

Significance
Diphthamide is a conserved modification on eukaryotic translation elongation factor 2 (eEF2). Analyses of genetically defined diphthamide-deficient cell lines indicate that this modification determines not only sensitivity of cells to the ADP-ribosylating toxins Pseudomonas exotoxin A and diphtheria toxin, but it also modulates nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) and TNF receptor signaling pathways.
Keywords: ADP-ribosylation of eEF2, Pseudomonas exotoxin, diphtheria toxin, translation, DPH gene knockout
Abstract
The diphthamide on human eukaryotic translation elongation factor 2 (eEF2) is the target of ADP ribosylating diphtheria toxin (DT) and Pseudomonas exotoxin A (PE). This modification is synthesized by seven dipthamide biosynthesis proteins (DPH1–DPH7) and is conserved among eukaryotes and archaea. We generated MCF7 breast cancer cell line-derived DPH gene knockout (ko) cells to assess the impact of complete or partial inactivation on diphthamide synthesis and toxin sensitivity, and to address the biological consequence of diphthamide deficiency. Cells with heterozygous gene inactivation still contained predominantly diphthamide-modified eEF2 and were as sensitive to PE and DT as parent cells. Thus, DPH gene copy number reduction does not affect overall diphthamide synthesis and toxin sensitivity. Complete inactivation of DPH1, DPH2, DPH4, and DPH5 generated viable cells without diphthamide. DPH1ko, DPH2ko, and DPH4ko harbored unmodified eEF2 and DPH5ko ACP- (diphthine-precursor) modified eEF2. Loss of diphthamide prevented ADP ribosylation of eEF2, rendered cells resistant to PE and DT, but does not affect sensitivity toward other protein synthesis inhibitors, such as saporin or cycloheximide. Surprisingly, cells without diphthamide (independent of which the DPH gene compromised) were presensitized toward nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) and death-receptor pathways without crossing lethal thresholds. In consequence, loss of diphthamide rendered cells hypersensitive toward TNF-mediated apoptosis. This finding suggests a role of diphthamide in modulating NF-κB, death receptor, or apoptosis pathways.
Eukaryotic translation elongation factor 2 (eEF2) is a highly conserved protein and essential for protein biosynthesis. EEF2 enables peptide-chain elongation by translocating the peptide–tRNA complex from the A- to the P-site of the ribosome (1, 2). The diphthamide modification at His715 of human eEF2 (or at the corresponding position in other species) is conserved in all eukaryotes (3) and in archaeal counterparts. It is generated by proteins that are encoded by seven genes (4). Proteins encoded by dipthamide biosynthesis protein (DPH)1, DPH2, DPH3, and DPH4 (DNAJC24) attach a 3-amino-3-carboxypropyl (ACP) group to eEF2. This intermediate is converted by the methyltransferase DPH5 to diphthine, which is subsequently amidated to diphthamide by DPH6 and DPH7 (5).
Diphthamide synthesis was previously described in yeast and other eukaryotes (4–6). However, the “complete picture” is (with the exception of the yeast pathway) to a large portion is composed of observations made in different cell types on single genes. Many reports related to diphthamide synthesis of mammalian cells describe “partial knockouts” and “partial phenotypes” (i.e., reduced levels but not complete loss of diphthamide modification or toxin sensitivities) (7–9). Because mammalian genomes are more complex than that of yeast, carrying extendend gene families, mammalian cells may compensate—at least to some degree—functional loss of genes that may be unique and essential in yeast. If and to what degree mammalian cells can compensate a partial or complete loss of DPH gene functionality (and with what consequences) is unknown to date.
So far, the function of diphthamide on eEF2 also remained rather elusive. Reports indicate that it contributes to translation fidelity (10–13). On the other hand, DPH genes or eEF2 can be mutated to prevent diphthamide attachment, yet cells carrying such mutations are viable (5, 11, 14, 15). Animals with heterozygous DPH knockouts (DPHko) can be generated, but homozygous DPH1ko, DPH3ko, and DPH4ko are embryonic lethal (13, 16–18). Because these studies are based on inactivation of individual genes, it is difficult to discriminate between phenotypes caused by gene loss and phenotypes as a consequence of loss of diphthamide.
Diphthamide-modified eEF2 is the target of ADP ribosylating toxins, including Pseudomonas exotoxin A (PE) and diphtheria toxin (DT) (19). These bacterial proteins enter cells and catalyze ADP ribosylation of diphthamide using nictotinamide adenine dinucleotide (NAD) as substrate (20, 21). This inactivates eEF2, arrests protein synthesis, and kills (14). Tumor-targeted PE and DT derivatives are applied in cancer therapies (22–28) and their efficacy depends on toxin sensitivity of target cells. Therefore, information about factors (and their relative contributions) that influences cellular sensitivities toward diphthamide-modifying toxins may predict therapy responses. For example, alterations in OVCA1 (human DPH1) were described for ovarian cancers (16, 29), yet it is not known if and to what degree such alterations would affect sensitivities of tumor cells toward PE-derived drugs.
Here we describe MCF7 breast cancer cell line derivatives with heterozygous or complete DPH gene inactivations. These cells are applied to analyze the contributions of individual DPHs not only to diphthamide synthesis and toxin sensitivity, but also to address gene dose effects. Because the set of knockout cell lines is derived from the same parent cell and provides loss of diphthamide as common consequence of inactivation of different genes, these cells can also shed light on the biological relevance of the diphthamide modification.
Results
Generation of MCF7 Cells with Heterozygous or Completely Inactivated DPH Genes.
Gene-specific zinc finger nucleases (ZFN) (30) were applied to generate MCF7 cells with inactivated DPH genes (SI Text S1–S3). Plasmids encoding ZFNs were transfected into MCF7. Forty-eight hours later, to enable ZFN binding, double-strand breaks, and mis-repair, mutated cells were identified by either phenotype selection or by genetic analyses, as described in detail in Fig. S1. For phenotype selection, cells were exposed to lethal doses of PE (100 nM) to kill all cells whose eEF2 is a substrate for the toxin. After an additional 48 h, dead cells were removed and the culture propagated in toxin-containing media. This procedure generated colonies of cells transfected with ZFNs for DPH1, -2, -4, and -5. No colonies were obtained under toxin selection in cells that were mock transfected, or with ZFNs that target DPH3, -6, and -7 (Table S1). Genetic analyses of single-cell clones revealed that all resistant isolates contained only defective (out-of-frame) copies of DPH1, DPH2, DPH4, or DPH5 genes. None of the toxin-resistant cells contained unaltered functional gene copies (SI Text S1).
Fig. S1.

Generation of MCF7 derivatives. (A) ZFN transfected cells are either exposed to PE followed by isolation and characterization of surviving cells (Left branch), or individually cloned and subjected to PCR-based genetic analyses without toxin exposure (Right branch and example for HRM analyses below). (B) ZFN target sequences and allele sequences of mutated MCF7 clones.
Table S1.
Summary of the individual clones that were obtained
| Gene | No. of transf. cells (t or g*) | No. of clones toxin selection | No. of clones genetic screen |
| DPH1 | 4 × 105 (t) | 3 (KO-KO) | 4 (WT-KO) |
| 1,6 × 106 (g) | 2 (KO-KO) | ||
| DPH2 | 4 × 105 (t) | 3 (KO-KO) | 4 (WT-KO) |
| 2 × 106 (g) | 0 (KO-KO) | ||
| DPH3 | 3.6 × 106 (t) | 0 | 5 (WT-KO) |
| 3.6 × 106 (g) | 0 (KO-KO) | ||
| DPH4 | 4 × 105 (t) | 3 (KO-KO) | 2 (WT-KO) |
| 1.2 × 106 (g) | 1 (KO-KO) | ||
| DPH5 | 4 × 105 (t) | 3 (KO-KO) | 3 (WT-KO) |
| 2.4 × 106 (g) | 1 (KO-KO) | ||
| DPH6 | 3.6 × 106 (t) | 0 | 4 (WT-KO) |
| 1.6 × 106 (g) | 0 (KO-KO) | ||
| DPH7 | 3.6 × 106 (t) | 0 | 3 (WT-KO) |
| 3.6 × 106 (g) | 0 (KO-KO) |
“t” or “g” indicates number of transfected cells subjected to toxin selection (t) and genetic screen (g). “No. of clones” lists the number of completely (KO-KO) or heterozygous (WT-KO) mutated individual clones (i.e., with different mutations) that were identified by sequence analyses of clones obtained by toxin selection or via HRM curve shapes in genetic screens.
To identify MCF7 mutants without toxin selection, single cells from each transfection were subjected to high-resolution melting (HRM) analyses genes (31). This technique identifies cells that contain two different alleles of the gene to be analyzed, as those generate biphasic or odd-shaped melting curves (SI Text S1). Analyses of candidate clones with bisphasic HRM profiles confirmed the presence of different DPH allele sequences. This approach delivered clones that had one gene copy inactivated and another functional wild-type copy for all DPH genes (Table 1 and SI Text S1). In addition, clones that had both genes inactivated with a different mutation on each allele were obtained for DPH4 and DPH5. For DPH3, DPH6, and DPH7, clones that had both genes inactivated could not be obtained in repeated attempts even though the ZFNs were effective (generated heterozygotes) and the number of colonies screened delivered several complete knockouts for other DPH genes.
Table 1.
DPH gene inactivation and toxin sensitivity
| DPHko | Alleles | IC50 nM PE | IC50 pM DT | IC50 nM CHX | IC50 pM TNFα |
| MCF7 | WT | 121 ± 12 | 12 ± 5 | 1,969 ± 454 | 1,403 ± 243 |
| DPH1 | WT-KO | 142 ± 124 | 14 ± 6 | 2,026 ± 1,238 | 1,220 ± 370 |
| KO-KO | >3,000 | >5,000 | 1,473 ± 244 | 193 ± 65 | |
| DPH2 | WT-KO | 95 ± 58 | 10 ± 2 | 2,158 ± 625 | 1,760 ± 579 |
| KO-KO | >3,000 | >5,000 | 1,945 ± 276 | 260 ± 81 | |
| DPH4 | WT-KO | 38 ± 27 | 14 ± 1 | 1,976 ± 1,122 | 1,765 ± 709 |
| KO-KO | >3,000 | >5,000 | 1,626 ± 594 | 330 ± 243 | |
| DPH5 | WT-KO | 133 ± 18 | 9 ± 3 | 1,769 ± 132 | 1,457 ± 599 |
| KO-KO | >3,000 | >5,000 | 1,030 ± 129 | 134 ± 37 | |
| DPH3 | WT-KO | 32 ± 19 | 11 ± 4 | 1,555 ± 23 | 1,217 ± 484 |
| DPH6 | WT-KO | 36 ± 32 | 18 ± 10 | 1,579 ± 1,014 | 985 ± 81 |
| DPH7 | WT-KO | 107 ± 80 | 16 ± 6 | 1,976 ± 1,002 | 891 ± 49 |
PE, DT, CHX, or TNF sensitivity was determined by BrdU incorporation assays. IC50 values calculated from dose–response curves shown in Fig. 2 and SI Text S4. Boldface entries indicate values of homozygous knockout cells.
DPH Gene Inactivation Influences H715 Modification and ADP Ribosylation.
Extracts of MCF7 and DPHko cells were subjected to Western blots with a rabbit mAb that we generated and which specifically detects unmodified eEF2 (see SI Text S2 and Fig. S2 for antibody generation). These analyses (Fig. 1A) revealed that unmodified eEF2 is virtually absent (below detection levels) in MCF7. Cells with complete inactivation of DPH1, DPH2, or DPH4 genes contained eEF2 without H715 modification as indicated by strong antibody signals. Unmodified eEF2 was also observed in cells with heterozygous DPH2 mutations, albeit to a much lower degree. All other heterozygous cell lysates generated only background signals. Thus, inactivation of DPH1, DPH2, and DPH4 interferes with H715 modification of eEF2.
Fig. S2.

Western blot identification of antibodies that specifically detect eEF2 without diphthamide.
Fig. 1.

Diphthamide modification and ADP ribosylation of eEF2. (A) Detection of unmodified eEF2 in parent and mutated MCF7 by a rabbit mAb that specifically detects eEF2 without diphthamide (SI Text S2). (B) Semiquantitative assessment of eEF2 modifications by MS (46) (SI Text S3). (C) ADP ribosylation of eEF2 was assessed in extracts exposed to PE Bio-NAD, followed by Western blot detection of Bio-ADPR-eEF2 (32).
Mass spectometry (MS) analyses were subsequently applied to extracts of DPH-mutated cell lines to determine the composition and eEF2 His715 modification in detail. This enabled the determination of relative levels of unmodified eEF2, of the ACP intermediate, and of diphthamide (SI Text S3 and Fig. S3). In wild-type cells, only diphthamide-modified eEF2 is detectable without evidence for unmodified eEF2 or diphthine or ACP modifications (Fig. 1B). Cells with complete inactivation of the DPH1, DPH2, DPH4, as well as DPH5 genes contained no diphthamide-modified eEF2. Thus, these genes are essential for diphthamide synthesis and their inactivation cannot be compensated by other genes. Complete inactivation of DPH1, DPH2, or DPH4 generated cells in which only unmodified eEF2 and no other modified form was detectable. Complete inactivation of DPH5 generated the ACP intermediate (eEF2 with this intermediate is not recognized by the antibody applied in the preceding Western blots) (Fig. 1A). The major eEF2 species in cells with one inactivated and one functional copy of DPH1 to -7 is diphthamide-modified eEF2. In contrast to parent MCF7, however, unmodified eEF2 was detectable in different amounts (up to 25% of the total eEF2) (Fig. 1B) upon heterozygous inactivation of DPH1 and DPH2. This finding demonstrates that gene-dose reduction by inactivation of one allele of DPH1 or DPH2 is insufficient to prevent diphthamide synthesis, but sufficient to modulate the amount of unmodified eEF2.
Fig. S3.

MS-based determination of eEf2 H715 modifications.
In vitro ADP ribosylation adressed the impact of partial or complete DPH inactivation: cell extracts were incubated with PE as enzyme and biotinylated NAD as substrate, followed by detection of Bio-ADPR-eEF2. This method reliably detects ADP ribosylation; however, it cannot be used for quantification of slight differences (32). Fig. 1C shows that eEF2 of parental MCF7, and of all seven heterozygote-inactivated MCF7 derivatives (DPH1–7) becomes ADP ribosylated by PE. In contrast, eEF2 from cells that have completely inactivated DPH1 or DPH2 or DPH4 or DPH5 genes is not amenable to ADP ribosylation. Only eEF2 with diphthamide, but not without modification (DPH1, -2, -4) or with partial modification (ACP in DPH5) serves as substrate for ADP ribosylating toxins. There is no other remaining toxic activity toward cells carrying these eEF2 forms.
Influence of Heterozygous and Complete DPH Inactivation on Toxin Sensitivity.
Susceptibilities toward PE and DT were determined for MCF7 derivatives with heterozygous or complete DPH gene inactivations. Fig. 2 and Table 1 show that complete inactivation of DPH1, DPH2, DPH4, or DPH5 genes confer absolute resistance of MCF7. Even toxin concentrations exceeding the doses that kill parent MCF7 by more than 10,000-fold did not influence cell growth or proliferation of mutant cells. Absolute resistance correlates with complete absence of diphthamide-modified eEF2 and lack of ADP ribosylation (Fig. 1). Thus, cytotoxicity inferred by PE and DT is solely a result of diphthamide-dependent ADP ribosylation of eEF2 without any other cytotoxic modality. In contrast to cells without functional DPH gene copy, MCF7 derivatives that contained one inactivated and one functional DPH gene (DPH1-DPH7) remained fully sensitive to PE and DT (Fig. 2). As all heterozygous DPH-inactivated MCF7 derivatives contain as the major species diphthamide-modified eEF2, toxin sensitivity correlates with the presence of diphthamide. Heterozygous DPH3, -4, -6, and -7 knockouts contain almost exclusively diphthamide-modified eEF2, indicating that loss of one functional allele of these genes does not affect efficacy of diphthamide synthesis. Heterozygous DPH1 or DPH2 cells contain detectable levels of unmodified toxin-resistant eEF2 (up to 25% in DPH2ko). Thus, DPH1 and DPH2 gene doses can influence diphthamide synthesis to some degree. Interestingly, despite harboring some toxin-resistant eEF2, such cells remained as toxin-sensitive as wild-type cells. Thus, DPH1 and DPH2 gene-dose modulates the relative amount of unmodified eEF2 in MCF7, but does not affect toxin sensitivity.
Fig. 2.
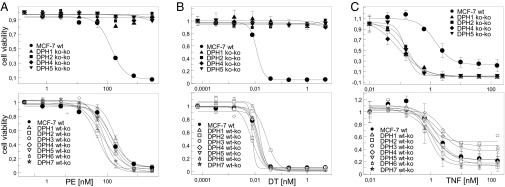
Influence of DPH inactivation on sensitivity to ADP ribosylating toxins and TNF. Dose–responses of cells with inactivation of DPH1, DPH2, DPH4, and DPH5 in comparison with parent MCF7 (bold line), exposed to PE (A), DT (B) or TNF (C). Complete knockouts (Upper) and partial knockous (Lower) are shown. Parent MCF7 (bold line) and partial knockouts show the same sensitivity to PE, DT, and TNF. Cells with complete inactivation of DPH1, DPH2, DPH4, and DPH5 are resistant to PE and DT and hypersensitive to TNF.
Loss of Diphthamide Has only a Minor Impact on Cell Growth and Does Not Alter Sensitivity Toward Protein Synthesis Inhibitors, Which Do Not Target eEF2.
The common denominator of all MCF7 derivatives with different DPH gene defects is loss of diphthamide. Thus, this cell line set cannot only elucidate the relative impact of each gene on toxin sensitivity, but also address the biological function of the diphthamide modification itself. To eliminate variations between individual clones, we have analyzed at least two independent clones for each knockout. Cytology indicated that morphology and chromosome composition of the individual clones did not diverge from that of parent MCF7 cells (SI Text S4). Because lack of diphthamide was achieved by inactivating different genes, common biological effects observed in these cell lines are attributable to loss of diphthamide, and not to loss of individual gene function or potential compensatory effects. Under normal growth conditions, we observed no impact of DPH inactivation on growth for all heterozygous clones (Fig. S4A). In addition, complete inactivation of DPH1, DPH2, or DPH4 did not cause significant reductions in cell growth or viability (Fig. S4A). We observed growth alterations for some clones, but these effects were not attributable to the gene itself because some clones showed differences but others with the same gene affected did not (SI Text S4). Cells with complete inactivation of DPH1, DPH2, and DPH4 harbor only unmodified eEF2. Thus, the exclusive presence of unmodified eEF2 by itself does not inhibit the growth of MCF7. Reduced growth rates were observed for all clones with completely inactivated DPH5. These contain ACP-modified eEF2, which occurs only in DPH5-deficient cells and not in other variants. Therefore, we cannot differentiate between growth reduction related to the presence of the eEF2-ACP intermediate or being a consequence of lost DPH5 function in other cellular processes.
Fig. S4.

(A) Growth, (B) susceptibility toward saporin and CHX, and (C) chromosome composition of MCF7 derivatives. (C) Images were captured with a Zeiss Plan-Neofluar microscope at 100× magnification.
Is the action of other protein synthesis inhibitors also affected in cells without diphthamide? Exposure of parent and DPH-inactivated MCF7 cells to saporin and cycloheximide (CHX) revealed no effect of DPH inactivation and loss of diphthamide on sensitivity (SI Text S4). Saporin and CHX inhibited the growth and killed DPH-inactivated cells to the same degree as wild-type MCF7 (Table 1, SI Text S4, and Fig. S4B). This finding indicates that stress posed upon cells by protein synthesis inhibition in general appears not to be aggravated by lack of diphthamide.
Loss of Diphthamide Activates Pathways That Resemble Nuclear Factor of Kappa Light Polypeptide Gene Enhancer in B Cells and Death Receptor Signaling, and Renders MCF7 Cells Hypersensitive to TNF.
ADP ribosylation of eEF2 stalls protein synthesis and induces apoptosis in MCF7 (33, 34). The CSE1L protein [identified as a toxin modulator (35–37)] influences not only PE and DT cytotoxicity in MCF7, but also sensitivity toward TNF-induced apoptosis (37). Thus, both processes (diphthamide ADP ribosylation and TNF apoptosis) could be linked. We analyzed if loss of diphthamide influences sensitivity toward TNF-mediated apoptosis. Cells that have at least one functional copy of each DPH gene, and therefore possess diphthamide, are as sensitive to TNF as parent cells (Fig. 2C). In contrast, cells with complete inactivation of DPH1, DPH2, DPH4, or DPH5 show increased TNF sensitivity (Fig. 2C). Hypersensitivity was observed for all clones carrying complete inactivation of DPH1 or DPH2 or DPH4 or DPH5. Because the common denominator of all derivatives with DPH defects is loss of diphthamide, hypersensitivity is attributable to loss of diphthamide. This effect is not a result of altered TNF-receptor (TNFR) expression (FACS analyses) (SI Text S5). Instead, whole transcriptome analyses by mRNA sequencing of parent and diphthamide-deficient (DPH2ko and DPH5ko) MCF7 cells revealed induction patterns that resemble “preinduction” of nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) death receptor signaling pathways (Fig. 3; see SI Text S5, Fig. S5, and Table S2 for experimental details). Thus, cells without diphthamide are presensitized to death receptor signaling, explaining their TNF-hypersensitivity.
Fig. 3.

Inactivation of DPH5 induces NF-κB and death receptor pathways. RNAseq data were obtained for untreated MCF7, TNF treated MCF7, and DPH5 inactivated MCF7 (SI Text S5). Genes that are significantly changed in their expression levels (MCF7 vs. TNF-MCF7, MCF7 vs. DPH5ko) were subjected to ingenuity upstream pathway analyses. Interestingly, differentially regulated pathways and regulators in both conditions overlap largely and share a common core of TNF- and IFNG-regulated genes, leading to a preactivation of death receptor signaling regulators as consequence of DPH5 deficiency and loss of diphthamide (orange: consequence of TNF treatment as well as DPH5ko; green: TNF treatment only; blue: DPH5ko only). Arrows indicate direct activating/inhibiting interactions between two nodes (based on literature findings) and dashed arrows indicate indirect interactions. Different protein types are represented by different symbols.
Fig. S5.

(A) Analyses of TNFR expression by FACS with TNFR specific antibodies. (B) Transcriptome (mRNAseq) comparisons of MCF7.
Table S2.
Top-inductions as a consequence of DPH2 and DPH5
| Gene | DPH2ko | DPH5ko | ||
| Rank | Value | Rank | Value | |
| TGFB2 | 4 | −4.9 | 2 | −9.5 |
| TNFSF15 | 6 | −4.8 | 1 | −10.3 |
| CLDN1 | 9 | −4.5 | 11 | −7.3 |
| Sulf1 | 10 | −4.4 | 18 | −6.5 |
| Slc2a12 | 17 | −4.1 | 28 | −5.9 |
| MCF2L2 | 21 | −4.0 | 37 | −5.7 |
| Krt87p (pseudogene) | 22 | −4.0 | 21 | −6.4 |
| PDE10A | 23 | −3.9 | 29 | −5.9 |
| S100A9 | 27 | −3.8 | 48 | −5.5 |
| GABARAPL1 | 40 | −3.5 | 46 | −5.5 |
| ALDH1A3 | 50 | −3.6 | 20 | −6.4 |
Top-inductions as a consequence of DPH2 as well as DPH5 inactivation were defined by calling transcripts that are among the 50 genes (rank) with highest induction levels (log2 value) compared with parent MCF7 in DPH2ko cells, as well as in DPH5ko cells. The most prominent markers for DPH2 and DPH5 inactivation in MCF7 are induction of TGFβ and TNFSF15.
SI Text S1: ZFN-Mediated Inactivation of DPH Genes in MCF7 Cells
The ZFN recognition sequences are derived from NM_001383.3 (DPH1-wt), NC_000001.11 (DPH2-wt), NC_000003.12 (DPH3-wt), NM_181706.4 (DPH4-wt), BC053857.1 (DPH5-wt), NM_080650.3 (DPH6-wt), and NC_000009.12 (DPH7-wt). ZFN were obtained from Sigma and transfected into MCF-7 cells in six-well dishes. Mutated clones were either obtained by isolating survivor clones following exposure to lethal doses of Pseudomonas exotoxin A, or by PCR-based HRM analyses (Fig. S1A).
For toxin selection, cells were treated 48 h after transfection with 100 nM PE and further propagated to generate toxin-resistant colonies. These were isolated and recloned from single cells. For the genetic screen, gene-specific PCR fragments were generated and subjected to HRM, which marks mutation containing clones by biphasic melting curves (Fig. S1A).
The sequences of the alleles in clones generated by both procedures were determined by characterizing plasmids containing PCR-fragments derived from genomic DNA encompassing the ZFN-target regions. Fig. S1B shows the ZFN binding sites on DPH genes with positions indicated in the WT sequence. ZFN cleavage sites are underlined. Uni-allelic knockouts (“i” and “ii” in the entries in Fig. S1B) contain one wild-type sequence and one with out-of-frame insertions or deletions. Complete knockouts (“iii” and “iv” in the entries in Fig. S1B) contain two mutated sequences (“a” and “b” in the entries in Fig. S1B) with out-of-frame insertions or deletions. Table S1 summarizes the individual clones that were obtained. Further characterizations of parent and knockout cells are described in SI Text S2 (growth analyses and chromosome composition) and SI Text S3 (transcriptome and TNF-pathway analyses by mRNAseq and FACS).
SI Text S2: A Monoclonal Rabbit Antibody That Specifically Detects Unmodified eEF2
A rabbit immunization and subsequent B-cell-cloning procedure was applied to generate antibodies that specifically bind eEF2 without diphthamide: Rabbits were immunized with a peptide spanning amino acids 708–724 of human eEF2 (TLHADAIHRGGGQIIPT, without diphthamide modification) coupled to KLH. After 10 rounds of immunization with adjuvant, B cells expressing peptide-binding antibodies were isolated by FACS (using fluorescence-labeled eEF2 peptide), and subsequently converted to recombinant antibody clones via B-cell cloning and PCR-mediated V-region extraction (47). ELISA and Biacore analyses with eEF2 peptide as antigen and scrambled peptides as controls were used to select antibody candidates. Western blot analyses (peptide and purified eEF2 and eEF2 detection in MCF7 cell extracts) were performed to evaluate eEF2 specificity and to identify those that selectively recognize eEF2 without diphthamide. Fig. S2 shows Western blots of eight different antibody candidates on extracts from parent MCF7 cells (contain only diphthamide-modified eEF2), and MCF7 DPH1ko-ko cells, which contain only eEF2 without diphthamide modification as determined by MS analyses (Fig. 1 and Fig. S3): of the eight analyzed antibodies, five detected eEF2 with low background in extracts of both cell lines. These bind eEF2 independent of presence or absence of the diphthamide and can be applied as eEF2 control reagents. Two antibodies (MGa and MGd) generate stronger eEF2 signals with extracts of DPH-inactivated cells than wild-type cells. These antibodies bind better to nondiphthamide eEF2 than to diphthamide eEF2 but are not specific for diphthamide-deficient eEF2. Finally, one of the eight antibodies did not generate signals in Western blots of wild-type MCF7, but revealed good signals without background in diphthamide-deficient MCF7. This antibody (MGb) can therefore specifically detect eEF2 without diphthamide modification.
SI Text S3: eEF2 Diphthamide Modification and ADP Ribosylation in MCF7 Cells
Cell extracts for analyses of eEF2 modifications were prepared by lysing cells (4 × 105 cells in six-well dishes) in RIPA buffer. For MS analyses, 100 μg of proteins were reduced with DTE and alkylated using iodoacetamide and further digested with trypsin. Resulting peptides were desalted using solid-phase extraction and ∼1 μg of the digest was analyzed by nanoLC-electrospray ionization-MS using Parallel Reaction Monitoring on a Thermo Q-Exactive Plus mass spectrometer (Thermo Electron). The most abundant precursor ions of the tryptic peptides F702-R716 containing the native or modified His715 at mass-over-charge 437.2234 (native peptide), 462.4854 (ACP-derivative), and 472.7511 (diphthamide-derivative) were selected and fragmented. The EF2 peptides T287-K298 (mass-over-charge 747.9047), V415-K425 (mass-over-charge 554.3241), G667-K675 (mass-over-charge 532.2928), and A785-R800 (mass-over-charge 900.4518) were coanalyzed to normalize EF2 amounts across preparations. All PRM chromatograms were processed using Skyline 2.6 (46).
For antibody-mediated determination of absence of eEF2 modification, an in-house generated (Roche Diagnostics) (SI Text S2) rabbit monoclonal antibody was applied. Western blot analyses of cell extracts with this antibody enables the determination of relative amounts of eEF2 without diphthamide. To detect actin and total human eEF2, monoclonal anti–β-Actin (AC-74, Sigma) and eEF2 (H-118, Santa Cruz) were applied. Secondary antibodies coupled to horseradish peroxidase were purchased from Dako. For detection of ADP ribosylation of eEF2, cell extracts were incubated with PE as toxin/enzyme and biotinylated NAD as substrate, followed by detection of Bio-ADPR-eEF2 in Western blots (32).
SI Text S4: Characterization of DPH-Deficient Cells
Growth of Parent and Mutated MCF7.
Growth of parent and mutated MCF-7 was assessed by seeding 10,000 cells in flat-bottom 96-well plates and incubation at 37 °C in humidified 5% CO2. Twenty-four hours after seeding, cells were exposed to toxins. To determine cell growth, a cell proliferation assay was performed according to the manufacturer’s specifications (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega). Proliferation (DNA replication) was addressed by BrdU incorporation assays (Roche Diagnostics, Mannheim FRG) 72 h after toxin exposure. Fig. S4A shows that heterozygous gene inactivation has no effect on cell growth and complete inactivation of DPH1, DPH2, and DPH4 does not affect growth or affects growth only slightly. Complete inactivation of DPH5 is tolerated by MCF7 cells, however, accompanied by a reduction of growth rate.
Sensitivity of MCF7 Derivatives to CHX and Saporin.
Sensitivity of MCF7 derivatives to CHX and saporin was adressed to analyze if inactivation of DPH genes (or loss of diphthamide) influences susceptibility toward protein synthesis inhibitors in a nonspecific manner. Fig. S4B shows that loss of diphthamide has no effect on cellular susceptibility toward non-ADP ribosylating protein synthesis inhibitors, such as CHX (Fig. S4B, Left) or saporin (Fig. S4B, Right).
Chromosome Composition.
Chromosome composition was analyzed by subjecting metaphase spreads to FISH analyses with BACs that contained human DPH genes: BAC RP11-694J09 for huDPH1, BAC RP11-662O03 for huDPH2, BAC RP11-608O8 for huDPH3, BAC RP11-627B05 for huDPH4, BAC RP11-376O20 for huDPH5, BAC RP11-879B24 for huDPH6, and BAC RP11-734A17 for huDPH7. Fig. S4C shows that the chromosome composition and DPH gene copy number remains constant, when comparing the parent and mutated MCF7 cells. Thus, the ZFN mutagenesis procedure addressed selectively the target genes and did not influence overall chromosome content, distribution, or DPH gene copy number. Suitable metaphase spreads for complete DPH5 knockout cells could not be obtained, possibly reflecting the “reduced growth” phenotype of complete DPH5 inactivation.
SI Text S5: Preactivation of Death Receptor Pathways in Diphthamide-Deficient MCF7
To analyze if TNF hypersensitivity upon DPH gene inactivation is caused by increased levels of TNF-receptors on their surfaces, parent MCF7 and derivatives with complete DPH2 or DPH5 knockouts were subjected to FACS analyses using TNFR-specific antibodies. Fig. S5A shows that the TNFR-specific cell surface signals are indistinguishable between parent and mutated MCF7 cells. Thus, TNF hypersensitivity is not explained by altered TNFR levels.
To analyze if DPH gene inactivation is accompanied by alterations in intracellular gene-expression patterns, parent MCF7 cells and derivatives with complete DPH2 or DPH5 knockouts were subjected to mRNA sequencing. Total RNA was extracted and purified using the High Pure RNA Isolation Kit (Roche) according to the manufacturer’s instructions. For all samples, high-quality RNA was obtained (RNA integrity number > 9.5). RNAseq libraries were prepared from 250 ng of total RNA using the TruSeq Stranded Total RNA preparation Kit (Illumina) following the manufacturer’s instructions. Sequencing libraries were quantified and quality controlled on Bioanalyzer using High Sensitivity chips (Agilent Technologies) and on Qubit using dsDNA HS Assay Kit (Life Technologies). All libraries were pooled and sequenced in four lanes on a HiSeq2500 sequencer (Illumina) for 2 × 50 cycles using version 3 cluster generation kits and version 3 sequencing reagents. As a sequencing control, 10% of PhiX control library (Illumina) was spiked into each lane. Reads were aligned to the human genome (hg19) using GSNAP (bioinformatics.oxfordjournals.org/content/26/7/873.abstract), with splice junctions and exons being defined based on Ensembl v73. Expression was then profiled using in-house tools and reads per kilobase and million mapped read values were computed as proposed by Mortazavi et al. (48). We achieved more than 20M aligned reads per sample of which more than 50% aligned to exons. Differential expression was computed using DESeq (www.genomebiology.com/2010/11/10/r106) and differentially expressed genes (|log2 ratio| > 1 and corrected p-Value < 0.01) were analyzed using Ingenuity Pathway Analysis. Expression patterns that are associated with diphthamide deficiency and cause TNF hypersensitivity should be similar in DPH5ko and DPH2ko, yet be different in parent MCF7. Therefore, we identified genes that become differently expressed (> log+1, <log−1, P < 0.01) between parent and DPH2ko cells, and between parent and DPH5ko cells. A comparison of both gene sets revealed an overlap of gene expression/pathway patterns that become induced upon inactivation of DPH2 and DPH5 (Fig. 3, Fig. S5B, and Table S2). Of 50 genes with highest induction levels upon inactivation of DPH2 or DPH5, 11 were identical (i.e., highly induced upon inactivation of diphthamide synthesis, irrespective of which gene has been compromised). The most prominent markers for DPH2 and DPH5 inactivation in MCF7 are strongly induced expression of TGFβ and TNFSF15.
Discussion
MCF7 cells exclusively contain diphthamide-modified eEF2; this provides a clean background, above which nonmodified or partially modified eEF2 can easily be detected. Therefore, our set of MCF7 derivatives enables a comprehensive assessment of complete or partial DPH gene inactivation, providing insights into gene dose effects, gene essentiality or redundancy, as well as relevance for diphthamide synthesis and toxin sensitivity. Because the MCF7 derivatives have different DPH genes affected but generate the common phenotype diphthamide deficiency, they can shed light on the biological function of the diphthamide and of diphthamide deficiency in mammalian cells. Of particular interest is the finding that cells without diphthamide (independent of which the DPH gene is being inactivated) are presensitized toward NF-κB and death-receptor associated-pathways, and are hypersensitive toward TNF.
Complete inactivation of DPH1, DPH2, and DPH4 lead to the accumulation of unmodified eEF2 and DPH5 inactivation generates the ACP intermediate. This finding agrees with the known synthesis pathway that assigns DPH1, -2, and -4 to initial modification and DPH5 to diphthine synthesis (4–6). Loss of DPH1, -2, -4, and -5 cannot be compensated by other genes, even though mammalian genomes harbor extended gene families. DPH1-, DPH2-, DPH4-, and DPH5-deficient cells are viable. Thus, these genes are not essential for cell propagation and survival, even though their function is nonredundant. We could not obtain cells with complete inactivation of DPH3, DPH6, or DPH7. These genes appear to be essential but likely in nondiphthamide-related cellular processes because diphthamide-loss per se is not lethal (viable DPH1, -2, -4, -5ko). Loss of DPH3, DPH6, or DPH7 is not compensated by other genes. Thus, the whole set of DPH genes and functionality appears to be nonredundant. Reduced expression or inactivation of DPH genes affects cellular sensitivity to eEF2-ribosylating toxins (7–9). We observed absolute toxin resistance in cells that lack diphthamide. In contrast, gene dose reduction by heterozygous gene knockout has only a minor (DPH1-2) or no (DPH3-7) impact on diphthamide synthesis and no impact on toxin sensitivity. Compensation of heterozygous gene inactivation suggests mechanisms that sense the presence/absence of diphthamide or DPH activity and regulate the expression of DPH genes. Diphthamide-eEF2 is the target for toxin-mediated ADP ribosylation and gene dose effects that influence diphthamidylation could be relevant for tumor therapy with targeted toxins. Alterations of the human DPH1 (OVCA1) gene are described for various cancers (16, 29, 38–40), but their impact on diphthamide modification and toxin sensitivity has not been quantified so far. Interestingly, partial DPH1ko and DPH2ko generated some gene dose-dependent modulation of the diphthamide content of the cellular eEF2 pool (up to 25% unmodified eEF2), yet without significant impact toward toxin sensitivity. This finding suggests that in addition to direct inactivation of the functionality of eEF2 in translation elongation, ADP ribosylation may trigger (possibly eEF2K-related) signaling events that interrupt protein synthesis, even though non-ADP ribosylated eEF2 is still present.
The set of MCF7 derivatives have the same genetic background, retain cell shape, and (with exception of complete DPH5 deficiencies) good growth properties, yet have different genes inactivated. Cells without DPH1, DPH2, DPH4, and DPH5 cannot produce diphthamide, a common phenotype of inactivating different genes. Common biological effects observed in these cell lines are therefore attributable to lack of diphthamide. This finding enables us to address the function of diphthamide. Inactivation of DPH1, DPH2, or DPH4 has no or only a minor impact on cell growth. Thus, loss of diphthamide or presence of unmodified eEF2 does not severely impact cell growth under normal conditions. Complete inactivation of DPH5 generates viable cells but with reduced growth. Lack of diphthamide in DPH5ko cells by itself does not pose a problem for cells (shown for DPH1ko, DPH2ko, and DPH4ko), indicating that growth reduction upon DPH5 inactivation is a gene-specific phenotype and not related to loss of diphthamide. This gene-specific effect may be explained by intermediate (ACP-eEF2) accumulation, or by a function of DPH5 in other biological processes. Because the diphthamide on eEF2 is highly conserved in all eukaryotes as well as in archea (41), it is surprising that lack of diphthamide synthesis has little overall impact on cell growth. Animals with homozygous DPHko, however, do not survive beyond embryonic stages (13, 16–18). This finding suggests that the diphthamide may be necessary for development.
An explanation for emryonal lethality of KPHko animals may be that NF-κB and death receptor signaling pathways (known to be involved in and necessary for development) are preactivated in diphthamide-deficient cells. These cells are nevertheless viable, as pathway induction does not pass thresholds sufficient to induce apoptosis without additional stimuli. Presensitization becomes phenotypically relevant upon triggering these preinduced pathways: all diphthamide synthesis-deficient cells (independent from target gene knockout) were hypersensitive to TNF-induced apoptosis. This finding indicates that the presence or absence of diphthamide affects NF-κB or death receptor pathways. Pathway preactivation and TNF hypersensitivity upon loss of diphthamide could be explained by modulation of eEF2-targeted stress responses [such as eEF2K-mediated phosphorylation (41–45)] in cells that exclusively contain diphthamide-free eEF2. Diphthamide may also modulate the primary function of eEF2 (i.e., translation elongation). Because eEF2 within MCF7 carries diphthamide, diphthamide-eEF2 enables translation of all essential proteins. eEF2 without diphthamide also supports the translation of essential proteins because cells that lack diphthamide are viable. Diphthamide loss may affect signaling pathways if eEF2 without diphthamide generates some defective or altered proteins (e.g., by allowing translational slippage) (10, 13) (but we did not find UPR genes induced in DPHko cells). It is also possible that translation of some nonessential proteins involved in NF-κB and death receptor pathways is different between modified and unmodified eEF2. This finding would explain preinduction of NF-κB and death receptor pathways and TNF-hypersensitivity that we observed in all diphthamide-deficient cells.
Materials and Methods
DPH Gene Inactivation.
DPH gene inactivation was achieved by ZFN mutagenesis followed by isolation of mutated clones via toxin selection (resistant homozygous DPH1, -2, -4, and -5ko) or by HRM analyses (31) (SI Text S1).
Diphthamide Modification and ADP Ribosylation.
Diphthamide modification and ADP ribosylation was determined by MS of trypsin-digested cell lysates (46) (SI Text S3), by Western blots with antibodies recognizing unmodified eEF2 (SI Text S2), or by detection of Bio-ADP ribosylated eEF2 (32).
Cell Proliferation/Cytotoxicity Assays.
Cell proliferation/cytotoxicity assays (SI Text S4) addressed the consequences of DPH inactivation on toxin and TNF sensitivity.
Acknowledgments
We thank Klaus Mayer, Hedda Herrmuth, Claudia Kirstenpfad, Andrea Herold, and Beatrix Bahle for excellent operative support. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and in part by Roche Pharma Research and Early Development (pRED). S.S. is a fellow of the Roche Postdoc Fund.
Footnotes
Conflict of interest statement: S.S., A.R.d.S.M.S., A.D., S.K.G., S.M., T.R., F.B., A.K.H., R.R., M.G., G.N., and U.B. are employed by Roche, and I.P. by the National Cancer Institute. All parties are interested in and hold patents (I.P.’s patent has been assigned to the NIH and he has a Cooperative Research and Development Agreement with Roche Pharmaceuticals) for the development of Pseudomonas exotoxin-derived entities in targeted cancer therapy.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512863112/-/DCSupplemental.
References
- 1.Merrick WC. Mechanism and regulation of eukaryotic protein synthesis. Microbiol Rev. 1992;56(2):291–315. doi: 10.1128/mr.56.2.291-315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaul G, Pattan G, Rafeequi T. Eukaryotic elongation factor-2 (eEF2): Its regulation and peptide chain elongation. Cell Biochem Funct. 2011;29(3):227–234. doi: 10.1002/cbf.1740. [DOI] [PubMed] [Google Scholar]
- 3.Van Ness BG, Howard JB, Bodley JW. Isolation and properties of the trypsin-derived ADP-ribosyl peptide from diphtheria toxin-modified yeast elongation factor 2. J Biol Chem. 1978;253(24):8687–8690. [PubMed] [Google Scholar]
- 4.Su X, Lin Z, Lin H. The biosynthesis and biological function of diphthamide. Crit Rev Biochem Mol Biol. 2013;48(6):515–521. doi: 10.3109/10409238.2013.831023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Milne GT, Kuremsky JG, Fink GR, Leppla SH. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol Cell Biol. 2004;24(21):9487–9497. doi: 10.1128/MCB.24.21.9487-9497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaffrath R, Abdel-Fattah W, Klassen R, Stark MJ. The diphthamide modification pathway from Saccharomyces cerevisiae—Revisited. Mol Microbiol. 2014;94(6):1213–1226. doi: 10.1111/mmi.12845. [DOI] [PubMed] [Google Scholar]
- 7.Wei H, et al. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proc Natl Acad Sci USA. 2012;109(18):6898–6903. doi: 10.1073/pnas.1204523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu X, et al. Methylation of the DPH1 promoter causes immunotoxin resistance in acute lymphoblastic leukemia cell line KOPN-8. Leuk Res. 2013;37(11):1551–1556. doi: 10.1016/j.leukres.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei H, et al. A modified form of diphthamide causes immunotoxin resistance in a lymphoma cell line with a deletion of the WDR85 gene. J Biol Chem. 2013;288(17):12305–12312. doi: 10.1074/jbc.M113.461343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argüelles S, Camandola S, Cutler RG, Ayala A, Mattson MP. Elongation factor 2 diphthamide is critical for translation of two IRES-dependent protein targets, XIAP and FGF2, under oxidative stress conditions. Free Radic Biol Med. 2014;67:131–138. doi: 10.1016/j.freeradbiomed.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimata Y, Kohno K. Elongation factor 2 mutants deficient in diphthamide formation show temperature-sensitive cell growth. J Biol Chem. 1994;269(18):13497–13501. [PubMed] [Google Scholar]
- 12.Ortiz PA, Ulloque R, Kihara GK, Zheng H, Kinzy TG. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem. 2006;281(43):32639–32648. doi: 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc Natl Acad Sci USA. 2012;109(34):13817–13822. doi: 10.1073/pnas.1206933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Liu S, Lajoie G, Merrill AR. The role of the diphthamide-containing loop within eukaryotic elongation factor 2 in ADP-ribosylation by Pseudomonas aeruginosa exotoxin A. Biochem J. 2008;413(1):163–174. doi: 10.1042/BJ20071083. [DOI] [PubMed] [Google Scholar]
- 15.Roy V, Ghani K, Caruso M. A dominant-negative approach that prevents diphthamide formation confers resistance to Pseudomonas exotoxin A and diphtheria toxin. PLoS One. 2010;5(12):e15753. doi: 10.1371/journal.pone.0015753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CM, Behringer RR. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev. 2004;18(3):320–332. doi: 10.1101/gad.1162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, et al. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol Cell Biol. 2006;26(10):3835–3841. doi: 10.1128/MCB.26.10.3835-3841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webb TR, et al. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J Cell Sci. 2008;121(Pt 19):3140–3145. doi: 10.1242/jcs.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Ness BG, Howard JB, Bodley JW. ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J Biol Chem. 1980;255(22):10717–10720. [PubMed] [Google Scholar]
- 20.Honjo T, Nishizuka Y, Hayaishi O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J Biol Chem. 1968;243(12):3553–3555. [PubMed] [Google Scholar]
- 21.Mateyak MK, Kinzy TG. ADP-ribosylation of translation elongation factor 2 by diphtheria toxin in yeast inhibits translation and cell separation. J Biol Chem. 2013;288(34):24647–24655. doi: 10.1074/jbc.M113.488783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassan R, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13(17):5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 23.Kelly RJ, Sharon E, Pastan I, Hassan R. Mesothelin-targeted agents in clinical trials and in preclinical development. Mol Cancer Ther. 2012;11(3):517–525. doi: 10.1158/1535-7163.MCT-11-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15(16):5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 26.Kreitman RJ, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18(8):1622–1636. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 28.Wayne AS, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: Preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16(6):1894–1903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CM, Behringer RR. OVCA1: Tumor suppressor gene. Curr Opin Genet Dev. 2005;15(1):49–54. doi: 10.1016/j.gde.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Flisikowska T, et al. Efficient immunoglobulin gene disruption and targeted replacement in rabbit using zinc finger nucleases. PLoS One. 2011;6(6):e21045. doi: 10.1371/journal.pone.0021045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liew M, et al. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem. 2004;50(7):1156–1164. doi: 10.1373/clinchem.2004.032136. [DOI] [PubMed] [Google Scholar]
- 32.Gupta PK, Liu S, Leppla SH. Characterization of a Chinese hamster ovary cell mutant having a mutation in elongation factor-2. PLoS One. 2010;5(2):e9078. doi: 10.1371/journal.pone.0009078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X, Youle RJ, FitzGerald DJ, Pastan I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degradation of Mcl-1. Mol Cell Biol. 2010;30(14):3444–3452. doi: 10.1128/MCB.00813-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morimoto H, Bonavida B. Diphtheria toxin- and Pseudomonas A toxin-mediated apoptosis. ADP ribosylation of elongation factor-2 is required for DNA fragmentation and cell lysis and synergy with tumor necrosis factor-alpha. J Immunol. 1992;149(6):2089–2094. [PubMed] [Google Scholar]
- 35.Brinkmann U. CAS, the human homologue of the yeast chromosome-segregation gene CSE1, in proliferation, apoptosis, and cancer. Am J Hum Genet. 1998;62(3):509–513. doi: 10.1086/301773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brinkmann U, Brinkmann E, Gallo M, Pastan I. Cloning and characterization of a cellular apoptosis susceptibility gene, the human homologue to the yeast chromosome segregation gene CSE1. Proc Natl Acad Sci USA. 1995;92(22):10427–10431. doi: 10.1073/pnas.92.22.10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkmann U, Brinkmann E, Gallo M, Scherf U, Pastan I. Role of CAS, a human homologue to the yeast chromosome segregation gene CSE1, in toxin and tumor necrosis factor mediated apoptosis. Biochemistry. 1996;35(21):6891–6899. doi: 10.1021/bi952829+. [DOI] [PubMed] [Google Scholar]
- 38.L’Allemain G. [Ovca1 gene, deleted in ovarian cancer is a special tumor suppressor] Bull Cancer. 2004;91(4):301–302. French. [PubMed] [Google Scholar]
- 39.Li AJ, Karlan BY. Genetic factors in ovarian carcinoma. Curr Oncol Rep. 2001;3(1):27–32. doi: 10.1007/s11912-001-0039-y. [DOI] [PubMed] [Google Scholar]
- 40.Nobukuni Y, Kohno K, Miyagawa K. Gene trap mutagenesis-based forward genetic approach reveals that the tumor suppressor OVCA1 is a component of the biosynthetic pathway of diphthamide on elongation factor 2. J Biol Chem. 2005;280(11):10572–10577. doi: 10.1074/jbc.M413017200. [DOI] [PubMed] [Google Scholar]
- 41.Greganova E, Altmann M, Bütikofer P. Unique modifications of translation elongation factors. FEBS J. 2011;278(15):2613–2624. doi: 10.1111/j.1742-4658.2011.08199.x. [DOI] [PubMed] [Google Scholar]
- 42.Hizli AA, et al. Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A-cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol Cell Biol. 2013;33(3):596–604. doi: 10.1128/MCB.01270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ovchinnikov LP, et al. Three phosphorylation sites in elongation factor 2. FEBS Lett. 1990;275(1-2):209–212. doi: 10.1016/0014-5793(90)81473-2. [DOI] [PubMed] [Google Scholar]
- 44.Sans MD, Xie Q, Williams JA. Regulation of translation elongation and phosphorylation of eEF2 in rat pancreatic acini. Biochem Biophys Res Commun. 2004;319(1):144–151. doi: 10.1016/j.bbrc.2004.04.164. [DOI] [PubMed] [Google Scholar]
- 45.Kruiswijk F, et al. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci Signal. 2012;5(227):ra40. doi: 10.1126/scisignal.2002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacLean B, et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seeber S, et al. A robust high throughput platform to generate functional recombinant monoclonal antibodies using rabbit B cells from peripheral blood. PLoS One. 2014;9(2):e86184. doi: 10.1371/journal.pone.0086184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]