

Abstract
Background:
Gliosarcoma (GS), a subtype of glioblastoma (GBM), is a rare primary neoplasm of the central nervous system. Certain features like temporal lobe affinity, tendency for extraneural metastasis and poorer outcome compared to GBM indicate that GS may indeed be a separate clinicopathologic entity. This led us to revisit this entity in our settings.
Materials and Methods:
Between 2009 and 2014, 16 cases of histologically proven GSs (14 primary, two secondary) were treated. Patient data were retrieved retrospectively. Statistical analysis was performed with? Statistical Package for Social Sciences, version 17.0. (Chicago, Illinois, USA). Survival was analyzed by Kaplan–Meier method.
Results:
GS predominantly affected males in their fifth decade of life. Raised intracranial pressure was the most common mode of clinical presentation. Temporal lobe was the most commonly affected part of the brain and majority of primary and all of secondary GBM were located peripherally. In 7 (43.8%) patients, tumor was radiologically well-demarcated and enhanced strongly and homogenously on contrast as compared to 9 (56.2%) patients where the tumor was ill-defined and showed heterogenous patchy or ring enhancement. Extent of excision was total in seven patients (43.8%), near total in 4 (25%) and subtotal in five patients (31.2%). Median survival was 6 months. Patients with well-demarcated, enhancing mass on imaging intraoperatively had firm tumors with a good plane of cleavage and had a better survival (8 months) compared to those in whom the tumor radiologically and intraoperatively mimicked GBM (2 months).
Conclusion:
GS is associated with poor survival (median survival 6 months). Radiological and intraoperative findings help categorize these tumors into GBM like GS and meningioma like GS. While the former histologically mimics GBM and has very poor survival (2 months), GS with meningioma like feature tends to have better survival (8 months).
Keywords: Chemotherapy, glioblastoma, gliosarcoma, immunohistochemistry, pathology, surgery, survival
Introduction
Gliosarcoma (GS), a variant of glioblastoma (GBM), is a rare primary malignant neoplasm of the central nervous system. GS characteristically displays bimorphic histopathological architecture consisting of both gliomatous (WHO grade 4) and sarcomatous components.[1,2,3,4] This tumor was first reported by Strobe in 1895. But it was Feigen and Gross (1955) who first described these tumors in details.[1,2] They share genetic, clinical, and prognostic similarities with GBM. Hence, it is not unusual that they are treated in a fashion similar to the GBM.[4,5] However, there are certain features which point toward a distinct clinicopathological behavior of GS namely peripheral location on cerebral lobes, tendency for dural attachment, resemblance to meningiomas, propensity to produce intra or extra cranial metastasis and a purportedly poor survival vis-à-vis GBM.[6] Due to the rarity of the disease and resultant lack of large prospective studies, the literature on GS is composed of smaller series with often conflicting findings, particularly related to the role of chemotherapy and survival outcome. In this paper, we have tried to analyze the clinical, radiological, histopathological, and treatment-related aspects of GS treated at our settings in the light of the existing literature.
Materials and Methods
This study included 16 consecutive patients (mean age: 45.2 years [range 17–70 years], M: F =7:1) of histopathologically proven GS affecting brain, operated at our institute over 5-year period (2009–2014). We analyzed both primary (n = 14) and secondary GS (n = 2). Patients operated elsewhere and referred to us for re-surgery or adjuvant therapy were excluded.
These patients were admitted and evaluated clinically. Apart from noting the duration of symptoms and symptomatology, performance status was determined using Karnofsky performance status (KPS) scale. They were radiologically investigated with contrast-enhanced computed tomography or magnetic resonance imaging scan of head or both. Radiological features noted were site, size, location, extent, and signal characteristics. Enhancement, calcification, edema, cystic changes, and dural/ependymal attachments were also looked for. All patients were operated with the intent of maximal resection. Extent of excision was labeled as complete if no residual enhancing tumor was seen in postoperative scan, near total if more than 90% tumor was removed, subtotal if more than 50% and <90% excision was done. Postoperative complications were noted. All patients were advised adjuvant chemo-radiotherapy. We divided the patients into two groups based on preoperative radiological characteristics and intraoperative findings. Patients in Group A (GS with meningioma like appearance) had radiologically well-circumscribed masses that showed more or less homogenous albeit strong contrast enhancement (with or without dural tail) and were firm, well-demarcated from surrounding brain at surgery. While Group B (GS with GBMs like appearance) comprised patients with heterogenous and ill-defined lesion with patchy/ring enhancement on imaging and soft, poorly demarcated tumor margin found out at the time of surgery. Histopathological examination was done with hematoxylin and eosin staining both at low and high power magnification as well as immunohistochemistry with markers like glial fibrillary acidic protein (GFAP), reticulin, vimentin, etc.
Patients were actively followed up, and their survival status was determined. For those who died, time of death since surgery was determined. Statistical analysis was performed using Statistical Package for Social Sciences, version 17.0, (Chicago, Illinois, USA) and survival was determined using Kaplan–Meier log-rank test.
Results
A total of 16 patients were treated. 14 patients (87.5%) had primary GS whereas two patients (12.5%) had secondary GS. During the same period of study, 290 patients of GBM were treated at our center. Hence, GS consisted 5.2% of GBM in our experience. The mean age of the patients with GS was 45.2 years (range 17–70 years) compared to 50.3 years (12–78 years) in GBM. There was a strong male predilection with 14 males as compared to 2 females (M: F-7:1). The sex ratio in GBM was 2.8:1. The mean duration of symptoms was 8.5 months (range 3 days to 36 months).
Majority of the patients had features of raised intracranial pressure (n = 11, 69%) and 3 (20.5%) patients had presented in an obtunded state requiring urgent surgery. 5 (31%) patients had a history of one or more episodes of seizures. One of the patients presented with symptomatic intracranial bleed. Angiogram was negative and hence the patient was followed up. But the patient presented later with features of progressive headache and hemiparesis. Neurological deficits were mostly in the form of hemiparesis (n = 10, 62.5%), aphasia (n = 3, 18.8%) and impaired vision/visual field cut (n = 3, 18.8%). Mean KPS was 65 (range 10–100). 9 (66.7%) patients had KPS score 70 or more while 7 (33.3%) patients had KPS <70. Table 1 depicts the demographic and clinical features of the patients in the current study.
Table 1.
The demographic, clinical, and radiologic characteristics of GS

Majority of the patients had involvement of one lobe only (n = 12, 75%) while remaining patients had tumor extending into adjoining lobe (two lobe involvement) (n = 4, 25%). Temporal lobe was the most common site of involvement (n = 8, 50%) followed by frontal and parietal lobes (n = 2, 12.5% each). Among multilobular tumors, two patients had tumor located at fronto parietal lobe, one patient each had tumor located at the peritrigonal region (parieto-temporo-occipital junction) and anterior corpus callosum with bifrontal extension (6.2%). There were 14 (87.5%) peripheral tumors while two patients had deep tumors (12.5%). Both secondary GS was superficially located (100%) over the cerebral lobes while 12 out of 14 primary GS showed a peripheral location (85.7%). There was no evidence of calcification inside the tumor. One patient had radiological evidence of bleed inside it [Figure 1]. Average tumor size was 5.1 cm [range: 3–6 cm]. Majority (n = 12, 75%) of the tumors were totally solid (with or without necrotic areas) and four patients (25%) had large cystic component, two of whom had the appearance of mural nodule (12.5%). All lesions were hypo intense on T1, heterointense on T2 and enhanced variably on contrast. Enhancement was intense and homogenous in 7 (43.8%), intense but heterogenous in 4 (25%) and patchy/ring type enhancement was seen in five patients (31.2%) [Figures 2 and 3]. The wall of the cystic component also showed enhancement with contrast. Although majority of the tumors were peripherally located, dural tail sign was present only in four patients (25%). All patients had peritumoral edema and mass effect. All lesions were single, and there were no evidences of multifocality. Angiographic study was available in one patient who presented with intratumoral bleed. However, no abnormality was detected. Similar to the primary GS, secondary GS also showed inconsistent imaging findings. One of the patients with secondary GS had superficial dural-based mass with dural tail sign [Figure 4] while the second patient had a subcortical ill-defined mass with patchy enhancement [Figure 5].
Figure 1.
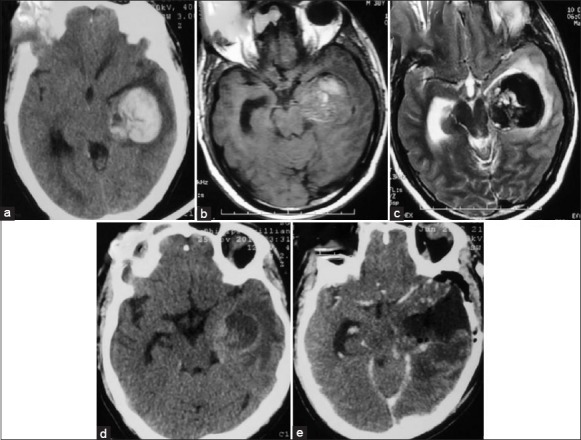
Hyperdensity suggestive of bleed in left subcortical temporal lobe (a). The lesion appeared hyperintense on T1 and hypointense on T2 further demonstrating bleed inside the lesion (b and c). After resolution of hematoma, a peripherally enhancing mass lesion can be seen on axial computed tomography (1-year after initial bleed) (d). Postoperative computed tomography scan showed complete excision of tumor (e)
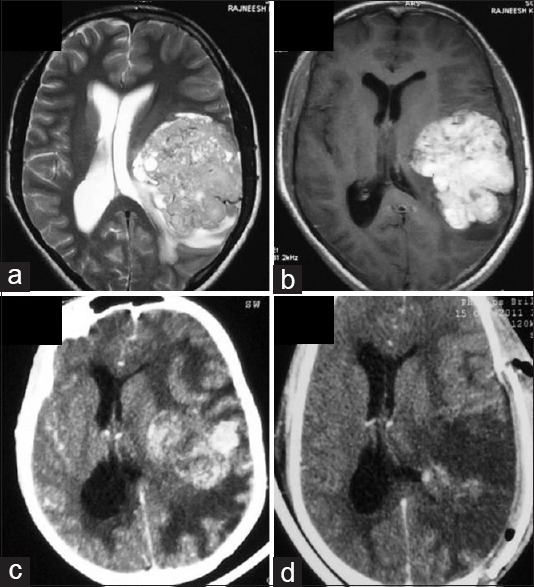
Figure 2.
Hyperdense right temporal cortical mass with extensive perilesional edema was seen on CT(a). The mass was heterogenously hypointense on T1 and hyperintense on T2 with heterogenous but strong postcontrast enhancement (b,c,d,e). Dural tail sign was seen in the axial section (d). Postoperative scan showed complete excision (f)
Figure 3.
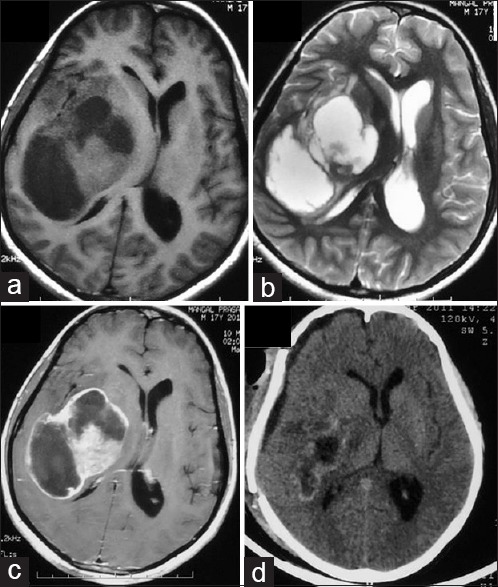
A T1 isointense to hypointense and T2 isointense to hyperintense large right frontoparietal heterogenous mass lesion showing peripheral ring-like enhancement with edema and mass effect (a-c). Postoperative scan shows subtotal excision with residual tumor abutting posterior limb of internal capsule (d)
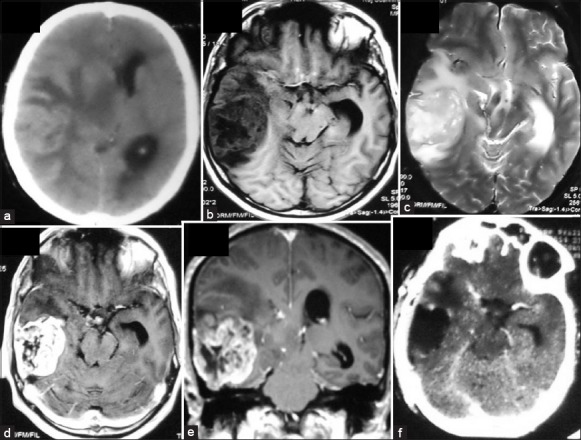
Figure 4.
Left frontoparietal glioblastoma which was heterogeneously hyperintense on T2 and showed intense enhancement (a and b). Contrast computed tomography head done 2 years after surgery and radiotherapy shows an ill-defined mass at the site of the original tumor and patchy enhancement (c). Postoperative image shows evidence of ear total excision (d)
Figure 5.
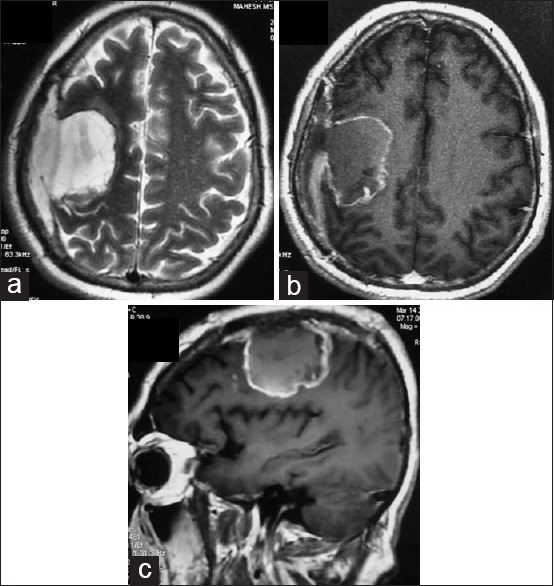
Dural-based T2 hyperintense (a) and sharply delineated homogenously enhancing right frontal secondary gliosarcoma displaying dural tail sign (b and c). Craniotomy defect of earlier surgery for glioblastoma can be seen in all the images
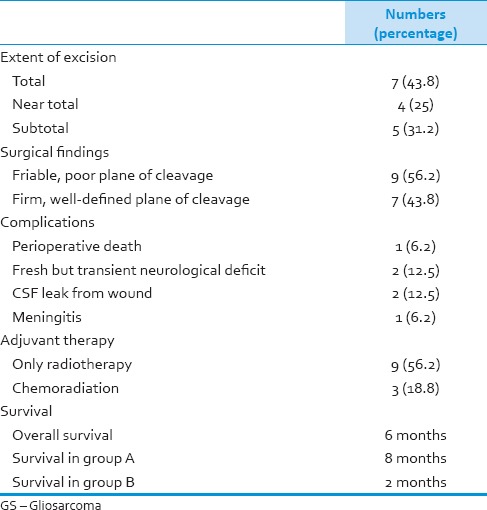
Total excision of the tumor was done in seven patients (43.8%), near total in 4 (25%) and subtotal in five patients (31.2%). Intraoperatively, these tumors were most often surfacing on the brain. The tumor appeared variegated in texture with soft suckable areas admixed with firm nonsuckable areas. At times, the plane of cleavage around the tumor was well-defined (n = 7, 43.8%) whereas in certain cases, the plane was poorly defined (n = 9, 56.3%). Most of the tumors were moderately vascular.
There was one perioperative death (6.2%). This patient was a 66-year-old male who presented in an unconscious state with extensor motor response. He had a huge right temporal lobar mass (6 cm × 6 cm) with extensive edema and uncal herniation. He failed to improve after an urgent surgery and succumbed to the disease after 3 days. In addition, operative complications were noted in three patients (18.8%). These included fresh but transient neurological deficit (n = 2, 12.5%), cerebrospinal fluid leak from wound (n = 2, 12.5%) and meningitis (n = 1, 6.2%). No patient had an operative site hematoma.
Pathologically, all patients showed a biphasic tumor, consisting of malignant gliomatous (Grade 4) areas admixed with sarcomatous areas showing a mostly spindle cell pattern. The gliomatous component displayed features of GBM with areas of necrosis, vascular proliferation, and stained avidly with GFAP. On the other hand, sarcomatous component was composed of spindle-shaped cells that failed to stain with GFAP [Figure 6]. Microscopic evidence of calcification was present in one patient. Two patients had myxoid degeneration of the sarcomatous area. Tumor giant cells were present in almost all of the patients (n = 15). Microscopic evidence of intratumoral hemorrhage was seen in two patients. Histology in one patient showed undifferentiated areas in addition to the gliomatous and sarcomatous components. Both patients of secondary GS developed sarcomatous change after 2 years of surgical resection of GBMs and chemoradiotherapy. Both patients had GBMs affecting temporal lobes and were located peripherally abutting dura. Histologically, they were indistinguishable from primary GS. We reviewed the histopathology reports to find out if there was any histological difference between the two groups (Group A and B). Although information regarding quantitative analysis of each component was not available in the reports, Group A (n = 7, 43.8%) revealed prominent areas of spindle cells with robust reticulin positivity interspersed with GFAP positive GBMs component. While Group B (n = 9, 56.3%) showed an abundance of strong GFAP staining cells with few often peripherally dispersed reticulin rich spindle cells. Hence, it appeared that the predominance of sarcomatous component imparted a meningioma like feature to a GS.
Figure 6.
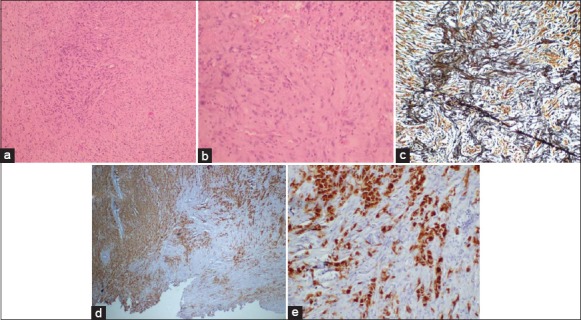
(a) Tumor disposed in sheets showing pleomorphic cells displaying anisomorphic nuclei, frequent mitosis (H and E, ×100). Areas of spindle tumor cells displaying anisonucleosis are also noted (b) (H and E, ×200). Foci of reticulin-rich tumor cells (c: Reticulin, ×40) suggest sarcomatous component. Glial fibrillary acidic protein (GFAP) positive tumor cells seen in glial component and interspersed glial GFAP negative tumor cells suggest sarcomatous component (d and e: GFAP, ×200)
Nine patients received radiotherapy (59.4 Gy/33 cycles). One patient could not complete the course and died after six cycles of radiation therapy. Available records showed that concurrent temozolomide was administered in only three patients (18.8%).
Outcome data were available for 11 out of 15 patients who were discharged after surgery. Eight patients had died while remaining three patients were surviving at mean follow-up of 9.6 months. Kaplan–Meier analysis revealed a median survival of 6 months [Figure 7]. Three patients survived 18 months or more with the maximum survival of 36 months. Although not significant, there was marked difference in survival between Group A and B (8 months vs. 2 months, P = 0.4) Table 2 shows extent of excision, operative findings, surgical complications, adjuvant therapy, and outcome of GS patients in our series.
Figure 7.
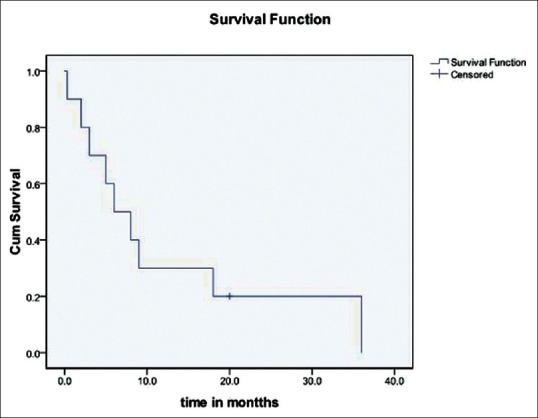
The survival curve. The median overall survival was 6 months
Table 2.
Extent of excision, operative findings, surgical complications, adjuvant therapy, and outcome in GS
Discussion
Gliosarcoma has been reported to constitute 1–8% of all GBM and <0.5% of all intracranial tumors.[3,4,7,8] In our experience, the incidence was 5.2%. GS can be primary or secondary. We encountered two cases of secondary GS (incidence 12.5%) with both cases occurring nearly 2 years after treatment of GBM. Both cases, interestingly, affected temporal lobe and were peripherally located. Secondary GS is rare, and only 50 odd cases have been reported worldwide.[9] The criteria used for diagnosis of radiation-induced tumors[10] in general include (1) the tumor appear in the area covered by irradiation, with a significant time interval (years) between irradiation and the appearance of the new tumor, (2) the tumor must be absent prior to irradiation, and (3) the new tumor has to be histologically different than the first tumor. Han et al.[11] reviewed tumors and distinguished two variants of nonprimary GS namely secondary GS and radiation-induced GS. Secondary GS developed usually within 1-year of treatment (surgery + radiation + chemotherapy) of GBM while radiation-induced GS developed many years (mean 5.2 years) after irradiation for other brain tumors like meningioma, low-grade glioma, medulloblastoma, etc. Therapeutic radiation has been consistently associated with the genesis of secondary GBMs. The management implication of these nonprimary GS are (1) poorer outcome than de novo GS and (2) more likelihood to present with intra/extra cranial metastasis.[11]
From a clinical standpoint, GS usually affects people in their fifth to sixth decade of life with a consistent male predominance,[4,7] a fact further substantiated at our study. However, the mean age of the patients (45.2 years) was relatively younger. Seven of our patients were younger than 40 years. The most common symptom was that of raised intracranial pressure and three patients presented in altered consciousness because of alarming mass effect. The reason for a high incidence of features of generalized mass effect in our experience, we believe, is because of the large size of tumors and peritumoral edema associated with them. Seizure was complained by only five patients. This assumes significance when one considers that GS has a tendency to involve juxtacortical cerebral lobes, particularly the temporal lobes. Interestingly, one of the patients presented with sudden headache with vomiting, and computed tomography showed evidence of intraparenchymal bleed. Angiographic evaluation at admission was normal.
Numerous reports have mentioned about the tendency of GS to affect supratentorial cerebral lobes in general and the temporal lobe in particular.[6,8,12] In our series, all patients had supratentorial tumors and temporal lobe was the most frequently involved site. In addition to temporal lobe affinity, GS have also been reported to affect peripheral parts of the brain.[6] In our series, 14 (87.5%) patients had tumors surfacing on the superolateral aspect of the brain. Damodaran et al.[6] have noted that 100% of secondary GBMs tended to be peripherally placed as compared to 67% of primary GSs.
From a neuroimaging point of view, GSs present heterogenous appearances on various imaging modalities. As noted by Salvati et al.,[13] we also found two basic patterns of imaging findings in GS. Seven patients had mostly homogenous, well-circumscribed, strongly enhancing masses often abutting dura simulating the appearance of meningiomas. On the other hand, there was another set of patients where the tumor was heterogenous, ill-defined in outline and showed either patchy or peripheral ring enhancement, so very characteristic of GBMs. This pattern was observed in primary as well as secondary GSs. Damodaran et al.[6] noticed two patterns on radiology. Peripherally located tumors tended to produce dural thickening whereas deeper tumors were associated with ependymal enhancement. They also noticed that deeper tumors were more frequently associated with satellite lesions than peripheral lesions. As far as the secondary GBMs are concerned, Damodaran et al.[6] noted that the majority of them showed mixed signal characteristic unlike primary GS and frontal lobes were most commonly affected by secondary GS. Angiography was done in one of our patients who presented with bleed, however, it failed to reveal any abnormality. In a study of patients, Jack et al.[14] have found that GSs have dual blood supply (dural and pial), show early cortical venous drainage, have irregular tumor vessels and prominent vascular stain within well-demarcated tumor margins.
The pathogenesis of GS has been a matter of debate. Earlier, it was believed that sarcomatous component in GS was due to neoplastic transformations of the endothelial cells in the vessels traversing the tumor.[2,15] However, this concept changed when studies could not consistently find endothelial markers inside GS. The current concept of the pathogenesis of GS points towards a monoclonal origin of gliomatous and sarcomatous cells.[16,17] This stems from the fact that similar genetic aberrations occur in these cell population namely identical p53 mutations,[16] homozygous p16 deletion, PTEN mutation, and co-amplification of MDM2 and CDK4.[17]
For histopathological diagnosis of GS, Meis[4] laid down certain (z). These include: (a) The tumor must be bimorphic that is composed of two morphologically distinct malignant cells population; (b) one component must be astrocytic with areas of necrosis, fulfilling the criteria for GBM; (c) the sarcomatous component must resemble a spindle cell sarcoma; and (d) a minimum of one confiuent sarcomatous area must fill one medium power field (10 objective with 10 eyepiece). The sarcomatous component is typically spindle cell sarcoma, but it can be either of fibrosarcoma, angiosarcoma, soft tissue sarcoma or rhabdomyosarcoma.[18] In addition, cartilage, bony mesenchymal components have also been described in these tumors.[19] Immunohistochemistry helps differentially visualize the bimorphic areas as GFAP stains gliomatous areas while reticulin positively stains sarcomatous areas. Salvati et al.[13] have stressed on the quantitative estimation of each component of GS. He found that tumors having >50% sarcomatous component tended to have better survival compared to those wherein sarcomatous component measures less than one-fourth of the tumor sample. They argued that the survival advantage was probably because of more complete excisions in sarcoma predominant GS, which tend to be firm and well-demarcated than anything else. As far as secondary or radiation-induced GS are concerned, there are hardly any histological differences apart from changes of radiotherapy and presence of fibrosarcoma in almost all cases.[11] At this point, it is very pertinent to remember that sampling errors during the first histopathological examination may miss sarcomatous component, thereby, a diagnosis of GBM rather than GS is wrongly made. In such a situation, when the tumor reappears after treatment, one may erroneously consider it to be secondary GS rather than just a recurrence. The possibility of such sampling error has been discussed by Han et al.[11] in their comprehensive review.
Gliosarcoma is managed in a fashion similar to GBM at present times. This includes maximal surgical resection followed by adjuvant chemoradiation. Because of frequent peripheral location, GS offers a better chance of gross total excision when compared with GBM. This is more applicable to sarcoma rich GSs. Numerous studies on GBMs have proven the efficacy of gross total excision in prolonging survival.[20,21] One must, however, agree that the data on GS are sparse, and there is a tendency to extrapolate results of GBM while managing GS patients. In our study, there was a difference of 6 months in survival between these two groups. There is hardly any controversy that maximal surgical resection followed by adjuvant radiation is the minimum treatment that a patient with GS should receive.[5] However, there is still a lot of disagreements regarding the role of chemotherapy in GSs. Morantz et al.[3] were the first to note a modest increase in terms of survival using chemoradiation compared with a control group treated only by radiation (36 vs. 33 weeks). Following the landmark publication by Stupp et al.,[20] adjuvant chemoradiation has become the standard in GBMs. In a recent study, Walker et al.[22] have shown that adjuvant chemoradiation with temozolomide resulted in an 20% 2-year overall survival as opposed to 10.2% among those not treated with this regimen (P = 0.68). They also pointed out that in the era of temozolomide, the 2-year survival in the SEER database between 2006 and 2008 was exactly double (24.2% vs. 12.1%) than the figure between 2000 and 2003 (P = 0.03). They attributed this difference to the widespread adoption of the Stupp[20] regimen (after 2005). However, it remains to be proven whether temozolomide is equally effective in sarcoma. This assumes further importance as people like Forshew et al.[23] have shown that in spite of a common monoclonal origin, both GS and GBM subsequently develop different genetic aberrations. This raises serious questions about the tendency to extrapolate the management guidelines of GBM in patients diagnosed with GS. However, it appears that GSs will continue to be managed in a fashion similar to the GBMs till the time we come up with molecular targeted therapies or better chemotherapeutic agents.
As far as the outcome of GS is concerned, most studies point toward a dismal survival in these patients which is worse compared to GBM. The median survival has been reported to range from 6 to 14.8 months.[3,5,21,24,25] The overall survival in our patients was only 6 months which is a further testimony to the fact that GS remains a highly malignant disease. As far as the factors affecting survival are concerned, numerous studies have identified patient age, preoperative KPS, (radiologic/intraoperative) impression of meningioma versus GBM, extent of excision, and adjuvant chemoradiation have been thought to be important. Younger age, good preoperative KPS (which ensures that the patient undergoes the entire treatment modalities) and gross total excision as opposed to lesser degree of excision are associated with better survival, facts that are similar to the GBMs. What is more relevant in GS is that GS with meningioma like features tends to have a better survival than those which resemble GBM. In the current series, the survival in the former was 8 months as compared to 2 months in the latter. Salvati et al.[13] were the first to recognize this when they noticed a significant difference in the survival between these two groups (71 ± 6 weeks in meningioma like GS vs. 63 ± 6 weeks in GBM like GS, P = 0.0417). The reason for this difference, according to them, was the better extent of excisions in meningioma like GS as compared to GBM like GS. One of our patients with secondary GS survived up to 2 years after surgery while the other was lost to follow-up after 6 weeks. Damodaran et al.[25,6] have noticed that secondary GS tends to have a poorer survival than de novo GSs. Although one of our patients survived for 2 years, the number is too little to draw any conclusion regarding prognosis of secondary GS.
Our study had certain limitations. The numbers of cases were few and analysis was retrospective. Hence, results did not reach statistical significance. Moreover, no quantitative estimation of histological components was carried out to convincingly prove that radiological and intraoperative findings could predict comparative histomorphology of GS. Another major limitation was that only three patients received chemotherapy with temozolomide which might have been one of the reasons for poorer survival in our series.
Nevertheless, the study attempted to provide some insight into the clinicoradiological, histolopathology-related aspects of GS, in general. Furthermore, it highlighted the current practice of management of these lesions and their continued dismal outcome in our settings. Further studies, especially exploring the role of chemotherapy (temozolomide [TMZ] or some new drugs) and molecular targeted therapies are needed to prolong survival in this category of patients.
Conclusion
Gliosarcoma affects younger patients compared to GBM with a stronger male predilection. Clinically, they are nearly indistinguishable from GBM. Radiological and intraoperative findings help categorize these tumors into GBM like GS and meningioma like GS, a fact that appeared to be applicable to both primary and secondary GSs. GS is associated with poor survival (median survival 6 months). GS with meningiomatous appearance tends to show better survival (8 months) than GS that resembles GBM (2 months). Maximal safe resection followed by radiotherapy and chemotherapy (TMZ) appears to be the best current treatment for these tumors.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
References
- 1.Stroebe H. Uber Entstehung und Bau der Gehirngliome. Beitr Pathol Anat Allg Pathol. 1895;18:405–86. [Google Scholar]
- 2.Feigen IH, Gross SW. Sarcoma arising in glioblastoma of the brain. Am J Pathol. 1955;31:633–53. [PMC free article] [PubMed] [Google Scholar]
- 3.Morantz RA, Feigen I, Ransohoff J. Clinical and pathological study of 24 cases of gliosarcoma. J Neurosurg. 1976;45:398–408. doi: 10.3171/jns.1976.45.4.0398. [DOI] [PubMed] [Google Scholar]
- 4.Meis JM, Martz Kl, Nelson JS. Mixed glioblastoma multiforme and sarcoma. A clinicopathologic study of 26 Radiation Therapy Oncology Group cases. Cancer. 1991;67:2342–9. doi: 10.1002/1097-0142(19910501)67:9<2342::aid-cncr2820670922>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 5.Perry JR, Ang LC, Bilbao JM, Muller PJ. Clinicopathologic features of prima- ry and postradiation cerebral gliosarcoma. Cancer. 1995;75:2910–18. doi: 10.1002/1097-0142(19950615)75:12<2910::aid-cncr2820751219>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.Damodaran O, van Heerden J, Nowak AK, Bynevelt M, McDonald K, Marsh J. Clinical management and survival outcomes of gliosarcomas in the era of multimodality therapy. J Clin Neurosci. 2014;21:478–81. doi: 10.1016/j.jocn.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 7.Lutterbach J, Guttenberger R, Pagenstecher A. Gliosarcoma: a clinical study. Radiother Oncol. 2001;61:57–64. doi: 10.1016/s0167-8140(01)00415-7. [DOI] [PubMed] [Google Scholar]
- 8.Zhang BY, Chen H, Geng DY, Yin B, Li YX, Zhong P, et al. Computed tomography and magnetic resonance features of gliosarcoma: a study of 54 cases. J Comput Assist Tomogr. 2011;35:667–73. doi: 10.1097/RCT.0b013e3182331128. [DOI] [PubMed] [Google Scholar]
- 9.Khalid AS, Mohamed O, Mohammed EM, Hassan S, Khalid H, Hamid M. Secondary gliosarcoma after the treatment of primary glioblastoma multiforme. North American Journal of Medical Sciences. 2011;3:527. doi: 10.4297/najms.2011.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schrantz JL, Araoz CA. Radiation induced meningeal fibro- sarcoma. Arch Pathol. 1972;93:26–33. [PubMed] [Google Scholar]
- 11.Han SJ, Yang I, Tihan T, Chang SM, Parsa AT. Secondary gliosarcoma: a review of clinical features and pathological diagnosis. J Neurosurg. 2010;112:26–32. doi: 10.3171/2009.3.JNS081081. [DOI] [PubMed] [Google Scholar]
- 12.Kozak KR, Mahadevan A, Moody JS. Adult gliosarcoma: epidemiology, natural history, and factors associated with outcome. Neuro Oncol. 2009;11:183–91. doi: 10.1215/15228517-2008-076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvati M, Caroli E, Raco A, Giangaspero F, Delfini R, Ferrante L. Gliosarcomas: analysis of 11 cases do two subtypes exist? J Neurooncol. 2005;74:59–63. doi: 10.1007/s11060-004-5949-8. [DOI] [PubMed] [Google Scholar]
- 14.Jack CR, Jr, Bhansali DT, Chason JL, Boulos RS, Mehta BA, Patel SC, et al. Angiographic features of gliosarcoma. AJNR Am J Neuroradiol. 1987;8:117–22. [PMC free article] [PubMed] [Google Scholar]
- 15.Haddad SF, Moore SA, Schelper RL, Goeken JA. Smooth muscle can comprise the sarcomatous component of gliosarco- mas. J Neuropathol Exp Neurol. 1992;51:493–8. doi: 10.1097/00005072-199209000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME. Identical mutations of the p53 tumor suppressor gene in the gliomatous and the sarcomatous components of gliosarcomas suggest a common origin from glial cells. J Neuropathol Exp Neurol. 1995;54:651–6. doi: 10.1097/00005072-199509000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Reis RM, Konu-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H. Genetic profile of gliosarcomas. Am J Pathol. 2000;156:425–32. doi: 10.1016/S0002-9440(10)64746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shintaku M, Miyaji K, Adachi Y. Gliosarcoma with angiosarcomatous features: a case report. Brain Tumor Pathol. 1998;15:101–5. doi: 10.1007/BF02478891. [DOI] [PubMed] [Google Scholar]
- 19.Alatakis S1, Stuckey S, Siu K, McLean C. Gliosarcoma with osteosarcomatous differentiation: review of radiological and pathological features. J Clin Neurosci. 2004;11:650–6. doi: 10.1016/j.jocn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62:753–64. doi: 10.1227/01.neu.0000318159.21731.cf. [DOI] [PubMed] [Google Scholar]
- 21.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 22.Walker GV, Gilbert MR, Prabhu SS, Brown PD, McAleer MF. Temozolomide use in adult patients with gliosarcoma: an evolving clinical practice. J Neurooncol. 2013;112:83–9. doi: 10.1007/s11060-012-1029-7. [DOI] [PubMed] [Google Scholar]
- 23.Forshew T, Lewis P, Waldman A, Peterson D, Glaser M, Brock C, et al. Three different brain tumours evolving from a common origin. Oncogenesis. 2013;2:e41. doi: 10.1038/oncsis.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cervoni L, Celli P. Cerebral gliosarcoma: prognostic factors. Neurosurg Rev. 1996;19:93–6. doi: 10.1007/BF00418077. [DOI] [PubMed] [Google Scholar]
- 25.Maiuri F, Stella L, Benvenuti D, Giamundo A, Pettinato G. Cerebral gliosarcomas: correlation of computed tomographic findings, surgical aspect, pathological features, and prognosis. Neurosurgery. 1990;26:261–7. [PubMed] [Google Scholar]