity (right panel), and results are expressed as fold-change over isopropanol-treated cells (Veh). The same conditions were employed to assess apoptotic nuclei and caspase 3/7 activity in KMCH cells (B), Mz-ChA-1 cells (C), and HuCCT cells (D). Each value represents the mean ± SEM of separate experiments (n = 6). *P < 0.001, compared to vehicle-treated cells. #P < 0.05 compared to vehicle-treated cells.
Caspase-Dependent Cholangiocyte Lipoapoptosis
We next examined whether inhibition of caspases using a cell permeable, pan-caspase inhibitor Z-VAD-fmk would prevent FFA-induced cell death. Addition of Z-VAD-fmk (50 μM) to PA- or SA-treated cells resulted in a significant reduction (although not complete elimination) of apoptotic nuclei in H69, KMCH, Mz-ChA-1, and HuCCT cells (Fig. 2A-D). In parallel, addition of Z-VAD-fmk to PA- or SA-treated cells resulted in complete prevention of caspase 3/7 activity induced by PA and SA treatments (Fig. 2A-D).
Fig. 2.

Caspase-dependent cholangiocyte lipoapoptosis. (A) H69 cells were treated for 24 hours with 800μM palmitate (PA) or stearate (SA), and 50 μM caspase inhibitor, Z-VAD-fmk (ZVAD), or dimethyl sulfoxide (DMSO) as a control for the measurement of apoptotic nuclei. Apoptotic nuclei were counted and expressed as a percent of total nuclei, at least 100 cells per replicate were counted (left panel). H69 cells were treated in parallel as above for 24 hours, followed by quantitation of caspase 3/7 activity (right panel). Results are expressed as fold change of caspase 3/7 activity compared to vehicle treatment (Veh). The same conditions were employed to apoptotic nuclei and caspase 3/7 activity in KMCH cells (B), Mz-ChA-1 cells (C), and HuCCT cells (D). (E) BDEneu cells were treated with 600 μM palmitate (PA) or PA plus 50 μM caspase inhibitor, Z-VAD-fmk (ZVAD) for 24 hours, followed by quantitation of apoptotic nuclei (left panel) and caspase 3/7 activity (right panel) as above. Each value represents mean ± SEM of separate experiments (n = 6). *P < 0.001, compared to vehicle, #P < 0.001, compared to 800 μM PA- or SA-treated cells.
To check the generality of cholangiocyte lipoapoptosis across species, we tested BDEneu cells, an immortalized tumorigenic rat cholangiocyte which has constitutive overexpression of the rat neu/her2 oncogene.12 BDEneu cells treated with vehicle showed 2.39 ± 6 0.8% apoptotic nuclei and 600 μM PA resulted in significantly increased levels of apoptotic nuclei to 35 ± 2.2% (Fig. 2E). Treatment with Z-VAD-fmk plus PA significantly decreased the percentage of apoptotic nuclei to 16 ± 0.7. In addition, BDEneu cells showed an increase in caspase 3/7 activity with 600 μM of PA which was blocked by Z-VAD-fmk (Fig. 2E).
FFAs Induced p38-MAPK, ERK1/2, and JNK Activation
Activation of MAPKs in patients with NAFLD has been reported.16 Here we tested whether activation of MAPKs was involved in cholangiocyte lipoapoptosis. Indeed, PA treatment resulted in activation of p38-MAPK, ERK, and JNK by way of phosphorylation. Increased levels of phospho-p38-MAPK were apparent after 1 hour of PA and remained elevated until after 24 hours of PA treatment (Fig. 3A). KMCH cells have some constitutively phosphorylated ERK, as seen in vehicle-treated cells, and 2 hours of PA treatment resulted in further activation of ERK. The increase in phospho-ERK levels with PA was time-dependent with peak activation time between 4-8 hours (Fig. 3A). Similarly, JNK is also activated by phosphorylation after 1 hour of PA treatment as compared to vehicle-treated KMCH cells. Phospho-JNK levels stayed elevated up to 16 hours of PA treatment, with peak activation observed at 8 hours of PA (Fig. 3A). PA-induced activation of p38-MAPK, ERK, and JNK was also evident in another cholangiocyte, Mz-ChA-1 cells. Here, PA-treated cells showed activation of p38-MAPK and ERK after 1.5 hours of treatment as compared with vehicle-treated cells. Both phospho-p38-MAPK and phospho-ERK levels stayed elevated until after 6 hours of PA-treatment and returned to control levels after 8 hours of PA treatment (Fig. 3B). Phospho-JNK levels were also increased after 1.5 hours of PA treatment and stayed elevated until after 3 hours in PA-treated Mz-ChA-1 cells (Fig. 3B). While all three MAPKs were detectable, total levels of each varied between cell lines (Fig. 3C).
Fig. 3.


FFAs induced p38-MAPK, ERK1/2, and JNK activation. (A) Cell lysates were prepared from KMCH cells treated with either 800 μM palmitate (PA) or vehicle (V) for different timepoints as indicated. Immunoblot analysis was performed for phospho p38-MAPK (P-p38), phospho-ERK (P-ERK), and phospho-JNK (P-JNK), and compared with total p38-MAPK (T-p38), total ERK (T-ERK), and total JNK (T-JNK), respectively. (B) Mz-ChA-1 cells were treated with either 800 μM PA (PA) or vehicle (V) at different timepoints. Immunoblot analysis was performed in Mz-ChA-1 cells for phosphorylated proteins and compared with total p38, ERK, and JNK, respectively. (C) Cell lysates were prepared from H69, Mz-ChA-1, KMCH, HuCCT, and BDEneu cells to show the expression levels of T-p38, T-ERK, T-JNK, FoxO3, PUMA, and actin. The images shown here are representative images. (D) H69, KMCH, Mz-ChA-1, HuCCT, and BDEneu cells were treated with vehicle (Veh) or 800 μM palmitate (PA), with or without 20 μM SB204580 (p38-MAPK inhibitor) for 24 hours, followed by caspase 3/7 activity and the results are expressed as fold change compared with vehicle-treated cells. (E) H69, KMCH, Mz-ChA-1, HuCCT, and BDEneu cells were treated with vehicle (Veh) or 800 μM palmitate (PA), with or without 30 μM FR180204 (selective ERK1/2 inhibitor) for 24 hours, followed by caspase 3/7 activity. (F) KMCH cells were treated with vehicle (Veh) or 800 μM palmitate (PA), with or without 50 μM SP600125 for 24 hours, followed by quantitation of apoptotic nuclei. Apoptotic nuclei were counted and expressed as a percent of total nuclei. (G) KMCH and Mz-ChA-1 cells were treated with vehicle (Veh) or 800 μM palmitate (PA), with or without 50 μM SP600125 (JNK inhibitor) for 24 hours, followed by caspase 3/7 activity. Vehicle was isopropanol (Veh <1% final) and control for SB204580, FR180204, and SP600125 was DMSO (<1% final). Each value represents the mean ± SEM of separate experiments (n = 6). *P < 0.001, compared to vehicle treated cells and n.s., nonsignificant.
We next tested whether inhibition of MAP kinases using small molecule inhibitors would prevent PA-induced cholangiocyte lipoapoptosis. First, treatment with SB204580 (20 μM), p38 inhibitor showed partial inhibition of PA-induced caspase activation in KMCH, Mz-ChA-1, HuCCT, and BDEneu cells but not in H69 cells (Fig. 3D). Treatment with FR180204 (30 μM), an ATP-binding pocket selective inhibitor of ERK1/2, also partially prevented PA-induced caspase activation in Mz-ChA-1, HuCCT, and BDEneu cells. FR180204 (30 μM) did not protect against PA-induced caspase activity in H69 and KMCH cells (Fig. 3E). SP600125, an inhibitor of JNK, did not alter cholangiocyte lipoapoptosis caused by PA in KMCH cells (Fig. 3F) or Mz-ChA-1 cells (Fig. 3G). These results suggest that FFAs induced activation of stress kinases p38-MAPK, ERK, and JNK and that inhibition of p38-MAPK and ERK altered cholangiocyte lipoapoptosis but inhibition of JNK did not alter cholangiocyte lipoapoptosis.
Palmitate Induced FoxO3 Nuclear Localization
We next tested whether FoxO transcription factors (FoxO1 and FoxO3) were involved in palmitate-induced cholangiocyte lipoapoptosis. Nuclear extracts were isolated to analyze FoxO levels in KMCH cells treated with 800 μM of PA. Increased FoxO3 was observed in the nucleus after 3 hours of PA as compared with vehicle-treated cells and nuclear FoxO3 levels stayed elevated until after 24 hours. Lamin B was used as a loading control (Fig. 4A). Nuclear levels of FoxO3 were also increased in H69 and Mz-ChA-1 cells treated with PA for 16 hours (Fig. 4B). FoxO1 expression in the nuclear extracts was high in H69 cells. However, FoxO1 levels were not altered with PA treatment in either H69 or Mz-ChA-1 cells (Fig. 4B). The levels of total FoxO3 in all the cholangiocytes were comparable (Fig. 3C). To check whether nuclear levels of FoxO3 were similar in both SA- and PA-treated cells, KMCH, H69, and HuCCT cells were treated with 800 μM PA or SA for 16 hours and nuclear extracts were prepared. Both PA and SA treatment resulted in similar increases in the nuclear levels of FoxO3 as compared with lamin B (Fig. 4C). Immunofluorescent analysis for FoxO3 in Mz-ChA-1 cells demonstrated clear nuclear localization after 16 hours of PA treatment as compared to the cytosolic staining observed in vehicle-treated cells (Fig. 4D). Quantitation of nuclear FoxO3 levels showed a 7-fold increase after 16 hours of PA treatment relative to vehicle-treated cells (Fig. 4E). To test posttranslational modifications, FoxO3 was immunoprecipitated from nuclear extracts and probed using an anti-acetyl-lysine antibody. We observed a dramatic decrease in levels of acetylated FoxO3 6 hours after treatment (Fig. 4F). At the same time, FoxO3 was also observed to be unphosphorylated (Fig. 4F). At a later timepoint, PA treatment of HuCCT and H69 cells resulted in an increase in the nuclear levels of acetylated FoxO3 and phosphorylated FoxO3 (Fig. 4G). These results suggest that FoxO3 was deacetylated and dephosphorylated at early timepoints, when it first translocated to the nucleus, and at the later timepoints FoxO3 is modified by way of acetylation and phosphorylation.
Fig. 4.


Palmitate induced FoxO3 nuclear localization. (A) Nuclear extracts were prepared from KMCH cells treated with either 800 μM palmitate (PA) or vehicle (Veh) for the indicated times. Immunoblot analysis was performed for FoxO3 and Lamin B was used as a loading control. Numbers below indicate the quantitation of relative band intensity, presented as the ratio of FoxO3 to Lamin B. (B) H69 and Mz-ChA-1 cells were treated with 800 μM palmitate (PA) or vehicle (V) for 16 hours. Nuclear extracts were analyzed for FoxO3, FoxO1, and Lamin B. (C) Nuclear extracts were prepared from KMCH, H69, and HuCCT cells treated with either vehicle (Veh), 800 μM stearate (SA), or 800 μM palmitate (PA) for 16 hours. Immunoblot analysis was performed for FoxO3 and Lamin B. Numbers below indicate the quantitation of relative band intensity, presented as the ratio of FoxO3 to Lamin B. (D) Immunofluorescence analysis of FoxO3 nuclear localization after 16 hours of 800 μM palmitate (PA) or vehicle treatment in Mz-ChA-1 cells and nuclei were counterstained with DAPI. (E) Quantified levels of nuclear FoxO3 after 16 hours of 800 μM palmitate (PA) or vehicle treatment in Mz-ChA-1 cells. Relative fluorescent intensity was quantified using ImageJ software. At least 30 cells per treatment were analyzed. *P< 0.001 compared to vehicle-treated cells, Student t test. (F) Immunoprecipitation of total FoxO3 from nuclear extracts of KMCH cells treated with palmitate (PA) or vehicle (V) for 6 hours. Immunoblot analysis was performed for acetylated FoxO3, phospho-FoxO3, and total FoxO3. Heavy chain of the IP antibody is indicated as IgG. (G) Immunoprecipitation of total FoxO3 from nuclear extracts of HuCCT, and H69 cells treated with 800 lM palmitate (PA) or vehicle (V) for 16 hours. Immunoblot analysis was performed for acetylated FoxO3, phospho-FoxO3, and total FoxO3. The images shown here are representative images.
FoxO3 Is Critical for Cholangiocyte Lipoapoptosis
FoxO3 shRNA transduction resulted in complete knockdown in H69 (Fig. 5A) and partial knockdown in KMCH cells (Fig. 5B). Control shRNA and FoxO3 shRNA-treated cells were then challenged with 800 μM of PA or SA for cholangiocyte lipoapoptosis. H69 cells transduced with FoxO3 shRNA showed dramatic protection against PA- or SA-induced apoptosis, measured by apoptotic nuclei or caspase 3/7 activity as compared with control shRNA-treated cells (Fig. 5C). Similarly, KMCH cells transduced with FoxO3 shRNA showed significant protection against PA- or SA-induced cholangiocyte lipoapoptosis as compared to control shRNA treated cells (Fig. 5D). Together, these results confirm that FoxO3 is critical for FFA-induced cholangiocyte lipoapotosis.
Fig. 5.
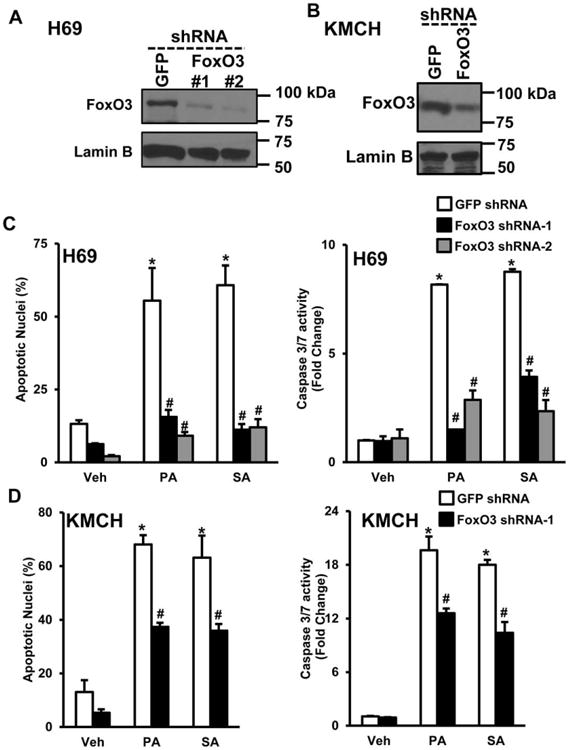
FoxO3 is critical for cholangiocyte lipoapoptosis. (A) H69 cells transduced with either control shRNA (GFP) or FoxO3 shRNA (#1 and #2). Immunoblot analysis of nuclear FoxO3 in H69 cells were performed. (B) Immunoblot analysis of FoxO3 in KMCH cells transduced with either control shRNA (GFP shRNA) or FoxO3 shRNA (#1). Lamin B was used as a loading control. (C) Control shRNA or FoxO3 shRNA transduced stable H69 cells were treated with either vehicle (Veh), 800 μM palmitate (PA), or 800 μM stearate (SA) for 24 hours. Apoptotic nuclei were counted and expressed as a percent of total nuclei (left panel). H69 cells were treated in parallel as above for 24 hours, followed by quantitation of caspase 3/7 activity (right panel), and results are expressed as fold-change over isopropanol-treated cells (Veh). The same conditions were employed to assess apoptotic nuclei and caspase 3/7 activity in KMCH cells (D). Each value represents the mean ± SEM of separate experiments (n = 6). *P < 0.001, compared to vehicle-treated cells and #P < 0.001, compared to PA- or SA-treated GFP shRNA control cells.
Palmitate Increases PUMA Protein Expression in Cholangiocytes
PUMA is a downstream target of FoxO3,8 thus we assessed the expression of PUMA protein in KMCH cells at various timepoints after treatment with 800 μM PA. PA-treated KMCH cells showed elevated levels of PUMA after 2 hours as compared to vehicle-treated cells and increased PUMA levels were sustained even after 48 hours (Fig. 6A). Similar to KMCH cells, the levels of PUMA were also found to be increased by both SA and PA treatment in H69, Mz-ChA-1, and HuCCT cells (Fig. 6B). To further validate the significance of PUMA in cholangiocyte lipoapoptosis, we transduced control or PUMA shRNA in H69 and HuCCT cells and confirmed the knockdown of PUMA protein expression (Fig. 6C,D). PUMA knockdown resulted in significant protection against 800 μM PA or SA treatment (Fig. 6C,D). Thus, the FoxO3 downstream target, PUMA is also critical in FFA-induced cholangiocyte lipoapoptosis.
Fig. 6.

Palmitate increased PUMA protein expression in cholangiocytes. (A) KMCH cells were treated with 800 μM palmitate (PA) or vehicle (Veh). Cell lysates were collected at different timepoints after PA treatment, as indicated. Immunoblot analysis was performed for PUMA and actin. (B) H69, Mz-ChA-1, and HuCCT cells were treated with either 800 μM of stearate (SA), palmitate (PA), or vehicle isopropanol (Veh). Cell lysates were prepared after 16 hours of SA or PA treatment for the immunoblot analysis of PUMA and actin was used as a loading control. Numbers below indicate the quantitation of relative band intensity, presented as the ratio of PUMA to actin. (C) Immunoblot analysis of PUMA were performed in stable H69 cells transduced with either control shRNA (GFP) or PUMA shRNA (left panel). Control shRNA or PUMA shRNA transduced H69 cells were treated with vehicle (Veh), 800 μM palmitate (PA), or 800 μM stearate (SA) for 24 hours, followed by quantitation of caspase 3/7 activity (right panel) and results are expressed as fold change. (D) Immunoblot analysis of PUMA in stable HuCCT cells transduced with control shRNA (GFP) or PUMA shRNA were performed (left panel). Control shRNA and PUMA shRNA stable HuCCT cells were treated with vehicle (Veh), 800 μM palmitate (PA), or 800 μM stearate (SA) for 24 hours, followed by quantitation of caspase 3/7 activity (right panel). Each value represents the mean ± SEM of separate experiments (n = 6). *P < 0.001, compared to vehicle-treated cells and #P < 0.001, compared to PA- or SA-treated cells.
Cholangiocytes Did Not Develop Steatosis With FFA Treatment
Treatment of hepatocytes with FFAs was previously shown to increase lipid storage as triglycerides in cytoplasmic lipid droplets.4 We tested cholangiocytes such as H69, Mz-ChA-1, KMCH, and hepatoma cells, Huh7 for lipid droplet formation after treatment with 600 μM PA, SA, or OA by staining with oil red O dye. H69 cells showed a background of minimal lipid droplet staining and this did not increase with PA or SA treatment, whereas OA-treated cells showed a slight increase in lipid droplet formation (Fig. 7). Mz-ChA-1 and KMCH cells did not show accumulation of lipid droplets with 600 μM of PA, SA, or OA after 24 hours. In contrast to cholangiocytes, Huh7 hepatoma cells showed a dramatic increase in the accumulation of lipid droplets with all of the FFAs. These results suggest that cholangiocytes do not develop steatosis, in contrast to hepatocytes.
Fig. 7.

Cholangiocytes do not develop steatosis with saturated FFA treatment. H69, Mz-ChA-1, KMCH cholangiocytes, or Huh7 hepatoma cells were treated with 600 μM palmitate (PA), stearate (SA), or oleate (OA) for 24 hours. Vehicle-treated cells were used as control (Veh). Red fluorescence was captured and images are displayed in grayscale. The images shown here are representative images and all micrographs were taken at the same magnification (scale bar = 25 μm).
Discussion
Circulating saturated FFAs are reported to be elevated in patients with metabolic syndrome as well as in patients with NAFLD or NASH.15 The principal finding of this study is that FFAs can induce biliary epithelial cell lipoapoptosis. Our data suggest four important findings in biliary cell damage induced by FFAs, termed cholangiocyte lipoapoptosis: 1) FFAs induced caspase activation and cholangiocyte apoptosis; 2) cholangiocyte lipoapoptosis was associated with the activation of FoxO3, a transcription factor known to induce apoptosis; 3) cholangiocyte lipoapoptosis appears to involve the activation of PUMA, a proapoptotic BH3-containing protein; and 4) unlike hepatocytes, cholangiocytes did not accumulate lipid droplets. The schematic representation of FFA-induced cholangiocyte lipoapoptosis is shown in Fig. 8. These results will be discussed below.
Fig. 8.

Schematic representation of FFA-induced cholangiocyte lipoapoptosis. FFAs resulted in the activation of p38-MAPK (p38) and ERK by way of phosphorylation. FFAs also induced the nuclear accumulation of FoxO3, a forkhead family member. Posttranslational modification of FoxO3 can further promote its nuclear localization for the induction of lipoapoptosis and this in part can be regulated by the activation of p38 and ERK. Increased nuclear FoxO3 levels potentially resulted in up-regulation of PUMA, a proapoptotic BH3-containing protein. Increased PUMA levels can trigger the activation caspases culminating in FFA-induced cholangiocyte lipoapoptosis.
Are cholangiocyte cell culture models a clinically relevant model to study biliary epithelial cell lipoapoptosis? In the present study we used the normal immortalized cholangiocyte cell line, H69, and cholangiocarcinoma-derived cell lines, KMCH, Mz-ChA-1, HuCCT, and BDEneu. H69 cells have been shown to share some characteristics with human primary cholangiocytes. H69 cells can produce primary cilia from the apical plasma membrane during in vitro culture conditions.17 In addition, H69 cells contain Cl-/HCO3- anion exchanger 2 (AE2) mRNA and protein and were shown to have AE2-mediated hydrocholeretic function similar to human cholangiocytes in 3D cultures.18 Proteomic studies from H69 cells have revealed the expression of Annexin A2, a Ca2+- and acidic phospholipid-binding protein and a marker for normal human cholangiocytes.19 This proteomic study also identified the expression of AE2 and several markers of cholangiocytes in H69 cells.19 Furthermore, mRNA and protein levels of G-protein-coupled receptor 55 were found to be similar in H69, Mz-ChA-1, HuCCT cells and human intrahepatic biliary epithelial cells.20 Both normal cholangiocytes and cholangiocarcinoma-derived cell lines used in this study were shown to express keratin 7, keratin 19, and gamma-glutamyl transpeptidase activity, similar to cholangiocytes in vivo and primary human cholangiocytes.21 Thus, the cell lines employed in the current study share phenotypic features with biliary epithelial cells and are a useful culture model of cholangiocytes, although we recognize that cultured cells may not fully recapitulate cholangiocyte physiology.
Saturated FFAs, like PA and SA, have been shown to cause lipoapoptosis due to the activation of caspases in pancreatic β-cells,10 hepatocytes,4,9 and other cell types. Hepatocytes and cholangiocytes have a common precursor cell, termed the bipotential hepatic progenitor cell.22 In the present study we report lipotoxicity due to saturated FFAs in biliary epithelial cells. This is a caspase-dependent process, consistent with previous studies in other cell types. Treatment with a pan-caspase inhibitor protected against cholangiocyte lipoapoptosis caused by saturated FFAs. Biliary injury has been reported in a subset of patients with NAFLD compared to a population with similar steatosis.22 Our data suggest that in patients with metabolic syndrome, higher circulating FFAs may contribute to biliary tree damage. Further studies will be needed to determine if cholangiocyte lipoapoptosis contributes to the progression of disease from simple steatosis to NASH to liver fibrosis, at least in a subset of patients.
JNK is one of the major cellular stress signaling kinases that is activated during FFA-induced apoptosis. In particular, hepatocyte lipoapoptosis is dependent on JNK activation.4,9 However, we found that inhibition of JNK using a small molecule inhibitor did not prevent cholangiocyte lipoapoptosis, suggesting a mechanism of apoptosis beyond JNK activation. A recent study in pancreatic β-cells showed that FFA-induced JNK activation is not critical for apoptosis, but JNK activation was involved in mediating ER-stress signaling pathways.23 Several studies have also demonstrated that p38-MAPK and ERK can also induce apoptosis in pancreatic β-cells,24,25 but the activation of p38-MAPK and ERK has been shown not to be critical for FFA-induced hepatocyte lipoapoptosis.4 ERK was described as a negative regulator of FoxO3 nuclear accumulation, and phosphorylation of FoxO3 by ERK was shown to increase its interaction with MDM-2, resulting in ubiquination and degradation of FoxO3.26 In the present study we observed a partial inhibition of cholangiocyte lipoapoptosis with an ERK-selective inhibitor, FR180204. However, FR180204 can also inhibit p38α at 30 μM, a dose similar to the present study.27 Inhibition of p38 by SB204580 caused partial protection of cholangiocytes, thus it is still not clear whether FR180204 treatment resulted in partial protection by way of inhibiting p38α. Further work is needed to elucidate this mechanism.
Our next focus was on FoxO3. FoxO3 has been shown to activate cell death pathways by inducing the expression of proapoptotic proteins such as PUMA, Bim, p27, and TRAIL.7,8 FoxO3 can also be regulated by the Akt signaling pathway, protein phosphatases, and protein deacetylases (Sirt1).29,30 Phosphorylation and acetylation of FoxO3 has been shown to alter cell death pathways towards cell survival by promoting export of FoxO3 from the nucleus to the cytoplasm.29 On the other hand, deacetylation of FoxO3 by Sirt1 in the nucleus decreased the proapoptotic transcriptional program.30 In the present study, deacetylation and dephosphorylation of nuclear FoxO3 were observed at the early timepoints after PA treatment in KMCH cells. However, after 16 hours of PA treatment the levels of acetylated nuclear FoxO3 and phosphorylated nuclear FoxO3 were increased, despite reports that phosphorylation promotes nuclear export. We conclude that in PA-treated cholangiocytes, phosphorylation of FoxO3 at Thr32 is not sufficient to cause nuclear export and FoxO3 inactivation. Previously, PA has been shown to decrease the expression of Sirt1 in pancreatic β-cells,10 and overexpression of Sirt1 protected against FFA-induced pancreatic β-cell apoptosis.31 Consistently, acetylated FoxO1 has been shown to induce Bim expression and promote apoptosis.32
As noted, PUMA is important in lipoapoptosis, and is a downstream target of FoxO3.8 Expression of PUMA was found to be up-regulated in human liver biopsies taken from NASH patients as compared to control subjects.9 In our experiments, PUMA expression was up-regulated by FFAs in cholangiocytes. Our time-course of PUMA expression revealed a rapid increase in PUMA protein that is sustained through 48 hours. Knockdown of PUMA demonstrated its critical role in FFA-induced cholangiocyte lipoapoptosis.
Furthermore, we measured cellular steatosis, i.e., accumulation of triglycerides in lipid droplets, which has been reported in many nonadipose tissues including pancreatic β-cells34 and hepatocytes.4 In the liver, there is controversy over whether lipid droplets in hepatocytes treated with FFAs promote lipotoxicity or serve as a protective mechanism for detoxifying FFAs.34-37 In the present study, FFA treatment of cholangiocytes did not induce cellular steatosis. These results are consistent with the clinical observation of hepatocyte lipid accumulation without biliary cell steatosis in NAFLD.38-40 Together, cholangiocytes undergo lipoapoptosis with FFA treatment, but unlike hepatocytes, cholangiocytes did not accumulate lipid droplets with FFA treatment.
In conclusion, our study shows that saturated FFAs induced cholangiocyte lipoapoptosis by way of caspase activation, nuclear translocation of FoxO3, and proapoptotic PUMA expression. Posttranslational modifications of FoxO3 may be involved in cholangiocyte lipoapoptosis. Further studies are required to elucidate the contribution of cholangiocyte lipoapoptosis to NAFLD or NASH in patients with metabolic syndrome.
Acknowledgments
We thank Crystal Cordes and John Davis for help with oil red O staining. We thank Carol Casey for advice and many helpful discussions.
Supported by the Fred and Pamela Buffett Cancer Center and the College of Medicine Student Summer Research Program, University of Nebraska Medical Center. The contents of the article are solely the responsibility of the authors.
Abbreviations
- BSA
bovine serum albumin
- DAPI
4′-6-diamidino-2-phenylindole
- FBS
fetal bovine serum
- FFA
free fatty acids
- FoxO
forkhead family of transcription factor
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- PUMA
p53-up-regulated modulator of apoptosis
- TRAIL
TNF-related apoptosis inducing ligand
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr. 2011;53:131–140. doi: 10.1097/MPG.0b013e31822578db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorrentino P, Tarantino G, Perrella A, Micheli P, Perrella O, Conca P. A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig Dis Sci. 2005;50:1130–1135. doi: 10.1007/s10620-005-2719-1. [DOI] [PubMed] [Google Scholar]
- 3.Chiba M, Sasaki M, Kitamura S, Ikeda H, Sato Y, Nakanuma Y. Participation of bile ductular cells in the pathological progression of nonalcoholic fatty liver disease. J Clin Pathol. 2011;64:564–570. doi: 10.1136/jcp.2011.090175. [DOI] [PubMed] [Google Scholar]
- 4.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 5.Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E, et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J Biol Chem. 2012;287:1545–1555. doi: 10.1074/jbc.M111.284224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282:27141–27154. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- 8.Amente S, Zhang J, Lavadera ML, Lania L, Avvedimento EV, Majello B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011;39:9498–9507. doi: 10.1093/nar/gkr638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu L, Zhou L, Lu Y, Zhang J, Jian F, Liu Y, et al. Activation of SIRT1 protects pancreatic beta-cells against palmitate-induced dysfunction. Biochim Biophys Acta. 2012;1822:1815–1825. doi: 10.1016/j.bbadis.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, et al. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J Hepatol. 2014;60:599–605. doi: 10.1016/j.jhep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 13.Sadowski HB, Gilman MZ. Cell-free activation of a DNA-binding protein by epidermal growth factor. Nature. 1993;362:79–83. doi: 10.1038/362079a0. [DOI] [PubMed] [Google Scholar]
- 14.Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011;286:39336–39348. doi: 10.1074/jbc.M111.280420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci. 2001;46:2347–2352. doi: 10.1023/a:1012338828418. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira DM, Castro RE, Machado MV, Evangelista T, Silvestre A, Costa A, et al. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia. 2011;54:1788–1798. doi: 10.1007/s00125-011-2130-8. [DOI] [PubMed] [Google Scholar]
- 17.Masyuk AI, Huang BQ, Radtke BN, Gajdos GB, Splinter PL, Masyuk TV, et al. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1013–1024. doi: 10.1152/ajpgi.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banales JM, Saez E, Uriz M, Sarvide S, Urribarri AD, Splinter P, et al. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology. 2012;56:687–697. doi: 10.1002/hep.25691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kido O, Fukushima K, Ueno Y, Inoue J, Jefferson DM, Shimosegawa T. Compensatory role of inducible annexin A2 for impaired biliary epithelial anion-exchange activity of inflammatory cholangiopathy. Lab Invest. 2009;89:1374–1386. doi: 10.1038/labinvest.2009.105. [DOI] [PubMed] [Google Scholar]
- 20.Huang L, Ramirez JC, Frampton GA, Golden LE, Quinn MA, Pae HY, et al. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab Invest. 2011;91:1007–1017. doi: 10.1038/labinvest.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphreys EH, Williams KT, Adams DH, Afford SC. Primary and malignant cholangiocytes undergo CD40 mediated Fas dependent apoptosis, but are insensitive to direct activation with exogenous Fas ligand. PLoS One. 2010;5:e14037. doi: 10.1371/journal.pone.0014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54:122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 23.Nemcova-Furstova V, Balusikova K, Sramek J, James RF, Kovar J. Cas-pase-2 and JNK activated by saturated fatty acids are not involved in apoptosis induction but modulate ER stress in human pancreatic beta-cells. Cell Physiol Biochem. 2013;31:277–289. doi: 10.1159/000343367. [DOI] [PubMed] [Google Scholar]
- 24.Yuan H, Zhang X, Huang X, Lu Y, Tang W, Man Y, et al. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS One. 2010;5:e15726. doi: 10.1371/journal.pone.0015726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K, Park M, Young Kim H. Ginsenoside Rg3 suppresses palmitate-induced apoptosis in MIN6N8 pancreatic beta-cells. J Clin Biochem Nutr. 2010;46:30–35. doi: 10.3164/jcbn.09-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 28.Natarajan SK, Becker DF. Role of apoptosis-inducing factor, proline dehydrogenase, and NADPH oxidase in apoptosis and oxidative stress. Cell Health Cytoskelet. 2012;2012:11–27. doi: 10.2147/CHC.S4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Natarajan SK, Zhu W, Liang X, Zhang L, Demers AJ, Zimmerman MC, et al. Proline dehydrogenase is essential for proline protection against hydrogen peroxide-induced cell death. Free Radic Biol Med. 2012;53:1181–1191. doi: 10.1016/j.freeradbiomed.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 31.Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, et al. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009;58:344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313–324. doi: 10.1593/neo.81358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong ML, Ding XZ, Adrian TE. Red oil A5 inhibits proliferation and induces apoptosis in pancreatic cancer cells. World J Gastroenterol. 2004;10:105–111. doi: 10.3748/wjg.v10.i1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 38.Contos MJ, Choudhury J, Mills AS, Sanyal AJ. The histologic spectrum of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:481–500, vii. doi: 10.1016/j.cld.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 39.Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37:1286–1292. doi: 10.1053/jhep.2003.50229. [DOI] [PubMed] [Google Scholar]
- 40.Xanthakos S, Miles L, Bucuvalas J, Daniels S, Garcia V, Inge T. Histo-logic spectrum of nonalcoholic fatty liver disease in morbidly obese adolescents. Clin Gastroenterol Hepatol. 2006;4:226–232. doi: 10.1016/s1542-3565(05)00978-x. [DOI] [PubMed] [Google Scholar]