

Significance
Since 2009, an emergent shrimp disease, acute hepatopancreatic necrosis disease (AHPND), has been causing global losses to the shrimp farming industry. The causative agent of AHPND is a specific strain of Vibrio parahaemolyticus. We present evidence here that the opportunistic V. parahaemolyticus becomes highly virulent by acquiring a unique AHPND-associated plasmid. This virulence plasmid, which encodes a binary toxin [V. parahaemolyticus Photorhabdus insect-related toxins (PirAvp and PirBvp)] that induces cell death, is stably inherited via a postsegregational killing system and disseminated by conjugative transfer. The cytotoxicity of the PirAvp/PirBvp system is analogous to the structurally similar insecticidal pore-forming Cry toxin. These findings will significantly increase our understanding of this emerging disease, which is essential for developing anti-AHPND measures.
Keywords: shrimp, AHPND, Vibrio parahaemolyticus, Pir toxin, virulence plasmid
Abstract
Acute hepatopancreatic necrosis disease (AHPND) is a severe, newly emergent penaeid shrimp disease caused by Vibrio parahaemolyticus that has already led to tremendous losses in the cultured shrimp industry. Until now, its disease-causing mechanism has remained unclear. Here we show that an AHPND-causing strain of V. parahaemolyticus contains a 70-kbp plasmid (pVA1) with a postsegregational killing system, and that the ability to cause disease is abolished by the natural absence or experimental deletion of the plasmid-encoded homologs of the Photorhabdus insect-related (Pir) toxins PirA and PirB. We determined the crystal structure of the V. parahaemolyticus PirA and PirB (PirAvp and PirBvp) proteins and found that the overall structural topology of PirAvp/PirBvp is very similar to that of the Bacillus Cry insecticidal toxin-like proteins, despite the low sequence identity (<10%). This structural similarity suggests that the putative PirABvp heterodimer might emulate the functional domains of the Cry protein, and in particular its pore-forming activity. The gene organization of pVA1 further suggested that pirABvp may be lost or acquired by horizontal gene transfer via transposition or homologous recombination.
Since its first outbreak in China in 2009 (1), the newly emergent shrimp disease acute hepatopancreatic necrosis disease (AHPND) (2), originally known as early mortality syndrome, has spread through Southeast Asia to Vietnam, Malaysia, and Thailand to reach as far as Mexico in early 2013 (3, 4). Shrimp production within the AHPND-affected region dropped to ∼60% compared with 2012, and the disease has caused global losses to the shrimp farming industry estimated at more than $1 billion per year (5). In 2013, Tran et al. showed that the causative agent of AHPND was a specific strain of the common Gram-negative halophilic marine bacterium Vibrio parahaemolyticus (6). Through some unknown mechanism, this strain had become virulent, and, in infected shrimp, it induced AHPND’s characteristic symptoms, i.e., a pale and atrophied hepatopancreas (HP) together with an empty stomach and midgut. Histological examination further showed that AHPND causes sloughing of the HP tubule epithelial cells into the HP tubule lumens (2, 6). Curiously, however, in the initial and acute stage of the disease, although a massive number of bacteria are sometimes found in the stomach chamber in some affected shrimp, there is no significant bacterial colonization of the HP tubule lumen (6). This led Tran et al. to propose that the distinctive pathology of AHPND was caused by a secreted toxin, and this suggestion was supported by the finding that injection of the bacteria-free supernatant of culture broth media into healthy shrimp by reverse gavage can induce the characteristic symptoms of AHPND (6).
Previous studies on AHPND have focused on isolate variations (7), appropriate farming practices (8), or comparisons of draft genome sequences of AHPND-causing strains vs. non-AHPND strains (3, 9–12). In our previous study (9), we used a next-generation sequencing (NGS) platform to sequence and compare three virulent strain and one nonvirulent strain, and found that a large (∼69-kbp) extrachromosomal plasmid was present in all AHPND V. parahaemolyticus strains but not in the non-AHPND strains. We also reported that one of the genes (in fact, the operon that comprised ORF50 and ORF51) on this plasmid encoded a homolog of the insecticidal Photorhabdus insect-related (Pir) binary toxin PirAB. This homology further suggested that, like PirAB, this plasmidic protein might also exhibit pore-forming activity.
In the present study, the identity of the AHPND virulence factor is determined and confirmed, and the role and importance of the V. parahaemolyticus PirAB (PirABvp) toxin in AHPND pathology is demonstrated in its ability to induce AHPND. We propose a structural model of how the PirABvp complex might act as a pore-forming toxin.
Results
Unique Characteristics of AHPND-Causing Strains of V. parahaemolyticus.
To identify the critical virulence factors associated with AHPND, we first sequenced the genomes of three AHPND-causing strains (3HP, 5HP, and China) and one non–AHPND-causing strain (S02; Table S1). The contigs were generated from the sequence reads by the de novo genomic assembler Velvet (13), and then Mugsy (14) was used to perform multiple sequence alignments on these closely-related whole genomes to search for AHPND-specific contigs. For this analysis, we also included the complete genome sequence of two other non–AHPND-inducing strains of V. parahaemolyticus: human clinical isolate V. parahaemolyticus RIMD 2210633 and environmental isolate V. parahaemolyticus BB22OP. We retrieved 315 contigs that were found in all three AHPND-causing strains but not in any of the non-AHPND strains. BlastN showed that many of these putative AHPND-specific contigs were homologous to sequences found in plasmids in other Vibrio spp. We inferred that these contigs very probably also originated from a plasmid rather than from the V. parahaemolyticus chromosomal DNA.
Table S1.
V. parahaemolyticus strains used in this study
| Bacterial strain | Origin | Country (Ref.) |
| AHPND-causing | ||
| 3HP | AHPND diseased P. vannamei | Thailand (7) |
| 5HP | AHPND diseased P. vannamei | Thailand (7) |
| China | AHPND diseased P. vannamei | China (7) |
| A/3 (NCBI 13–028A3) | AHPND diseased P. vannamei | Vietnam (6) |
| D/4 | AHPND diseased P. vannamei | Mexico (4) |
| PD2 | AHPND diseased P. vannamei | Taiwan |
| M1-1 | Water in AHPND shrimp pond | Vietnam |
| Non-AHPND | ||
| S02 | Shrimp pond sediment collected from Phang-Nga province before EMS/AHPND emerged | Thailand (7) |
| M2-36 | Water in AHPND shrimp pond | Vietnam |
| BCRC12865 | Blue crab hemolymph | Taiwan |
| BCRC12959 | Shrimps | Taiwan |
| BCRC12864 | Shrimps involved in food poisoning | United States |
EMS, early mortality syndrome.
Analysis of Plasmids Purified from AHPND and Non-AHPND Strains.
To confirm that the unique sequences of the AHPND-causing strains were of plasmidic origin, we purified plasmids from six AHPND and four non-AHPND strains. All the AHPND-causing strains contained from one to five extrachromosomal plasmids (Fig. 1A), whereas Southern blot hybridization with a probe (AP2) (9) derived from one of the largest AHPND-specific contigs (12.2 kb) detected a plasmid of ∼70 kbp in each of the AHPND-causing strains (Fig. 1B). As this strongly suggested that AHPND virulence in V. parahaemolyticus is conferred by a common plasmid, we next sequenced the purified plasmids from two strains of AHPND-causing V. parahaemolyticus (3HP and M1-1). Our sequencing data revealed that both strains contained a plasmid with a size of ∼70 kbp. This APHND-associated plasmid was designated pVA1. Sequence identity of the two pVA1s was 98–99%, and the size also varied slightly.
Fig. 1.

Analysis of plasmids purified from AHPND and non-AHPND strains. (A) Uncut plasmids from various AHPND-causing strains (lanes 1–6) and non-AHPND strains (lanes 7–10) after separation in 0.8% agarose gel. The size marker lane (marked “M”) shows uncut plasmids from Pantoea stewartii SW2 (29). (B) Southern blot hybridization signals of the same uncut plasmids after they were transferred to a nylon membrane and probed with alkaline phosphatase-labeled DNA fragments derived from the AP2 amplicon. Asterisks indicate corresponding plasmid bands.
Sequence Analysis of the APHND-Associated Plasmid pVA1.
Fig. S1A shows one of the AHPND-causing strains of V. parahaemolyticus (3HP). This strain contains chromosome I (3.3 Mbp), chromosome II (1.9 Mbp), the plasmid pVA1 (70,452 bp), and a 64-kbp non–AHPND-associated plasmid. The pVA1 plasmid was assembled into a single contiguous sequence with a size of 70,452 bp and a G + C content of 45.95%. With the Rapid Annotations using Subsystems Technology online service (15), a total of 59 predicted ORFs were found on the forward strand, whereas 31 predicted ORFs were found on the reverse strand, with sizes ranging from 120 bp to 2,415 bp. The average length of the ORFs was approximately 652 bp, and the gene density was 0.78 per kb (Fig. S1B).
Fig. S1.

Full sequence of the AHPND-associated plasmid pVA1. (A) Genomic material in the V. parahaemolyticus strain 3HP. Circular (B) and linear (C) diagrams of the plasmid pVA1 generated by DNAPlotter (42). ORFs were predicted by RAST (15) and annotated manually by BlastP using the National Center for Biotechnology Information Web site. In B, the innermost circle shows a GC plot, with dark yellow representing below-average GC content and brown indicating above-average GC content.
The predicted ORFs were annotated by using a protein BLAST (BLASTP) search. Among the 45 putative ORFs with known function (Fig. S1 B and C, orange), one putative ORF (ORF7) showed homology to the toxin-antitoxin gene pndA (16), which is associated with a postsegregational killing (PSK) system. Five putative ORFs resembled transposase (Fig. S1 B and C, green; ORF15, ORF48, ORF55, ORF57, and ORF68), and three of these (ORF15, ORF48, and ORF55) were identical (ORF48 is in reverse orientation). pVA1 also includes an operon that encodes ORF50 and ORF51, which are homologs (∼30% identity) to the pore-forming Pir toxins PirA and PirB (17). We note that, before the emergence of AHPND, this binary insecticidal toxin had not been reported in a marine organism to our knowledge.
PirABvp Toxin Leads to Shrimp HP Cell Death and to the Characteristic Sloughing of the Dead Epithelial Cells into the HP Tubules.
Previous reports have shown that, even though AHPND induces massive sloughing of HP cells into HP tubules, there is no sign of any bacteria in the lesions (4, 6, 18). This has led to the suggestion that the observed necrotic effects are caused by some unidentified toxin (6), and we hypothesized that the secreted form of the pVA1-encoded Pirvp toxin was very likely to be responsible.
To test this hypothesis, we first investigated whether AHPND-causing V. parahaemolyticus secretes PirAvp and PirBvp into the culture medium. By Western analysis with antibodies against PirAvp and PirBvp, we detected secreted PirAvp (12 kDa) and PirBvp (50 kDa) in the AHPND-causing V. parahaemolyticus culture medium 1 h after cultivation (Fig. 2A).
Fig. 2.

PirABvp toxin leads to shrimp HP cell death, the characteristic sloughing of the dead epithelial cells into the HP tubules, and mortality within 24 h. (A) Western blot analysis with anti-PirAvp and anti-PirBvp antibodies detected PirAvp and PirBvp proteins in the medium of a V. parahaemolyticus culture during the log phase of growth (1–4 h). (B) PCR confirmation of the AP1, AP2, pirAvp, and pirBvp sequences in the ΔpirABvp and ΔpirABvp+pirABvp strains. (C) Western blot analysis with anti-PirAvp and anti-PirBvp antibodies shows the presence or absence of PirAvp and PirBvp proteins in the ΔpirABvp and ΔpirABvp+pirABvp strains. (D) Virulence assay shows cumulative mortalities of shrimp immersed in the WT, ΔpirABvp, and the complemented strains. Shrimp immersed in TSB were used as a negative control. Significant differences were determined by unpaired Student t test (**P< 0.005). (E) Sections of the HP from shrimp in the virulence assay were stained with H&E and subjected to histological examination. The typical AHPND signs of sloughed epithelial cells (arrows) and hemocytic encapsulation (HE) were induced by the WT and the complemented strains, whereas shrimp immersed in the ΔpirABvp strain or the TSB control exhibited normal B- and F-cells (broken arrows) without any sloughing of the tubule cells. (Scale bar: 100 μm.) (F) To confirm the toxin content used in the per os challenge, Western blot with anti-PirA and anti-PirB probes was used to detect the purified recombinant Pir toxins rPirAvp, rPirBvp, rPirAvp+rPirBvp, and the E. coli recombinants BL21 (ppirAvp), BL21 (ppirBvp), and BL21 (ppirAvp) + BL21 (ppirBvp). WT AHPND strain 3HP was used as the positive control, and BSA and the empty vector BL21 (pET21b) were used as the negative controls. (G and H) Results of per os challenge show cumulative mortalities of shrimp fed with pellets soaked for 10 min in solutions containing (G) purified recombinant Pir toxin rPirAvp, rPirBvp, and rPirAvp+rPirBvp and (H) the E. coli recombinants BL21 (ppirAvp), BL21 (ppirBvp), and BL21 (ppirAvp) + BL21 (ppirBvp). Shrimp fed with pellets soaked in BSA or in the E. coli recombinant with the expression vector only, BL21 (pET21b), was used as the negative controls. Significant differences were determined by unpaired Student t test (**P< 0.005).
For the next experiment, we constructed a pirAB operon deletion mutant (Fig. 2B) of AHPND-causing V. parahaemolyticus (ΔpirABvp AHPND-causing V. parahaemolyticus) by conjugation of AHPND-causing V. parahaemolyticus with Escherichia coli S17-1λpir that had been transformed with recombinant pIT009 with the pirABvp operon deletion allele. We also constructed a complementation mutant (Fig. 2B) by introducing a recombinant pIT009 with the pirABvp operon into the ΔpirABvp AHPND-causing V. parahaemolyticus by conjugation for in trans expression of PirABvp toxin. We confirmed by Western blot analysis that ΔpirABvp AHPND-causing V. parahaemolyticus did not produce PirAvp or PirBvp (Fig. 2C). The results of challenge tests revealed that the isogenic ΔpirABvp mutant showed a dramatic decrease in virulence and caused no AHPND-like histopathology, whereas both effects were recovered by the complementation mutant that exogenously expressed PirABvp (Fig. 2 D and E). These results suggest the PirABvp toxin plays a critical role in producing the characteristic symptoms of AHPND.
To further explore the toxicity of PirAvp, PirBvp, and the PirABvp complex, shrimp were challenged with the recombinant proteins themselves or with two E. coli BL21 recombinants that respectively expressed recombinant PirAvp (rPirAvp) and rPirBvp (Fig. 2F). When reverse gavage was used to directly inject 100 μL of 2 μM rPirAvp, rPirBvp, or premixed rPirAvp+rPirBvp into shrimp (body weight 1 g), histological examination of HP tissue sections showed that, although rPirAvp caused only minor histological changes, the premixed rPirAvp+rPirBvp and the rPirBvp alone induced typical signs of AHPND, including sloughing of the epithelial cells into the HP tubules (Fig. S2A). However, when shrimp were challenged by the more natural per os pathway in feeding trials, rPirBvp alone showed no histological effect, whether administered as a protein or via E. coli BL21 (Fig. S2 B and C), whereas mortality was significantly increased only by a mixture of both toxins and by a mixture of both toxin-expressing E. coli BL21 recombinants (Fig. 2 G and H).
Fig. S2.

H&E-stained sections of the HP from shrimp challenged by reverse gavage (A) and per os (B and C). For reverse gavage, shrimp were injected with PBS solution or 100 μL 2 μM rPirAvp, rPirBvp, or premixed rPirAvp/rPirBvp. Normal B- and F-cells without any sloughing of the tubule cells were seen in the shrimp injected with PBS solution. Injection of rPirAvp+rPirBvp or PirBvp produced epithelial sloughing (arrows). For the per os feeding challenge, after 1 d of starvation, shrimp were fed with pellets soaked in the indicated protein(s) or E. coli recombinant(s). Epithelial sloughing (arrows) was observed only when challenged with pellets containing both rPirAvp and rPirBvp or with E. coli BL21 recombinants that expressed both rPirA and rPirB. (Scale bar: 100 μm.)
Natural Deletion/Insertion of pir Genes.
Further evidence of the importance of the PirABvp toxin in causing AHPND comes from V. parahaemolyticus strain M2-36, which was isolated from a culture pond in Vietnam after an outbreak of AHPND. Although M2-36 gave positive PCR results with primer sets derived from AP1 and AP2 (Fig. S3), it failed to induce AHPND in shrimp that were challenged by immersion (Fig. S4). PCR amplification of a region of the plasmid between AP1 and AP2 produced an amplicon of only ∼3,701 bp instead of the expected ∼9,116 bp. Southern hybridization confirmed that the amplified region was from an M2-36 plasmid (Fig. S5), and sequencing revealed that the entire pirAB operon was missing from this amplicon (Fig. 3). Interestingly, the missing pirAB fragment was flanked by two mobile elements: upstream, there was a transposable element protein (transposase) with terminal inverted repeats, whereas the transposase downstream shared 100% sequence identity but was oriented in the opposite direction as the transposase upstream, suggesting that the acquisition of pirABvp by pVA1 or the deletion of pirAB from M2-36 might have resulted from a horizontal gene transfer between microorganisms via gene transposition or homologous recombination.
Fig. S3.

PCR detection of the AP1, AP2, pirAvp, and pirBvp sequences in AHPND and non-AHPND strains by PCR. PCR was performed on the indicated strains after they had been cultured from stock for 2 h at 37 °C with agitation. The PCR products were then separated by electrophoresis in a 2.0% (wt/vol) agarose gel, and the appearance of a band of the predicted size was interpreted as a positive signal. Strain 3HP was the AHPND-positive control. Strain S02 contains no plasmids and was used as the non-AHPND negative control.
Fig. S4.

Strain M2-36 failed to induce AHPND in shrimp. (A) Challenge with the WT 3HP strain induced 80% mortality within 24 h, whereas no shrimp died when challenged with strain M2-36 or the negative control (culture medium only). (B) No AHPND-like symptoms were seen in the apparently healthy HP of an M2-36–challenged shrimp collected at 24 h. Both histopathology samples were fixed with Davidson AFA fixative and stained with H&E.
Fig. S5.

Demonstration of absence of pirBvp from the plasmids of V. parahaemolyticus strain M2-36 by Southern hybridization. (Left) Uncut plasmid from strains 3HP and M2-36 separated in a 0.8% agarose gel. (Right) After the bands were transferred to a nylon membrane, hybridization was performed with alkaline phosphatase-labeled probes derived from pirBvp (A) and a sequence common to pVA1 and the 3,701-bp M2-36 deletion (B) (i.e., within identical region I in Fig. 3).
Fig. 3.
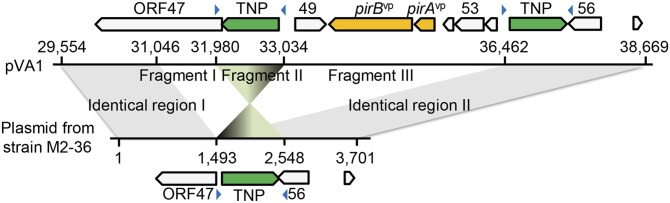
Comparison of pVA1 and the non–AHPND-causing plasmid from strain M2-36 shows the location of the missing DNA fragments. Putative pVA1 ORFs are indicated by the boxes marked with arrows. In the sequence amplified by the AP1-F16/AP2-R1 primer set (Table S2), fragments I and III are absent from the plasmid in the M2-36 strain, whereas fragment II is in reverse orientation. The blue arrowheads represent inverted repeats.
The pVA1 Plasmid Is the Only Source of the AHPND-Causing Toxin.
To confirm that the pVA1 plasmid was the only source of pirABvp, the plasmid needed to be cured, i.e., removed, from the bacterium. However, in our preliminary trials, we failed to produce any cured clones from more than 20,000 clones that were sensitive to the chloramphenicol acetyltransferase marker. We determined that this was a result of the pndA toxin-antitoxin gene, which provides the pVA1 plasmid with a PSK system. When a bacterium contains a PSK+ plasmid, any of the bacterium’s progeny that do not inherit this plasmid will die because the stable pndA mRNA will be translated to the bactericidal PndA toxin (16). To prevent the PSK effect, pndA was therefore deleted by allelic exchange under sucrose stress conditions via conjugation of an AHPND-causing V. parahaemolyticus (3HP) with E. coli S17-1λpir that had been transformed with the recombinant suicide vector pDS132-ΔpndAvp. After pndA deletion, the pVA1 plasmid was successfully cured from the pVA1 ΔpndAvp 3HP mutant by using a high concentration of the plasmid curing agent acridine orange. As shown in Fig. 4A, the 64-kb non-AHPND plasmid was still present in the pVA1-cured strain, and the bacterial growth of the cured strain was unaffected after undergoing the curing process (Fig. S6). However, the virulence of the pVA1-cured derivative was greatly impaired (Fig. 4 B–D), and histopathological examination of shrimps infected with this pVA1-cured strain showed normal HP tissue morphology (Fig. 4E). We also found that reintroduction of the PirABvp toxin via exogenous expression (by introducing a recombinant pIT009 with pirABvp operon into the pVA1-cured strain) restored virulence and the cytopathic effects (Fig. 4 B–E). These data suggested that pirABvp, irrespective of other plasmidic factors of pVA1, was sufficient to produce the symptoms associated with AHPND.
Fig. 4.

AHPND virulence is abolished in the pVA1-cured derivative and recovered by in trans expression of PirABvp. (A) Uncut plasmid profiles of the 3HP AHPND WT strain and its pVA1-cured derivative. Plasmid DNAs extracted from the respective strains were separated in a 0.8% agarose gel. The band corresponding to the plasmid pVA1 is indicated by an asterisk. M, uncut plasmids from P. stewartii SW2 as size markers. (B) PCR confirmation of the AP1, AP2, pirAvp, and pirBvp sequences in the pVA1-cured and pVA1-cured+pirABvp strains. (C) Western blot analysis with anti-PirAvp and anti-PirBvp antibodies to show the presence or absence of PirAvp and PirBvp proteins in the pVA1-cured and pVA1-cured+pirABvp strains. (D) Virulence assay shows cumulative mortalities for shrimp immersed in the WT, pVA1-cured, and pVA1-cured+pirABvp strains. Shrimp immersed in tryptic soy broth (TSB) were used as a negative control. Significant differences compared with the negative control were determined by unpaired Student t test (**P < 0.005). (E) Sections of the HP from shrimp in the virulence assay were stained with H&E and subjected to histological examination. The typical AHPND signs of sloughed epithelial cells (arrows) and hemocytic encapsulation (HE) of the hepatopancreatic tubules were observed in the shrimp immersed in the WT and pVA1-cured+pirABvp strains, whereas normal B- and F-cells without any sloughing of the tubule cells (broken arrows) were seen in the shrimp immersed in the pVA1-cured strain and TSB control. (Scale bar: 100 μm.)
Fig. S6.

Bacterial growth curves of various V. parahaemolyticus strains in LB broth. The overnight-cultured WT, pVA1-cured, pVA1-cured+pirABvp, ΔpirABvp, and ΔpirABvp+pirABvp strains were cultured overnight, diluted 1:100 with fresh LB broth, and then cultured again at 37 °C with agitation. Bacterial growth during the second culture period was estimated by measuring the turbidity of the culture at OD600 at the indicated time points.
The Combined Structural Topology of PirAvp/PirBvp Resembles That of the Cry Insecticidal Toxins Even Though the Shared Sequence Identity Is Low.
For the original Photorhabdus PirAB insecticidal toxin to be effective, both components needed to be coexpressed (17), which suggested that the two proteins might form a binary complex. We found that PirAvp and PirBvp interacted to form a complex in vitro (Fig. 5A), and this PirAvp/PirBvp complex was detected in the supernatant of a bacterial culture in midlog phase (Fig. 5B). In the same supernatant, we also observed a strong signal for PirBvp but not PirAvp.
Fig. 5.

Formation of the PirABvp complex in vitro and in vivo, and a proposed binding model that suggests a functional similarity between PirABvp and the Cry proteins. (A) When purified PirAvp, PirBvp, and a PirAvp/PirBvp mixture were analyzed separately by using a Superose 16 gel filtration column (GE Healthcare), we found that, for the PirAvp/PirBvp mixture, both proteins were eluted faster than the individual proteins on their own. This suggests that PirAvp and PirBvp have formed a complex. (B) In the supernatant from a midlog-phase culture of the WT and the pVA1-cured+pirABvp (CP) strains, PirBvp was found in a complex with PirAvp and as a monomer, whereas PirAvp was only detected in the complex. Immunoblotting was performed with anti-PirAvp or anti-PirBvp antibody under undenatured, native PAGE conditions (Left). The denatured SDS/PAGE (β-mercaptoethanol; Right) confirmed that PirAvp and PirBvp were present in the culture medium. (C) Structural comparison of PirAvp and PirBvp with Cry. (Left) PirAvp (magenta) is superimposed on domain III of Cry (yellow). (Right) PirBvp (magenta) is compared with domains I and II of Cry. PirAvp has an rmsd of 3.2 Å for 88 matched Cα atoms in CRY domain III, and PirBvp superimposes with Cry domain I and II with an rmsd of 2.8 Å for 320 matched Cα atoms. (PDB ID codes: PirAvp, 3X0T; PirBvp, 3X0U). (D) A putative model of the PirABvp heterodimer was constructed by superimposing PirAvp on domain III of Cry and PirBvp on domains I and II. (Left) Cry toxin shown for comparison (PDB ID codes for Cry, 1CIY and ref. 30).
As the crystal structures of the components of the Photorhabdus binary PirAB insecticidal toxin are unknown and because, in any case, there is genetic distance between the pirAB coding region of the V. parahaemolyticus strains vs. Photorhabdus and the other pirAB-harboring bacterial species (Fig. S7), we crystallized PirAvp and PirBvp and used X-ray crystallography to determine their respective structures (Fig. S8 and Table S3). The overall structural topology of PirAvp/PirBvp is very similar to that of the Bacillus Cry insecticidal toxin-like proteins, even though their shared sequence identity is less than 10% (Fig. 5C). This similarity suggested how the putative PirABvp heterodimer might emulate the functional domains of the Cry protein (Fig. 5D), with the N terminal of PirBvp corresponding to Cry domain I (pore-forming activity), the C-terminal corresponding to Cry domain II (receptor binding), and PirAvp corresponding to Cry domain III, which is thought to be related to receptor recognition and membrane insertion (19–21). If this predicted functionality is correct, PirABvp might induce cell death by forming ionic pores in the cell membrane by using a Cry insecticidal toxin-like mechanism.
Fig. S7.

Phylogenetic analysis of the pirAB coding region from V. parahaemolyticus and several other bacterial species. Multiple alignments of PirAvp and PirBvp protein sequences were performed by using BioEdit (43). The tree was inferred by using MEGA6 and the neighbor-joining method. The robustness of the tree was tested by using bootstrap analysis (1,000×). Percentage values are indicated at the nodes. The following species (GenBank accession numbers of PirAvp and PirBvp amino acid, respectively, in parentheses) were included: Alcaligenes faecalis (WP_003801867 and WP_003801865), Photorhabdus asymbiotica (WP_015835800 and WP_015835799), V. parahaemolyticus 13–028A3 (AIL49948 and KM067908), Photorhabdus luminescens subsp. luminescens (ABE68878 and ABE68879), Xenorhabdus doucetiae FRM16 (CDG18638 and CDG18639), Xenorhabdus cabanillasii JM26 (CDL79383 and CDL79384), and Xenorhabdus nematophila (WP_013183676 and WP_010845483).
Fig. S8.

Overall structure of PirAvp and PirBvp. The two asymmetric monomers of PirAvp (Left) and PirBvp (Right) are superimposed and colored in green and cyan. Arrows and cylinders represent β-strands and α-helices, respectively. All secondary structural elements are labeled, and the N and C termini are shown. PirAvp folds into an eight-stranded antiparallel β-barrel with jelly-roll topology as seen in viral capsid proteins, and composed of the BIDG and CHEF β-sheets. A large β-bulge was found in strand C. PirBvp folds into an N domain with seven α-helices and a C domain with 10 β-strands. Helix α1 is short, and helices α2, α4, and α7 are kinked. A large loop insertion was found in the middle of helix α2. Strands β1–β4 and β5–β8 of the C domain constitute two antiparallel sheets in a wedge-like formation, and the long β9–β10 ribbon intercalates between the two β-sheets on the proximal side near the N domain. The N and C domains are connected by a long loop of more than 40 residues, which includes a short helix (α8). The N-terminal segments of both PirAvp and PirBvp are flexible. Except for the N terminals, the two monomers of PirAvp show an rmsd of 0.25 Å for 101 Cα atoms, and those of PirBvp differ by an rmsd of 0.76 Å for 412 Cα atoms. The inserted loop in α2 deviates by 3.3–5.4 Å.
Table S3.
Data collection and refinement statistics of PirAvp and PirBvp crystals
| Hg soaking PirAvp (Highreomte) | Native PirAvp crystal | Hg soaking PirBvp (Highreomte) | Native PirBvp crystal | |
| Data collection | ||||
| Beamline | NSRRC13C | NSRRC13C | NSRRC13B | NSRRC13B |
| Wavelength, Å | 0.976 | 0.976 | 0.993 | 1.000 |
| Space group | C2 | C2 | C2221 | C2221 |
| Unit cell a, b, c, Å | 78.3, 67.4, 53.7 | 78.4, 67.3, 53.7 | 121.7, 128.2, 117.9 | 120.9, 128.6, 117.1 |
| α, β, γ, ° | 90.0, 122.9, 90.0 | 90.0, 122.8, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution, Å | 50–1.49 (1.54–1.49) | 50–1.17 (1.21–1.17) | 50–2.05 (2.12–2.05) | 50–1.70 (1.76–1.70) |
| Unique reflections | 37,421 (3,660) | 78,388 (7,597) | 57,899 (5,694) | 99,668 (9,812) |
| Redundancy | 8.3 (8.3) | 4.0 (3.4) | 8.1 (8.1) | 4.7 (4.6) |
| Completeness, % | 98.1 (96.5) | 99.3 (96.9) | 99.9 (100.0) | 99.7 (99.3) |
| I/σ(I) | 59.1 (7.7) | 33.8 (2.8) | 37.3 (2.8) | 24.8 (2.5) |
| Rmerge, % | 2.8 (20.8) | 2.3 (24.2) | 5.0 (50.3) | 3.4 (50.9) |
| Refinement | ||||
| Rwork, % | 13.1 | 14.9 | ||
| Rfree, % | 16.9 | 18.1 | ||
| Bond rmsd, Å | 0.022 | 0.019 | ||
| Angle rmsd, ° | 2.5 | 1.7 | ||
| Ramachandran plot, % | ||||
| Most favored, % | 98.48 | 96.08 | ||
| Allowed, % | 1.52 | 3.44 | ||
| Outliers, % | 0.48 |
Discussion
We have shown here that the characteristic symptoms of AHPND are caused by the binary insecticidal toxin homologs PirAvp and PirBvp, and that these two proteins are encoded by the pVA1 plasmid. This plasmid was found in all six of the AHPND-causing strains isolated from diseased shrimp in several Asian countries and Mexico (Fig. 1). Although virulence plasmids have been reported in several Vibrio spp. that are pathogenic for aquatic animals (22–24), plasmid prevalence in V. parahaemolyticus is usually less than 25%, and plasmid size typically ranges from 2.4 kb to 23.0 kb (25), which is much smaller than pVA1 and the other non-AHPND plasmids that were found in the AHPND-causing strains. Another interesting difference is that, unlike other fish-pathogenic Vibrio species, in which the plasmids contribute to virulence via a plasmid-encoded iron acquisition system (22) or confer resistance against the serum killing effect to overcome host defenses (23), the virulence plasmids of V. parahaemolyticus produce toxins that immediately destroy the host cells. We also note that the insecticidal Pir toxins/homologs currently described from bacterial species such as Photorhabdus and Xenorhabdus have all been found in the bacterial chromosomes (FM162591.1, FN667742.1, and FO704550.1), whereas PirABvp is the only toxin to be encoded by a plasmid.
It was reported previously that both PirA and PirB needed to be present to induce mortality in insect larvae (17), and now that this binary insecticidal toxin homolog has been found in a marine organism, it likewise seems that both PirAvp and PirBvp are necessary to produce the characteristic symptoms of AHPND (Fig. 2 G and H and Fig. S2 B and C).We note, however, that, when challenged by reverse gavage, the PirBvp toxin alone was able to induce AHPND-like histological signs (Fig. S2A). Reverse gavage is a challenge method commonly used in shrimp to analyze toxicity because, even though it is stressful to the animal, the amount of injected toxin can be quantified, and the mortalities associated with each dosage can be precisely compared. In the present case, in which our per os feeding protocol also ensured that the experimental animals were challenged with comparable dosages, the reason for the observed discrepancy in the toxicity of PirBvp is not clear, but it is presumably related to the different physiological conditions, especially the different enzymes, that would be encountered on the two respective challenge pathways. Meanwhile, as ingestion of AHPND-causing V. parahaemolyticus is probably the natural route of infection, it seems likely that the results of the oral feeding challenge are more reliable and more closely represent reality compared with the reverse gavage results, and we therefore tentatively conclude that PirAvp and PirBvp are both required to induce AHPND.
Our results show that the pVA1 plasmid contains a cluster of conjugative transfer genes and two plasmid mobilization genes (Fig. S1 B and C), which suggests that pVA1 may be a self-transmissible plasmid. When combined with the pndA PSK system, this ensures that the plasmid is inherited during bacterial replication, and, in this way, the pVA1 plasmid is passed on to subsequent generations of AHPND-causing V. parahaemolyticus. Indeed, as noted earlier (Results), we could not obtain any plasmid-cured derivative until the pndA gene was deleted. (This result also confirmed that the pndA in pVA1 was functional.) Taken together, these two features, i.e., the pndA PSK system and the conjugative transfer gene cluster, suggest that the AHPND virulence plasmid might be disseminated via conjugation and then become permanently inheritable in the recipient. The high homology (99∼100% identity) of the nucleotide sequences between pVA1 (KP324996) and two corresponding plasmids from the AHPND strains M1-1 and A/3, pVAM1-1 and pVPA3-1 (NC_025152), respectively, further supports the notion that these plasmids—and by extension, all the APHND-causing plasmids—might have originated from the same ancestor.
As for the bacterium itself, Kongrueng et al. found that several AHPND V. parahaemolyticus strains isolated from southern Thailand were serologically similar, had closely related pulsed field gel electrophoresis DNA profiles, and possessed the same repertoire of virulence factors (26). This led them to postulate that all these strains might have originated from a single clone. However, it is hard to generalize from this evidence alone because, although all of these strains were isolated from different locations, these locations were all in a single geographic area and lacked geographic diversity. Other reports that have focused on characterizing different AHPND-causing strains have also isolated samples from farms within a single locality (7, 18). To properly address this question, a phylogenetic analysis of various AHPND strains with a wide geographic diversity will be necessary. Until then, it remains unclear whether the different AHPND strains arose from the acquisition of pVA1 by a single V. parahaemolyticus ancestor (monophyletic origin) or by several different V. parahaemolyticus ancestors (polyphyletic origin).
We postulate that the cytotoxicity of PirAvp and PirBvp can be explained by their crystal structures. Structural alignment shows that PirAvp corresponds to domain III of the Bacillus thuringiensis Cry toxin and PirBvp corresponds to domains I and II (Fig. 5D). Functionally, Cry toxin induces cell death by undergoing a series of processes that include receptor binding, oligomerization, and pore forming (19, 20). In the case of B. thuringiensis Cry1A toxin, domain III first interacts with the GalNAc sugar on the aminopeptidase N (APN) receptor and facilitates further binding of domain II to another region of the same receptor (19–21). This binding promotes localization and concentration of the activated toxins. The APN-bound Cry toxin subsequently binds to another receptor, cadherin, which facilitates the proteolytic cleavage of its domain Iα1 helix. This cleavage induces the formation of Cry oligomer, which has pore-forming activity (19–21). Interestingly, when Cry toxin binds only to cadherin, it triggers an alternative signal transduction pathway. By activating protein G and adenylyl cyclase, cellular cAMP concentration is increased and protein kinase A is activated. This will destabilize the cytoskeleton and ion channels on the membrane, and induce cell death (27, 28). As the structural similarities suggest that the PirAvp/PirBvp system uses a similar strategy to kill cells, the first step to explore this possibility will be to identify cell receptors that might interact with the PirAvp/PirBvp heterodimer. Given that PirABvp toxin induces cell death in the HP but not in the stomach, it seems very likely that these putative PirABvp receptors will be found exclusively in the HP and any other PirABvp toxin-sensitive organs.
Materials and Methods
The detailed protocols for NGS, multiple sequence alignment, plasmid isolation, Southern blotting, construction of mutants and complemented strains, plasmid-cured derivative isolation, immersion challenge test, histopathology examination, reverse gavage injection, per os challenge, purification of recombinant Pir protein, gel filtration, protein crystallization, native PAGE analysis, and Western blotting as well as the information of bacterial strains and primers used, are provided in SI Materials and Methods.
SI Materials and Methods
Vibrio parahaemolyticus Strains Used in This Study.
A total of 12 Vibrio parahaemolyticus AHPND or non-AHPND strains isolated from Thailand, China, Vietnam, Mexico, the United States, and Taiwan were used in this study (Table S1). For plasmid purification and mutant and complemented strain construction, all strains were grown in TSB or Luria broth (LB) and incubated at 37 °C. For the immersion challenge test, the bacteria were grown at 30 °C.
NGS of the Bacterial Genomes of Strains 3HP, 5HP, China, and S02 and Plasmids pVA1 and pVAM1-1.
Genomic DNA was extracted from V. parahaemolyticus 3HP, 5HP, China, and S02 by using a QIAamp DNA mini kit (Qiagen). The extracted DNA samples (3 μg) were used to construct paired-end libraries with an Illumina TruSeq DNA HT Sample Prep Kit (average insert length, 634–645 bp). The sequencing was performed by an Illumina MiSeq sequencer (read length, 250 bp). The NGS reads were mapped to two reference genomes (RIMD 2210633 and BB22OP; GenBank accession nos. NC_004603, NC_004605, NC_019955, and NC_019971). The unmapped reads were imported into CLC Genomics Workbench 7.0 software (CLC Bio) for de novo sequence assembly. The plasmids pVA1 and pVAM1-1 were respectively extracted from strains 3HP and M1-1 using a TENS solution (Tris, EDTA, NaOH, and SDS; the extraction method is described below), and further purified by CsCl gradient as described previously (31). Plasmid DNA was subjected to sequence determination on an Illumina MiSeq sequencer (insert length, ∼450 bp; read length, ∼250 bp). The plasmid sequences were assembled into 105 and 76 contigs, respectively, and overlapping end sequences were used to join the contigs together. The plasmid sequences were further confirmed by the Sanger method. The ORF predictions were performed by the RAST (15) online program and annotated by BLASTP search.
Use of Multiple Sequence Alignment to Identify Specific Sequences Found Only in the AHPND-Causing Strains.
Four raw read sets (3HP, 5HP, China, and S02) were de novo assembled by the assembler Velvet (13). The parameter k used in constructing the de Bruijn graphs was tuned to deliver the best assembly quality, which was determined based on the N50 statistic of the assembled sequences with length >200. After assembly, the four draft genomes were aligned with the two reference genomes (RIMD 2210633, BB22OP) by using a multiple whole-genome alignment tool (14). A total of 315 contigs were found in all three pathogenic genomes (3HP, 5HP, China) and in none of the non-AHPND strains.
Isolation of Plasmids from the AHPND and Non-AHPND V. parahaemolyticus Strains.
The plasmid DNA from the bacteria cells was extracted by using a TENS solution (0.09 N NaOH and 0.45% SDS in Tris⋅EDTA buffer). Briefly, bacterial cells were cultured overnight, a 1-mL sample was taken and centrifuged at 14,000 × g for 2 min, and, after the supernatant was discarded, the residue was mixed with 300 μL TENS solution followed by 150 μL of sodium acetate (3 M, pH 5.2). The mixture was then centrifuged again at 14,000 × g for 5 min and the supernatant was transferred to a new tube, mixed with an equal volume of phenol–chloroform (1:1), and centrifuged at 14,000 × g for 10 min to separate the two liquids. The upper layer was transferred to a new tube, and DNA was precipitated by prechilled 100% ethanol. After the precipitate was air dried, the resulting DNA pellet was dissolved in TE buffer (pH 8.0, supplemented with RNase) and analyzed on a 0.8% agarose gel. A 1-kb plus DNA ladder (Invitrogen) and uncut plasmids from P. stewartii SW2 were used as size markers.
Southern Blotting.
DNA fragments of AP1, AP2, pirBVP, and pndA were amplified by the primer sets AP1F/AP1R, AP2F/AP2R, PirB400F/PirB400R, and pnd-9/pnd-10 (Table S2), respectively, and labeled by Amersham AlkPhos Direct Labeling and Detection Systems (GE Healthcare) according to the user manual. Plasmid DNA extracted from each strain was separated in a 0.8% agarose gel, denatured, transferred onto a Hybond-N+ nylon membrane, and hybridized with the labeled probes as previously described (23). The hybridized plasmids were visualized with a Gene Image CDP-Star detection module (GE Healthcare) and exposed to an X-ray film (Super RX; Fuji).
Table S2.
Primers used in this study
| Primer | Sequence (5′-3′) |
| pir-1 | GAGTCGACGGCATCAGTTACGTTAG |
| pir-2 | GAGGTACCCGATGAATTTGGTACAG |
| pir-3 | GTGGTACCTCGACAGTCCAATCGTGAG |
| pir-4 | GAGGTACCGTCAGAGAACCACGGTTG |
| pir-5 | GAGAGCTCCGTTCACAGTCGCTACAG |
| pir-8 | CTGAGCTCCCTTAGTGCCAGCGATC |
| pnd-1 | GCCGATACGGTGAATGAATC |
| pnd-2 | GCATCGCTTGCCTCCAGTC |
| pnd-3 | CAGGAAGGGCGGTTACAG |
| pnd-4 | GTCATAGTATGGCGATGAC |
| PirA_1F | ATGAGTAACAATATAAAACATG |
| PirA_336R | TTAGTGGTAATAGATTGTACAG |
| PirA_1F_NdeI | GCGCATATGAGTAACAATATAAAACATG |
| PirA_336R_XhoI | GCGCTCGAGGTGGTAATAGATTGTACAG |
| PirB_1F | ATGACTAACGAATACGTTGTAAC |
| PirB_1317R | CTACTTTTCTGTACCAAATTCAT |
| PirB_1F_NdeI | GCGCATATGACTAACGAATACGTTGTAAC |
| PirB_1317R_XhoI | GCGCTCGAGCTTTTCTGTACCAAATTCAT |
| PirB400F | GAGCCAGATATTGAAAACATTTGG |
| PirB400R | CCACGCAGCGAGTTCTGTAATGTA |
| VP tdh F | GTAAAGGTCTCTGACTTTTGGAC |
| VP tdh R | TGGAATAGAACCTTCATCTTCACC |
| VP trh F | TTGGCTTCGATATTTTCAGTATCT |
| VP trh R | CATAACAAACATATGCCCATTTCCG |
| VP tlh F | AAAGCGGATTATGCAGAAGCACTG |
| VP tlh R | GCTACTTTCTAGCATTTTCTCTGC |
| VP ldh F | GTTCTTCGCCAGTTTTGCGT |
| VP ldh R | GACATTACGTTCTTCGCCGC |
| AP1F | CCTTGGGTGTGCTTAGAGGATG |
| AP1R | GCAAACTATCGCGCAGAACACC |
| AP2F | TCACCCGAATGCTCGCTTGTGG |
| AP2R | CGTCGCTACTGTCTAGCTGAAG |
| pnd-9 | GCCTCTTCACTGACTGGA |
| pnd-10 | GAGCGGCTTCCTGTAACC |
| AP1-F16 | GTAGTAAAGAGGACTTGAGGA |
| AP2-R1 | ACACTAAGTCTTGCCGTTTC |
The restriction enzyme cutting sites used for cloning purpose are underlined.
Construction of the pirABvp and pndA Deletion Mutants and the Complemented PirABvp Strain.
The pirAB deletion mutant, ΔpirABvp, was constructed by in vivo allelic exchange as described in Vibrio vulnificus (32). Briefly, DNA fragments from the pirABvp down- and upstream regions were amplified with the primer pairs pir-1/pir-2 and pir-3/pir-4 (Table S2), respectively, and cloned into pGEMT-easy vector (Promega) in the correct orientation to produce a recombinant fragment containing a 1,587 bp-deletion in the pirABvp operon. This DNA fragment was removed from the pGEMT-easy vector by enzyme digestion with SalI, and then cloned into the suicide vector pDS132. The recombinant suicide plasmid was then used to produce the mutant by allelic exchange. The isolated mutant was checked by PCR (Fig. 2B), and its DNA sequence was determined to confirm the deletion was in-frame.
To construct the pirABvp-complemented strain, the entire pirABvp operon together with its putative promoter was amplified with primers pir-5 and pir-8 and then cloned into a broad-host range plasmid, pIT009 (23), at the SacI site. The resultant plasmid was transformed into Escherichia coli S17-1λpir, from which it was transferred into the ΔpirABvp by conjugation as described previously (23). We confirmed that the ΔpirABvp mutant and the complemented strain exhibited growth in LB broth that was similar to that of the WT strain (Fig. S6). The same pirABvp-expressing plasmid was also transferred by conjugation into the pVA1-cured derivative, and, again, the pVA1-cured strain and the pVA1-cured+pirABvp strain exhibited growth that was similar to that of the WT (Fig. S6). Expression of the PirAvp and PirBvp toxins in each of these experimentally manipulated strains was confirmed by Western blot analysis.
Isolation of Plasmid-Cured Derivative.
First, the pndA deletion mutant, ΔpndA, was constructed by using the same in vivo allelic exchange methods as described earlier except that the primer pairs pnd-1/pnd-2 and pnd-3/pnd-4 (Table S2) were respectively used to amplify fragments from the up- and downstream regions and the recombinant fragment was cloned into pDS132 between SphI and SacI sites. The plasmid pVA1 was then cured from the strain by the plasmid curing method (23). Briefly, an AP1 DNA fragment was cloned into pDS132 (33), which is a suicide vector that has chloramphenicol resistance. The resulting construct was then integrated into pVA1 by homologous recombination via AP1 to obtain a strain with chloramphenicol-resistant pVA1. This strain was cultured in LB broth containing 5 μg/mL of acridine orange (Sigma) at 37 °C with agitation (230 rpm) overnight and then plated on LB agar. Individual colonies were then screened for loss of antibiotic resistance to chloramphenicol to identify strains that had been cured of the pVA1 plasmid. To confirm the absence of pVA1 in colonies sensitive to chloramphenicol, we analyzed the plasmid profiles by extracting the plasmids and separating them in a 0.8% agarose gel (Fig. 4A). We also performed PCR analysis by using primers derived from AP1, AP2, pirAvp, and pirBvp (Fig. 4B). Finally, we confirmed the viability of the cured strain by checking that the growth of the plasmid-cured strain in LB broth was similar to that of the WT (Fig. S6).
Immersion Challenge Test.
Before the challenge test, juvenile (∼1 g) specific pathogen-free white shrimp (Penaeus vannamei) were acclimated in 40-L tanks containing artificial seawater (30‰ salinity, 28 °C) for 3 d. For the immersion challenge, individual colonies of the WT and the pVA1-cured, pVA1-cured+pirABvp, ΔpirABvp, and ΔpirABvp+pirABvp strains of V. parahaemolyticus were selected and cultured overnight in TSB+ medium [containing 2% (wt/vol) sodium chloride] at 30 °C. The cultures were then diluted 1:100 with fresh TSB+ medium, and cultured again at 30 °C with agitation until the bacterial density reached an OD600 of 0.8∼0.9. The bacterial suspension was then adjusted to a bacterial density of ∼1 × 107 cfu⋅mL−1 (with TSB+), and groups of 15 shrimp were immersed in 200 mL of this suspension for 15 min. The shrimp and bacterial suspension were then poured into tanks containing 20 L of seawater to give a final bacterial density of ∼1 × 105 cfu⋅mL−1. The control shrimp were immersed in TSB+ medium for 15 min as described earlier. The mortality of each group was recorded every 12 h. Moribund shrimp and shrimp that survived until 96 h post infection were fixed with Davison's alcohol formalin acetic acid (AFA) fixative for histopathological examination. All experiments were done in triplicate.
Histopathology Examination.
At selected time points after the shrimp were challenged with different V. parahaemolyticus strains as described earlier, random individual samples were taken and fixed with Davidson AFA fixative for 48 h. Tissue was sectioned and stained with hematoxilin and eosin-phloxine (H&E) following standard histological methods (34). The specimens were analyzed and photographed by a light microscopy system.
Reverse Gavage Injection of Pir Toxin into Shrimp.
The reverse gavage injection protocols followed those of Aranguren et al. (35). Briefly, 100 μL of the 2 μM purified PirAvp or PirBvp recombinant protein or rPirAvp/rPirBvp protein mixture were delivered into juvenile (∼3 g body weight) P. vannamei by reverse gavage through the anal cavity. At 24 h post injection, the moribund shrimp were collected and fixed with Davidson AFA fixative for histopathological examination. Shrimp receiving 100 μL of PBS solution by reverse gavage were used as the negative control.
Purification of the Recombinant PirAvp and PirBvp Proteins.
The full-length encoding sequence of pirAvp and pirBvp genes were amplified by PCR with the primer sets Vp_PirA_1F/Vp_PirA_336R and PirB_1F/PirB_1317R, respectively (Table S2). After the sequences were confirmed, pirAvp and pirBvp amplicons were separately ligated into a pET21b expression vector (Novagen). The resultant vectors were transformed into E. coli BL21 (DE3) cells, and expression of the recombinant, C-terminal His6-tagged PirAvp and PirBvp proteins, respectively, was induced by the addition of 1 mM isopropyl-b-d-thiogalactopyranoside (IPTG). After expression had proceeded at 16 °C for 16 h, the recombinant proteins were purified by resuspending the IPTG-induced BL21 cells in binding buffer (20 mM Tris⋅base, pH 8.0; 100 mM NaCl for PirAvp and 300 mM NaCl for PirBvp; 20 mM imidazole). After cell disruption with a french press, the lysate was centrifuged at 15,000 × g for 10 min to separate the soluble and insoluble proteins. The soluble protein fraction was loaded into a 5-mL Ni-NTA column and the column was then washed with the same binding buffer (200 mL). The PirAvp or PirBvp proteins that bound to the column were eluted by using a 20–500 imidazole gradient. The proteins were then suspended in a gel filtration buffer [for PirAvp, 30 mM Tris⋅HCl, pH 7.4, 100 mM NaCl, 1 mM DTT, and 5% (vol/vol) glycerol; for PirBvp, 20 mM Tris⋅base, pH 8.0, 300 mM NaCl] and passed through a Superdex 200 gel filtration column (GE Healthcare) to further improve the purity.
Per Os Challenge with Pir Toxins.
Groups of 15 shrimp (mean weight, ∼3 g) were acclimated in 40-L tanks containing seawater. The shrimp starved for 1 d before being challenged with shrimp pellets equal to 3% of total shrimp body weight (i.e., 1.35 g of pellets per tank) that had been soaked for 10 min in 1.35 mL of PBS solution containing 17.53 μM purified recombinant PirAvp, PirBvp, PirAvp+PirBvp, or BSA (negative control). The mortality of each group was recorded every 24 h. All experiments were performed in triplicate.
Per Os Challenge with E. coli Recombinants Expressed Pir Toxins.
As with the Pir toxin challenge, groups of 15 acclimated shrimp (∼3 g) in 40-L tanks were starved for 1 d before challenge, and the challenge was administered by using the same quantity of shrimp pellets, i.e., 3% of the total shrimp body weight per tank. Expression of the recombinant, C-terminal His6-tagged PirAvp and PirBvp proteins was induced in E. coli BL21 (DE3) cells by the addition of 1 mM IPTG at 16 °C for 16 h as described earlier. Culture medium (1.35 mL) containing the IPTG- induced E. coli strains BL21 (ppirAvp), BL21 (ppirBvp), and BL21 (ppirAvp) + BL21 (ppirBvp), or BL21 (pET21b; expression vector only; negative control) was then used to soak the shrimp pellets, and the pellets were immediately fed to the shrimp. The mortality of each group was recorded every 24 h. All experiments were performed in triplicate.
Use of Gel Filtration to Confirm the Formation of the PirAvp/PirBvp Complex.
For PirAvp and PirBvp analysis, 0.5 mg purified C-terminal His6-tagged PirAvp or 1 mg of C-terminal His6-tagged PirBvp in 500 μL gel filtration buffer [30 mM Tris⋅base, pH 8.0, 100 mM NaCl, 1 mM DTT, and 5% (vol/vol) glycerol] were loaded into a Superose 16 gel filtration column (GE Healthcare). For the PirAvp/PirBvp complex analysis, 0.5 mg purified C-terminal His6-tagged PirAvp was mixed with 1 mg of C-terminal His6-tagged PirBvp in 500 μL gel filtration buffer and incubated for 15 min at room temperature. The mixture was then analyzed by using the same gel filtration column. The flow rate was 0.5 mL/min, and 0.5-mL fractions were collected. The fractions from each peak were analyzed by SDS/PAGE and stained with Coomassie blue (Sigma-Aldrich).
Recombinant PirAvp and PirBvp Crystallization, Data Collection, and Structure Determination.
For PirAvp crystallization, 2 µL of the purified C-terminal tagged PirAvp (10 mg/mL) were mixed with a 2-µL reservoir of 20% (wt/vol) PEG 3350 and 0.2 M potassium nitrate. Before flash-cooling, ethylene glycol at a final concentration of 15% (wt/vol) was added as a cryoprotectant. X-ray diffraction data for native and HgCl2-soaked PirAvp crystals were collected on beamline 13C at the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan.
For PirBvp crystallization, two different conditions were used. The first PirBvp crystal was obtained by mixing 2 µL of the purified C-terminal tagged PirBvp (15 mg/mL) with a 2 µL reservoir of 20% (vol/vol) 1,4-butanediol, and 0.1 M sodium acetate, pH 4.5. After 15% (vol/vol) 2,3-butanediol was added as the cryoprotectant, this crystal was used to collect native PirBvp X-ray diffraction data. Another PirBvp crystal was obtained by screening for a crystal of the PirAvp/PirBvp complex. For this crystal, 2 µL of the purified PirAvp/PirBvp complex (12 mg/mL) was mixed with a 2-µL reservoir of 0.2 M potassium sodium tartrate tetrahydrate, 0.1 M sodium citrate tribasic dihydrate, pH 5.6, and 2.0 M ammonium sulfate. The resulting crystal was soaked in 5 mM HgCl2, and 15% (wt/vol) ethylene glycol was used as cryoprotector. X-ray diffraction data of the HgCl2-soaked crystal was collected on NSRRC beamline 13B, and, instead of a PirAvp/PirBvp crystal, we determined that the resulting crystal was in fact PirBvp.
The collected X-ray diffraction data were processed by using HKL2000 software (36). The structures of PirAvp and PirBvp were determined by using the single-wavelength anomalous diffraction phasing method and the program Shelx CDE (37). Both crystals contained two protein molecules in an asymmetric unit. The major heavy atom sites were found at His72 (chain A), His81 (chain A and B), and His111 (chain A and B) of PirAvp, and Cys175, Cys239, and Cys407 in both chains (A and B) of PirBvp, plus a minor site at Met40 (chain A). The initial models were produced by the program BUCCANEER (38). Both PirAvp and PirBvp phases were further extended to high resolution by using the native data sets, and the programs COOT (39) and Refmac (40) were used for the subsequent refinement. Statistics for the data collection and refinement are shown in Table S3. The CCP4 package (41) and Pymol program (version 1.2r3pre) were used for the structural analyses and also for figure production.
Native PAGE and SDS/PAGE Western Blots of WT and CP Culture Supernatants.
After the WT strain and the pVA1-cured+pirABvp (CP) derivative were cultured separately at 37 °C for 6 h, 10 mL of the culture was collected, centrifuged at 15,300 × g for 10 min to remove the cell debris, and concentrated into 500 μL by using Amicon 10-kDa filter tubes. For native PAGE separation, each lane was loaded with 16 μL of supernatant mixed with 4 μL of 5× sample buffer [0.25 M Tris⋅HCl, pH 6.8, 50% (vol/vol) glycerol, 0.2% bromophenol blue]. Separation was performed by using 12% (vol/vol) resolving gel [3.3 mL ddH2O, 4.0 mL 30% (wt/vol) acrylamide mix, 2.5 mL 1.5 M Tris⋅HCl, pH 8.8, 0.1 mL 10% (wt/vol) ammonium persulfate, 0.004 mL TEMED (Tetramethylethylenediamine)] and stacking gel [2.7 mL ddH2O, 0.67 mL 30% (wt/vol) acrylamide mix, 0.5 mL 1 M Tris⋅HCl, pH 6.8, 0.04 mL 10% (wt/vol) SDS, 0.04 mL 10% (wt/vol) ammonium persulfate, 0.002 mL TEMED]. The separated proteins were transferred to PVDF membrane (400 mA, 1.5 h) and, after blocking overnight with blocking buffer [3% (wt/vol) skim milk in 0.5% TBST buffer (0.5 M NaCl, 50 mM Tris⋅HCl, pH 7.5, 0.5% Tween 20)] at 4 °C, the membrane was incubated with appropriate antibody for 1 h at room temperature. The membrane was then washed three times with 0.5% TBST buffer and incubated with a secondary antibody conjugated with HRP for 1 h at room temperature. The chemiluminescent signal was detected by using an ImageQuant LAS 4000 Mini imaging system (GE Healthcare). SDS/PAGE was performed by using the same procedures except that the sample buffer included the reducing agent β-mercaptoethanol (10 mM), the supernatant and buffer mixture was boiled for 5 min before loading, the buffer contained 10% (wt/vol) SDS, and the resolving gel contained 0.1 mL 10% (wt/vol) SDS.
Acknowledgments
The four V. parahaemolyticus strains, 3HP, 5HP, China, and S02 were provided by Dr. Timothy W. Flegel, Dr. Kallaya Sritunyalucksana, and Dr. Siripong Thitamadee. We thank the staff of beamlines BL13B1 and 13C1 at the Radiation Research Center in Hsinchu, Taiwan, for their help in X-ray crystal data collection and Paul Barlow for his helpful criticism of the manuscript. This work was financially supported by the Ministry of Education, Taiwan, Republic of China, under National Cheng Kung University Aim for the Top University Project Promoting Academic Excellence and Developing World Class Research Grant D103-38A01; National Science Council (MSC) of Taiwan Grants NSC 103-2321-B-006-014 and NSC 103-2622-B-006-008; Ministry of Science and Technology (MOST) Grants MOST 103-2313-006-005-MY2, MOST 103-2622-B-006-012-CC1, and MOST 103-2633-B-006-004; Council of Agriculture, Executive Yuan Grants G103-B032 and G103-b329; and the Chen Jie Chen Scholarship Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3X0T (PirAvp) and 3X0U (PirBvp)]. The sequence of plasmid pVA1 has been deposited in the GenBank database (accession no. KP324996).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1503129112/-/DCSupplemental.
References
- 1.NACA-FAO . Quarterly Aquatic Animal Disease Report (Asia and Pacific Region), 2011/2, April-June 2011. NACA; Bangkok: 2011. [Google Scholar]
- 2.Lightner DV, Redman RM, Pantoja CR, Noble BL, Tran L. Early mortality syndrome affects shrimp in Asia. Global Aquaculture Advocate. 2012;15:40. [Google Scholar]
- 3.Gomez-Gil B, Soto-Rodríguez S, Lozano R, Betancourt-Lozano M. Draft genome sequence of Vibrio parahaemolyticus Strain M0605, which causes severe mortalities of shrimps in Mexico. Genome Announc. 2014;2(2):e00055-14. doi: 10.1128/genomeA.00055-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunan L, Lightner D, Pantoja C, Gomez-Jimenez S. Detection of acute hepatopancreatic necrosis disease (AHPND) in Mexico. Dis Aquat Organ. 2014;111(1):81–86. doi: 10.3354/dao02776. [DOI] [PubMed] [Google Scholar]
- 5.FAO Fisheries and Aquaculture Report of the FAO/MARD Technical Workshop on Early Mortality Syndrome (EMS) or Acute Hepatopancreatic Necrosis Syndrome (AHPNS) of Cultured Shrimp (under TCP/VIE/3304) 2013 rep. no. 1053, Hanoi, Viet Nam, 25–27 June 2013. Available at www.fao.org/docrep/018/i3422e/i3422e.pdf. Accessed July 28, 2015.
- 6.Tran L, et al. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis Aquat Organ. 2013;105(1):45–55. doi: 10.3354/dao02621. [DOI] [PubMed] [Google Scholar]
- 7.Joshi J, et al. Variation in Vibrio parahaemolyticus isolates from a single Thai shrimp farm experiencing an outbreak of acute hepatopancreatic necrosis disease (AHPND) Aquaculture. 2014;428–429:297–302. [Google Scholar]
- 8.De Schryver P, Defoirdt T, Sorgeloos P. Early mortality syndrome outbreaks: A microbial management issue in shrimp farming? PLoS Pathog. 2014;10(4):e1003919. doi: 10.1371/journal.ppat.1003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang YT, et al. Draft genome sequences of four strains of Vibrio parahaemolyticus, three of which cause early mortality syndrome/acute hepatopancreatic necrosis disease in shrimp in China and Thailand. Genome Announc. 2014;2(5):e00816-14. doi: 10.1128/genomeA.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo H, et al. Draft genome sequences of six strains of Vibrio parahaemolyticus isolated from early mortality syndrome/acute hepatopancreatic necrosis disease shrimp in Thailand. Genome Announc. 2014;2(2):e00221–e14. doi: 10.1128/genomeA.00221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez-Jimenez S, et al. High-quality draft genomes of two Vibrio parahaemolyticus strains aid in understanding acute hepatopancreatic necrosis disease of cultured shrimps in Mexico. Genome Announc. 2014;2(4):e00800–e00814. doi: 10.1128/genomeA.00800-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han JE, Tang KFJ, Tran LH, Lightner DV. Photorhabdus insect-related (Pir) toxin-like genes in a plasmid of Vibrio parahaemolyticus, the causative agent of acute hepatopancreatic necrosis disease (AHPND) of shrimp. Dis Aquat Organ. 2015;113(1):33–40. doi: 10.3354/dao02830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angiuoli SV, Salzberg SL. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27(3):334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aziz RK, et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen AK, Thorsted P, Thisted T, Wagner EGH, Gerdes K. The rifampicin-inducible genes srnB from F and pnd from R483 are regulated by antisense RNAs and mediate plasmid maintenance by killing of plasmid-free segregants. Mol Microbiol. 1991;5(8):1961–1973. doi: 10.1111/j.1365-2958.1991.tb00818.x. [DOI] [PubMed] [Google Scholar]
- 17.Waterfield N, Kamita SG, Hammock BD, ffrench-Constant R. The Photorhabdus Pir toxins are similar to a developmentally regulated insect protein but show no juvenile hormone esterase activity. FEMS Microbiol Lett. 2005;245(1):47–52. doi: 10.1016/j.femsle.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 18.Soto-Rodriguez SA, Gomez-Gil B, Lozano-Olvera R, Betancourt-Lozano M, Morales-Covarrubias MS. Field and experimental evidence of Vibrio parahaemolyticus as the causative agent of acute hepatopancreatic necrosis disease of cultured shrimp (Litopenaeus vannamei) in Northwestern Mexico. Appl Environ Microbiol. 2015;81(5):1689–1699. doi: 10.1128/AEM.03610-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soberón M, et al. Pore formation by Cry toxins. Adv Exp Med Biol. 2010;677:127–142. doi: 10.1007/978-1-4419-6327-7_11. [DOI] [PubMed] [Google Scholar]
- 20.Xu C, Wang BC, Yu Z, Sun M. Structural insights into Bacillus thuringiensis Cry, Cyt and parasporin toxins. Toxins (Basel) 2014;6(9):2732–2770. doi: 10.3390/toxins6092732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins JL, Lee MK, Valaitis AP, Curtiss A, Dean DH. Bivalent sequential binding model of a Bacillus thuringiensis toxin to gypsy moth aminopeptidase N receptor. J Biol Chem. 2000;275(19):14423–14431. doi: 10.1074/jbc.275.19.14423. [DOI] [PubMed] [Google Scholar]
- 22.Crosa JH. A plasmid associated with virulence in the marine fish pathogen Vibrio anguillarum specifies an iron-sequestering system. Nature. 1980;284(5756):566–568. doi: 10.1038/284566a0. [DOI] [PubMed] [Google Scholar]
- 23.Lee CT, et al. A common virulence plasmid in biotype 2 Vibrio vulnificus and its dissemination aided by a conjugal plasmid. J Bacteriol. 2008;190(5):1638–1648. doi: 10.1128/JB.01484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Roux F, et al. Virulence of an emerging pathogenic lineage of Vibrio nigripulchritudo is dependent on two plasmids. Environ Microbiol. 2011;13(2):296–306. doi: 10.1111/j.1462-2920.2010.02329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vadivelu J, et al. Hemolysins and plasmid profiles of Vibrio parahaemolyticus. Southeast Asian J Trop Med Public Health. 1996;27(1):126–131. [PubMed] [Google Scholar]
- 26.Kongrueng J, et al. Characterization of Vibrio parahaemolyticus causing acute hepatopancreatic necrosis disease in southern Thailand. J Fish Dis. 2014 doi: 10.1111/jfd.12308. [DOI] [PubMed] [Google Scholar]
- 27.Soberón M, Gill SS, Bravo A. Signaling versus punching hole: How do Bacillus thuringiensis toxins kill insect midgut cells? Cell Mol Life Sci. 2009;66(8):1337–1349. doi: 10.1007/s00018-008-8330-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pigott CR, Ellar DJ. Role of receptors in Bacillus thuringiensis crystal toxin activity. Microbiol Mol Biol Rev. 2007;71(2):255–281. doi: 10.1128/MMBR.00034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coplin DL, Rowan RG, Chisholm DA, Whitmoyer RE. Characterization of plasmids in Erwiniastewartii. Appl Environ Microbiol. 1981;42(4):599–604. doi: 10.1128/aem.42.4.599-604.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grochulski P, et al. Bacillus thuringiensis CryIA(a) insecticidal toxin: crystal structure and channel formation. J Mol Biol. 1995;254(3):447–464. doi: 10.1006/jmbi.1995.0630. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed Cold Spring Harbor Lab Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 32.Shao CP, Hor LI. Metalloprotease is not essential for Vibrio vulnificus virulence in mice. Infect Immun. 2000;68(6):3569–3573. doi: 10.1128/iai.68.6.3569-3573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid. 2004;51(3):246–255. doi: 10.1016/j.plasmid.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Lightner DV. A Handbook of Shrimp Pathology and Diagnostic Procedures for Diseases of Cultured Penaeid Shrimp. World Aquaculture Society; Baton Rouge, LA: 1996. [Google Scholar]
- 35.Aranguren FL, Tang KFJ, Lightner DV. Quantification of the bacterial agent of necrotizing hepatopancreatitis (NHP-B) by real-time PCR and comparison of survival and NHP load of two shrimp populations. Aquaculture. 2010;307(3–4):187–192. [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 9):1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vagin AA, et al. REFMAC5 dictionary: Organisation of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2284–2295. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]