

Significance
Mutations in genes encoding proteins that function in splicing of mRNA precursors occur frequently in myelodysplastic syndromes (MDS) and certain leukemias. However, the mechanism by which the mutated splicing factors function has begun to be elucidated only recently. Here we use genome-editing techniques to introduce a common MDS mutation in the gene Serine/arginine-rich splicing factor 2 (SRSF2), which encodes an RNA-binding splicing regulator, in cultured blood cells. We show that splicing of several hundred transcripts, including some with possible relevance to disease, is altered. We further show that mutant SRSF2 is sufficient to induce these changes and does so by binding to RNA sequence elements in the misregulated mRNAs with altered specificity.
Keywords: spliceosome, pre-mRNA splicing, serine/arginine-rich proteins, myelodysplastic syndromes, leukemia
Abstract
Serine/arginine-rich splicing factor 2 (SRSF2) is an RNA-binding protein that plays important roles in splicing of mRNA precursors. SRSF2 mutations are frequently found in patients with myelodysplastic syndromes and certain leukemias, but how these mutations affect SRSF2 function has only begun to be examined. We used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease to introduce the P95H mutation to SRSF2 in K562 leukemia cells, generating an isogenic model so that splicing alterations can be attributed solely to mutant SRSF2. We found that SRSF2 (P95H) misregulates 548 splicing events (<1% of total). Of these events, 374 involved the inclusion of cassette exons, and the inclusion was either increased (206) or decreased (168). We detected a specific motif (UCCA/UG) enriched in the more-included exons and a distinct motif (UGGA/UG) in the more-excluded exons. RNA gel shift assays showed that a mutant SRSF2 derivative bound more tightly than its wild-type counterpart to RNA sites containing UCCAG but bound less tightly to UGGAG sites. Thus in most cases the pattern of exon inclusion or exclusion correlated with stronger or weaker RNA binding, respectively. We further show that the P95H mutation does not affect other functions of SRSF2, i.e., protein–protein interactions with key splicing factors. Our results thus demonstrate that the P95H mutation positively or negatively alters the binding affinity of SRSF2 for cognate RNA sites in target transcripts, leading to misregulation of exon inclusion. Our findings shed light on the mechanism of the disease-associated SRSF2 mutation in splicing regulation and also reveal a group of misspliced mRNA isoforms for potential therapeutic targeting.
Myelodysplastic syndromes (MDS) are a heterogeneous group of hematopoietic disorders characterized by ineffective production of myeloid blood cells, which have various risks of progression into acute myeloid leukemia (AML) (1, 2). The most frequently occurring mutations found in patients with MDS involve genes encoding pre-mRNA splicing factors, including Splicing factor 3B, subunit 1 (SF3B1), Serine/arginine-rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1), and U2 small nuclear ribonucleoprotein auxiliary factor 35 kDa subunit-related protein 2 (ZRSR2) (3–6), suggesting that altered RNA splicing may play a critical role in the pathogenesis of MDS. Despite some recent advances (e.g., ref. 7; see Discussion), the molecular mechanisms by which the mutated splicing factors misregulate pre-mRNA splicing have not been studied thoroughly. However, it is now well established that splicing deregulation contributes to multiple diseases, especially cancer (8, 9).
SRSF2 is a well-studied serine/arginine-rich splicing factor (SR protein). SR proteins play important roles in the regulation of both constitutive and alternative pre-mRNA splicing, functioning, for example, to facilitate spliceosome assembly, most frequently by binding to RNA motifs known as exonic splicing enhancers (ESEs) (10). SRSF2 and other SR proteins carry out two major functions through two domains: the RNA recognition motif (RRM) domain at the N terminus involved in sequence-specific RNA binding (11) and the arginine/serine-rich (RS) domain at the C terminus involved in interactions with other splicing factors (10, 12, 13).
Heterozygous mutations in SRSF2 occur frequently in patients with MDS or chronic myelomonocytic leukemia (CMML) and generally are associated with a less favorable prognosis (3–6, 14). The great majority of SRSF2 mutations found in these patients occur at Pro-95, and frequent changes to histidine, arginine, or leucine have been observed (3–6, 14). Pro-95 is located in the linker region between the RRM and RS domains. Recent NMR studies of SRSF2 revealed that Pro-95, despite lying outside the canonical RRM, in fact plays a role in sequence-specific RNA binding (15). Therefore, mutations affecting this residue may lead to changes in the binding affinity of SRSF2 to cognate RNA sites in target transcripts, and in turn these changes could affect alternative splicing, for example by influencing the efficiency of exon inclusion.
Here we describe experiments providing insight into the functional mechanism of mutant SRSF2 (mutSRSF2). We first introduced the most frequently occurring SRSF2 mutation (P95H) into K562 cells and then identified changes in pre-mRNA splicing using high-throughput RNA-sequencing (RNA-seq). A number of these changes, which included both increased exon inclusion and increased exon exclusion, were confirmed by RT-PCR, and transient transfection assays in heterologous cells showed that expression of mutSRSF2 was sufficient to induce the splicing changes. We then used RNA gel shift assays to show that the P95H mutation increases or decreases SRSF2 affinity for target sites in a sequence-specific manner and that this change in affinity correlates with the nature of the splicing changes observed. Finally, we found that interactions of mutSRSF2 with other key components of the spliceosomal complex were unaffected. We conclude that the SRSF2 P95H is a gain-of-function mutation that alters SRSF2 affinity for target ESEs.
Results
Mutant SRSF2 Misregulates Hundreds of Splicing Events.
We first wished to obtain SRSF2 mutant cells for analysis of splicing changes that were otherwise isogenic with control cells, so that any changes could be attributed to the mutSRSF2 protein. To this end, we used Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) technology to knock in the SRSF2 P95H hotspot mutation in K562 leukemia cells (Materials and Methods). We isolated four independent CRISPR cell clones with mutant SRSF2, named P9, P15, P22, and P38 (Fig. S1). SRSF2 mutations found in patients are heterozygous (3–6, 14), suggesting that one WT allele may be required for cellular function. Therefore, we maintained at least one WT SRSF2 allele in each of the mutant CRISPR clones (note that K562 cells can have two or three SRSF2 alleles) (Fig. S1). To obtain WT SRSF2 controls, we knocked in synonymous mutations and obtained four independent CRISPR clones with WT SRSF2 (W19, W33, W36, and W42) (Fig. S1). We then performed RNA-seq of poly(A)+ RNAs from these four mutant and four WT SRSF2 cell clones. Expression from the P95H alleles in the four mutant clones varied from ∼27–58% of total SRSF2 mRNA (Fig. 1A, Lower Left), which was within the physiological range. Importantly, total mRNA levels of SRSF2 and 11 other SR proteins and 14 of 16 major heterogeneous nuclear ribonucleoprotein (hnRNP) splicing factors were comparable in mutant and WT SRSF2 CRISPR clones (Fig. S2). To identify the splicing alterations associated with mutSRSF2, we analyzed the RNA-seq data using the computational tool rMATS (16). Using a cutoff of >10% for splicing differences and a false-discovery rate of <10%, we found that mutSRSF2 misregulated 548 splicing events, including 374 cassette exons, 68 retained introns, 66 mutually exclusive exons, 25 alternative 5′ splice sites, and 15 alternative 3′ splice sites (Dataset S1). It is noteworthy that these regulated events comprise a very small fraction (<1%) of the splicing events examined (Table S1). Interestingly, inclusion of cassette exons was either increased (206) or decreased (168) (Dataset S1).
Fig. S1.

Confirmation of SRSF2 mutations in CRISPR clones by DNA sequencing. P95H and synonymous mutations were introduced to SRSF2 in K562 cells by electroporation of the SRSF2 gRNA CRISPR/Cas9 vector along with either the P95H mutant or WT SRSF2 ssODN. Synonymous mutations were used to create restriction enzyme sites [NaeI (C→G) in WT cells and HaeII (C→G) in mutant cells], to facilitate identification of knock-in clones by restriction digest, or to disrupt the protospacer-adjacent motif (PAM) site (G→A) to prevent gRNA/Cas9 from recognizing and cutting the newly introduced mutant allele. Genomic DNAs were extracted from positive clones, and the SRSF2 mutations were confirmed using PCR and DNA sequencing. The P95H mutation (C→A) and synonymous mutations are indicated above the sequencing chromatograms. (A) K562 parental cells. (B) CRISPR clones with WT SRSF2. (C) CRISPR clones with P95H mutSRSF2.
Fig. 1.

Validation of splicing targets of mutSRSF2. (A, Lower Left) P95H allele expression in the mutant CRISPR clones. (Lower Right) Expression of SRSF2 in 293T cells either mock transfected (–) or transfected with plasmid encoding HA-tagged WT (W) or P95H mutSRSF2 (P). In the immunoblot analysis, the mAb104 antibody was used to detect endogenous and HA-tagged SRSF2. (Upper) RT-PCR products of exon inclusion and exclusion isoforms of ATF2. (B–G) RT-PCR products of splicing isoforms of MELK (B), PFKM (C), CDK5RAP2 (D), ARMC10 (E), DGUOK (F), and WDR45 (G). In all panels, each CRISPR clone was considered as one independent experiment. Because we had four WT and four mutSRSF2 CRISPR clones, n was 4 (as indicated). For 293T cells, three independent transfection experiments were performed (n = 3). Rounded percentages of exon inclusion or exclusion and SDs are shown as indicated. *P < 0.05, **P < 0.01, and ***P < 0.001.
Fig. S2.

Total mRNA levels of SR proteins and hnRNP splicing factors were comparable in WT and mutSRSF2 CRISPR clones. (Upper) Total mRNA levels of SRSF2 and 11 other SR proteins in K562 parental cells and WT and mutSRSF2 CRISPR cells. (Lower) Total mRNA levels of 16 major hnRNP splicing factors. Except that there were small but significant differences for HNRNPA2B1 (∼7% decrease in mutant vs. WT SRSF2 cells, P = 0.043) and HNRNPC (∼19% decrease, P = 0.008), the mRNA levels of the other 14 hnRNP splicing factors and all 12 SR proteins did not show significant changes between WT and mutant SRSF2 cells. Note that expression of SRSF8 and SRSF12 was barely detectable.
Table S1.
Only a small fraction of splicing events are differentially spliced in P95H mutant versus WT SRSF2 cells
| Splicing type | Splicing events | Included* | Excluded† | % included | % excluded |
| A3SS | 5,094 | 8 | 7 | 0.16 | 0.14 |
| A5SS | 3,520 | 16 | 9 | 0.45 | 0.26 |
| MXE | 16,179 | 46 | 20 | 0.28 | 0.12 |
| RI | 3,927 | 35 | 33 | 0.89 | 0.84 |
| SE | 69,685 | 206 | 168 | 0.30 | 0.24 |
A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exons; RI, retained intron; SE, cassette exon.
More included in P95H mutant vs. WT SRSF2 cells.
More excluded in P95H mutant vs. WT SRSF2 cells.
The splicing differences we observed between WT and mutSRSF2 were typically quite modest, between 10–30%. To confirm these subtle differences, we selected a dozen targets for experimental validation, based mainly on their potential relevance to disease (Discussion and Table 1) (17–26). We examined these splicing events by 32P RT-PCR using total RNA extracted from all eight CRISPR cell lines and from K562 parental cells. We successfully validated 10 splicing targets (Fig. 1, Table 1, and Fig. S3 A–E), reflecting an ∼80% validation rate. With an 11th, Armadillo repeat containing 10 (ARMC10), rMATS indicated that only exon 5 was more included in mutSRSF2 cells, but RT-PCR indicated that double inclusion of exon 5 and exon 6 was promoted by mutSRSF2 (Fig. 1E). We noticed that the splicing differences of all validated exons were slightly greater in mutant clone P9 than in the other three mutant clones, possibly reflecting higher mutant allele expression in P9 (see above).
Table 1.
Summary of target validation by RT-PCR
| Gene | Validated? | SRSF2 site | Potential disease relevance |
| More included | |||
| ATF2 | Yes | uggUCCAGca | Tumorigenesis and DNA repair, see review (17) |
| DGUOK | Yes | auaUCCAGgc | Mitochondrial DNA depletion (18) |
| ARMC10 | Yes/no (see text) | uguUCCAGgu | One protein isoform binds p53, see Discussion |
| CRAT | Yes | uccUCCAGcc | Key metabolic enzyme in mitochondria (19) |
| CDK5RAP2 | Yes | ugaUGGAGau | Doxorubicin resistance (20) |
| ABI1 | Yes | uaaUGGAGgu | Transduces signals from Ras to Rac (21) |
| CDC45 | No | Initiation of DNA replication (22) | |
| More excluded | |||
| MELK | Yes | aauUGGAGuc | Tumorigenesis and hematopoiesis, see review (23) |
| WDR45 | Yes | uggUGGAGau | Iron accumulation in brain (24) |
| FYN | Yes | gucUGGAGaa | Tyrosine kinase proto-oncogene, see Discussion |
| PFKM | Yes | augUGCAGag | Associated with breast cancer (25) |
| SLC25A26 | Yes | ggcUGCAGag | Transports S-adenosylmethionine into mitochondria (26) |
Target exons chosen for RT-PCR validation are all cassette exons (either more included or more excluded in mutant versus WT SRSF2 cells), except for the mutually exclusive exons of FYN. The SRSF2 site of FYN is in the more excluded exon of the mutually exclusive exons. The 5-nt motif sequences are capitalized and are shown in bold font.
Fig. S3.

Validation of additional splicing targets of mutSRSF2. Experiments were performed as in Fig. 1. The panels show the RT-PCR products of splicing isoforms of CRAT (A), SLC25A26 (B), ABI1 (C), FYN (D), CDC45 (E), and EZH2 (F, see ref. 7). *P < 0.05, **P < 0.01, and ***P < 0.001.
We next wished to extend these results by determining whether exogenous expression of mutSRSF2 in another cell type was sufficient to induce splicing changes. To this end, we transiently transfected plasmids encoding HA-tagged WT and mutSRSF2 into 293T cells (expression levels are shown in Fig. 1A, Lower Right). RT-PCR revealed that the splicing differences in all validated targets [except Solute carrier family 25 member 26 (SLC25A26)] were recapitulated in the transfected 293T cells (Fig. 1 and Fig. S3 A–E). Note that the magnitude of the splicing changes was smaller in the transfected cells than in CRISPR cells, perhaps reflecting a background of preexisting mRNAs. Indeed, cotransfection of the SRSF2-expressing plasmids with minigenes containing two of the regulated exons [from Maternal embryonic leucine zipper kinase (MELK) and Activating transcription factor 2 (ATF2)] revealed larger splicing differences in minigene transcripts (see the first two lanes of Fig. 5 A and B).
Fig. 5.
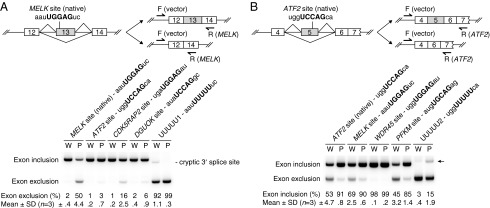
Exon inclusion/exclusion in minigene reporter assays. Splicing reporter minigenes were cotransfected with plasmid expressing WT (W) or P95H mutSRSF2 (P) into 293T cells. Exon inclusion and exclusion isoforms were examined by RT-PCR. Three independent experiments were performed (n = 3). (A) MELK cassette exon minigene containing the native 10-nt site and its mutant minigenes containing mutated 10-nt sites as indicated. The 5-nt motif sequences were capitalized. Note that a cryptic 3′ splice site was detected in the UUUUU1 minigene (last two lanes). (B) The ATF2 cassette exon minigene containing the native 10-nt site and its mutant minigenes containing mutated 10-nt sites as indicated. The 5-nt motif sequences were capitalized. In the last two lanes, the PCR products of the exon inclusion isoform (indicated by an arrow) appeared to migrate slightly more slowly than the PCR products in other lanes. However, DNA sequencing indicated that the PCR products in the last two lanes had exactly the same 5′ and 3′ splice sites as the PCR products in the first two lanes.
Together, our results indicate that mutSRSF2 misregulates hundreds of splicing events. A majority of these events involve relatively subtle changes in the inclusion of cassette exons, which also is the most common form of alternative splicing in humans. Notably, mutSRSF2 induced both increased exon inclusion and increased exon exclusion.
Mutant SRSF2-Regulated Exons Show Distinct Sequence Features.
Next we set out to determine the underlying basis for the splicing differences we detected. Because SRSF2 regulates splicing by binding to ESEs, we first examined the target exons for conserved sequence motifs. To facilitate this search, we compiled a list of 109 SRSF2 sites previously identified by in vitro systematic evolution of ligands by exponential enrichment (SELEX), functional selection, or individual studies (11, 27–36). Using the program MEME (37), we identified a conserved 10-nt motif containing a USSWG (S = G or C, W = A or U) sequence in 88 of the 109 sites (Fig. S4). With the position-specific scoring matrix of this motif, we searched for sequence matches in the 10 validated splicing targets and in ARMC10 exon 5 using the program FIMO (37). In each regulated exon we obtained the best-scoring 10-nt RNA site that had a USSWG sequence (Table 1). Note that the best-scoring site in the MELK exon was gauUGGUGug (P = 0.00195), but another site with a comparable score (aauUGGAGuc, P = 0.00561) is shown in Table 1, because mutation of this site to aauUUUUUuc almost completely abolished exon inclusion in assays using minigene splicing reporters (Fig. 5A). These initial findings suggest that the validated transcripts are likely direct targets of mutSRSF2. Significantly, we observed a conserved UCCAG sequence in four of the six more-included exons (mutant vs. WT) and a UGGAG sequence in three of the five more-excluded exons (the other two contained UGCAG; see below).
Fig. S4.

Sequence logo of the conserved motif in SRSF2-binding sites. A list of 109 published SRSF2-binding sites was compiled (see text). A 10-nt motif was identified in 88 of the 109 sites using MEME. A sequence logo of the motif was generated using WebLogo (weblogo.berkeley.edu/logo.cgi).
We next wished to identify conserved sequence features in all the 374 cassette exons identified by rMATS. To identify conserved sequences in an unbiased manner, we performed k-mer enrichment analysis. We computationally scanned all the regulated cassette exons for all 4-mers and 5-mers. A short list of enriched 4-mers and 5-mers (the top approximately 6%) is shown in Dataset S2. We found that the top enriched 4-mers were CCWG and GCWG in the more-included exons and GGWG in the more-excluded exons (Fig. 2 and Table 2). Similarly, the top enriched 5-mer in the more-excluded exons was UGGAG (Fig. 2 and Table 2), consistent with our finding in the validated targets. In the more-included exons, UCCWG was highly enriched, consistent with the enriched 4-mer CCWG (Fig. 2 and Table 2). It is important to note that the locations of UCCWG/UGGWG sequences relative to exon boundaries did not show an obvious uneven distribution (Fig. S5). Significantly, SRSF2 makes multiple direct contacts with CCAG, GCAG, or GGAG when binding to RNA substrates containing these sequences (15). Thus it is possible that the distinct sequence features in the mutSRSF2 deregulated exons underlie the splicing changes observed and do so by altered binding by the mutant protein.
Fig. 2.

Distinct sequence motifs are enriched in the regulated cassette exons. The relative frequencies of occurrence of highly enriched 4-mers (A) and 5-mers (B) in the more-included or more-excluded exons (mutant vs. WT) are shown.
Table 2.
Enriched 4-mers and 5-mers in the cassette exons
| 4-mers | 5-mers | ||||||
| Included* | Rank† | Excluded‡ | Rank | Included | Rank | Excluded | Rank |
| CCUG | 1 | GGAG | 1 | UCCUG | 3 | UGGAG | 1 |
| GCUG | 2 | CCUG | 7 | UGCAG | 23 | UGCUG | 3 |
| CCAG | 3 | GCUG | 8 | UGCUG | 29 | UGGUG | 5 |
| GCAG | 7 | GGUG | 15 | UCCAG | 61 | ||
More included exons in mutant vs. WT SRSF2 cells.
Rank by frequency of occurrence (relative to random).
More excluded exons in mutant vs. WT SRSF2 cells.
Fig. S5.
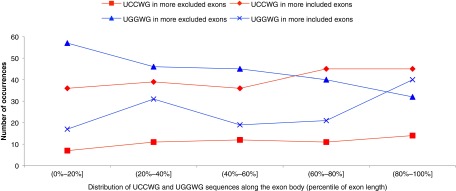
Enriched sequence motifs do not show obvious positional effects. Locations of UCCWG and UGGWG sequences relative to exon boundaries do not show an obvious uneven distribution. Data are shown as percentile of exon length.
Mutant SRSF2 Binds UCCAG and UGCAG Sites More Tightly but Binds UGGAG Sites Less Tightly.
We next wished to examine the binding activity of mutant and WT SRSF2 to target sequences from the deregulated exons. To do so, we first purified His6-tagged mutant and WT SRSF2 RRM derivatives (amino acids 1–101; see ref. 15) from Escherichia coli to apparent homogeneity (Fig. S6). We then performed RNA gel shift assays with the purified proteins and 10-nt RNAs from seven of the validated exons, including three UCCAG-containing sites, three UGGAG-containing sites, and one UGCAG-containing site (Fig. 3). Strikingly, the results showed that mutSRSF2 bound more tightly than WT SRSF2 to all three UCCAG-containing sites (Fig. 3 A, C, and E) and the UGCAG-containing site (Fig. 3G) but bound less tightly to all three UGGAG-containing sites (Fig. 3 B, D, and F). In all seven cases, the differences in RNA-binding affinities were small (less than twofold), consistent with the subtle differences detected in splicing. Additionally, the binding results were consistent with the enrichment of UCCWG and UGCAG in the more-included exons and the enrichment of UGGWG in the more-excluded exons (Fig. 2 and Table 2).
Fig. S6.

Purified His6-tagged WT and mutSRSF2 (amino acids 1–101). Two micrograms of His6-tagged WT (W) or P95H mutant (P) SRSF2 (amino acids 1–101) were subjected to 15% SDS/PAGE and Coomassie staining.
Fig. 3.

The P95H mutation alters binding of SRSF2 to distinct RNA sites. The indicated SRSF2-binding sites (10-nt RNAs) from target transcripts were incubated with increasing concentrations of His6-tagged WT or mutant (P95H) SRSF2 (amino acids 1–101), and protein–RNA complexes were resolved from free oligonucleotides by gel electrophoresis. Experiments were performed with two independent protein preparations. Final concentrations of recombinant SRSF2 proteins in the gel shift assays were (A) 0, 0.18, 0.24, 0.30, 0.36, 0.42, and 0.48 μM; (B) 0, 0.4, 0.5, 0.6, 0.7, 0.8, and 0.9 μM; (C) 0, 0.06, 0.12, 0.18, 0.24, 0.30, and 0.36 μM; (D) 0, 0.12, 0.18, 0.24, 0.30, 0.36, and 0.42 μM; (E) 0, 0.36, 0.48, 0.60, 0.72, 0.84, and 0.96 μM; (F) 0, 0.12, 0.18, 0.24, 0.30, 0.36, and 0.42 μM; and (G) 0, 0.3, 0.4, 0.5, 0.6, 0.7, and 0.8 μM. The apparent Kds are shown in each panel.
Factors in Addition to the Core SRSF2-Binding Site Can Influence the Effect of Mutant SRSF2.
In five of the seven exons whose RNA sites we tested for SRSF2 binding, the pattern of exon inclusion or exclusion correlated with stronger or weaker RNA binding, respectively. In all of the 374 deregulated cassette exons, we found that about one third (119) had only one UCCWG or UGGWG site and, in ∼85% of cases, exon inclusion or exclusion correlated with the presence of a UCCWG or UGGWG site, respectively (Fig. 4). To investigate whether binding of mutSRSF2 to individual sites can be the sole determinant of splicing outcome, we generated minigene reporter plasmids using the two regulated exons from MELK and ATF2 as backbones and then replaced the native sites with different SRSF2 sites containing UCCAG, UGGAG, or UGCAG. We used naturally occurring 10-nt RNA sites whose binding to both mutant and WT SRSF2 were confirmed in Fig. 3. We then cotransfected these minigenes with plasmid expressing HA-tagged WT or mutSRSF2 into 293T cells and analyzed splicing by RT-PCR. As noted above, the splicing differences brought about by mutSRSF2 on minigene transcripts containing native MELK and ATF2 sites were larger than, but consistent with, those seen in CRISPR cells (first two lanes of Fig. 5 A and B). Mutation of the 5-nt motif sequences of the native sites to UUUUU greatly reduced exon inclusion (last two lanes of Fig. 5 A and B), suggesting that binding of SRSF2 to these sites is necessary to induce exon inclusion. Replacing the native UGGAG-containing site in MELK with either of two UCCAG-containing sites [from ATF2 or deoxyguanosine kinase (DGUOK)] greatly reduced exon exclusion in cells expressing mutSRSF2, from 50% to 3% and 6%, respectively, but had little effect in cells expressing WT SRSF2 (Fig. 5A), confirming the contribution of the UGGAG motif to increased mutSRSF2-mediated exon exclusion. However, replacing the native UCCAG-containing site in ATF2 with UGGAG-containing sites [from MELK or WD repeat domain 45 (WDR45)] or with a UGCAG-containing site [from phosphofructokinase (PFKM)] had almost no effect on the level of mutSRSF2-mediated exon inclusion (Fig. 5B). In neither case did swapping of sites completely switch the pattern from more exon exclusion to more exon inclusion, or vice versa, suggesting that the effect of mutSRSF2 can depend on sequence context in addition to binding affinity to a single site. In the MELK exon, for example, we found an additional UGGUG-containing site (mentioned above), which also may account for the residual exon inclusion seen with the UUUUU1 minigene (Fig. 5A). Indeed, about 40% (142) of the 374 cassette exons we identified have two or more UCCWG and/or UGGWG sites (Fig. 4). Given that there may be divergent SRSF2-binding sites and/or sites for other splicing regulators, it is likely that in some cases combinatorial effects determine the ability of mutSRSF2 to alter splicing.
Fig. 4.
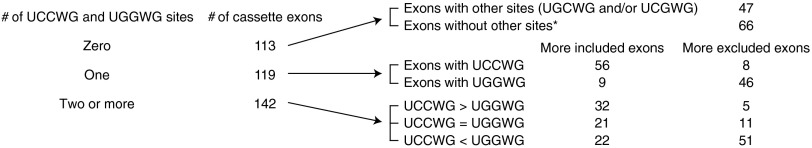
Occurrences of putative SRSF2 sites in the cassette exons. Numbers of cassette exons (either more included or more excluded in mutant vs. WT SRSF2 cells) harboring UCCWG, UGGWG, and/or other putative sites are shown. Note that 66 exons do not harbor UCCWG, UGGWG, UGCWG, or UCGWG sites. These may be false positives of rMATS (66/374 = 18%), as is consistent with the ∼80% validation rate (∼20% false rate).
The P95H Mutation Does Not Affect Protein–Protein Interactions of SRSF2 with Other Spliceosomal Components.
In addition to RNA binding, SRSF2 regulates splicing by interacting with other splicing factors, via interactions involving the RS domain (12). Because, as mentioned above, Pro-95 is situated in the linker region between the RRM and RS domains, it was conceivable that the mutation might affect protein–protein interactions as well as protein–RNA interactions. To test this possibility, we expressed HA-tagged WT or mutSRSF2 in 293T cells by transient transfection and used the cell extracts to perform coimmunoprecipitation in the presence of RNase (Fig. 6). The previously observed interactions of SRSF2 with the spliceosomal components U2AF1 and Small nuclear ribonucleoprotein 70 kDa (snRNP70) (12) were unaffected by the mutation of Pro-95 (rows 2 and 3 in Fig. 6). Notably, SF3B1 also coimmunoprecipitated with both WT and mutSRSF2 (row 1 in Fig. 6). Because SF3B1 does not have an apparent RS domain, this interaction was likely indirect, perhaps bridged by the U2AF1/U2AF2 heterodimer (12, 38). In any event, these results provide evidence that SRSF2 protein–protein interactions are unaffected by the Pro-95 mutation, supporting the view that alterations in splicing are caused solely by changes in RNA binding affinity.
Fig. 6.

Interactions of SRSF2 with U2AF1, snRNP70, and SF3B1 are unaffected by the P95H mutation. Coimmunoprecipitation was performed, in the presence of RNase, with anti-HA rabbit polyclonal antibody using extracts of 293T cells either mock-transfected (–) or transfected with plasmid expressing HA-tagged eGFP (G), WT (W), or P95H mutSRSF2 (P). The heavy and light chains of anti-HA polyclonal antibody were detected in the anti-ACTIN (rabbit) and anti-U2AF1 (rabbit) immunoblots due to the use of the anti-rabbit secondary antibody. The mAb104 antibody was used to detect endogenous and HA-tagged SRSF2 proteins.
Discussion
The results presented here show that an SRSF2 mutation found in MDS and certain leukemias alters SRSF2 RNA-binding affinities both positively and negatively, leading to the increased or decreased inclusion of numerous target exons in a sequence-dependent manner. Below we discuss the significance of these findings, with respect both to protein–RNA interactions and splicing mechanisms and to how splicing factor mutations may lead to disease.
The most frequent class of mutations found in patients with MDS involves genes encoding splicing factors, suggesting that splicing alterations play significant roles in the pathogenesis of MDS and likely other neoplasias. However, the mechanism by which these mutated factors misregulate pre-mRNA splicing in disease has only begun to be elucidated. Recently, Kim et al. (7) analyzed the effects of disease-associated SRSF2 mutations and reached conclusions in many ways consistent with ours. For example, they found that mutSRSF2 led to misregulation of a small fraction of splicing events in mouse models, AML and CMML patients, and K562 cells infected with SRSF2-expressing viruses; that splicing changes typically were very small; that C/GCNG motifs were enriched in included exons, and C/GGNG motifs were enriched in excluded exons; and that in vitro binding affinities of SRSF2 mutant derivatives (including P95H) for four artificial 6-nt RNA sites (USSAGU) increased or decreased in a manner consistent with the enriched SSNG variants in the promoted or repressed exons, respectively. Although this study provided considerable insight into the mechanism of SRSF2 mutations in MDS, several issues were unresolved. For example, SSNG motifs occur once in every 16 nt of random sequence. In an average human exon (145 bp) (39), there can be nine SSNG sites by chance. From this limited consensus, it would be difficult to determine which exons are true targets of mutSRSF2. Also, in the binding assays four artificial 6-nt RNA sites were used instead of naturally occurring sites, and it was not shown how many of the putative target exons actually harbored these ad hoc sites. Thus, it was unclear what percentage of the targets identified by RNA-seq were primary (direct) targets rather than secondary (or indirect) targets.
Our work not only confirms the elegant studies of Kim et al. (7), but also extends them in important ways. For example, our studies used multiple CRISPR-generated K562 cell clones. Thus the splicing changes detected could be attributed conclusively to mutSRSF2. Of the hundreds of altered splicing events detected, we validated a subset in CRISPR cells and obtained an ∼80% validation rate (not including ARMC10 exon 5), which is consistent with the 86% and 94% validation rates in the RNA-seq analyses performed by the developers of rMATS (16, 40). Because of the high validation rate, a large majority of the mutSRSF2 target exons we identified are very likely to be true positives. Among these exons, we observed an enriched UCCWG sequence in more-included exons and UGGWG in more-excluded exons, a more stringent consensus than determined by Kim et al. Our use of naturally occurring RNA sites in our RNA-binding assays confirmed both that these sites are direct targets and that mutSRSF2 indeed alters RNA-binding preferences in accordance with the enriched sequence motifs in the regulated exons. Finally, our minigene assays confirmed that the binding of mutSRSF2 to its target sites is sufficient to induce exon inclusion, and we also demonstrated that mutSRSF2 interactions with other splicing factors are unaffected. Taken together, these results strongly suggest that the effects of mutSRSF2 on splicing are direct and that the protein is fully functional in splicing and support the view that mutSRSF2 is a gain-of-function mutation reflecting its altered RNA-binding specificity.
How does the P95H mutation mechanistically alter RNA binding? Based on our results and the structural studies of Daubner et al. (15), we propose the following model (illustrated in Fig. S7). Pro-95 in WT SRSF2 forms a stacking interaction with both the second cytosine in the UCCAGU site and the second guanine in the UGGAGU site (15). Mutation of Pro-95 to His-95 brings a side chain with hydrogen bond (H-bond) donors and acceptors, and His-95 may form an H-bond with the second cytosine in UCCWG sites. Because an H-bond is generally stronger than a stacking interaction, this change might explain why mutSRSF2 displays increased binding to UCCWG sites. The situation with UGGWG sites is different. Daubner et al. (15) showed that the second guanine in the UGGAGU site is in syn-conformation, i.e., the guanine ring is rotated about the glycosidic bond and therefore the guanine's H-bond donors and acceptors face away from Pro-95. Thus in mutSRSF2 it is not possible for His-95 to form an H-bond with the guanine, yielding weaker binding to UGGWG sites.
Fig. S7.
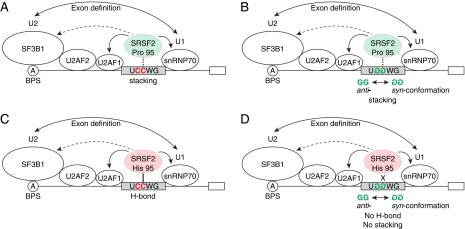
A hypothetical model of the functional mechanism of mutSRSF2. Pro-95 in WT SRSF2 (shown as a light green oval) forms a stacking interaction (dashed vertical line) with both the second cytosine in UCCWG sites (A) and the second guanine in UGGWG sites (B). His-95 in mutSRSF2 (shown as a light red oval) may form an H-bond (solid vertical line) with the second cytosine in UCCWG sites (C). An H-bond is generally stronger than a stacking interaction. Because the second guanine in the UGGWG sites is in syn-conformation, it is not possible for His-95 in mutSRSF2 to form an H-bond, yielding weaker binding to UGGWG sites (shown as an X) (D).
A critical question is which splicing change(s), if any, are important for disease causation. One possibility is that none of them are and that disease is caused by another function of SRSF2 unrelated to splicing. However, this scenario seems very unlikely for two reasons. First, mutations in multiple splicing factors, including SF3B1, SRSF2, U2AF1, and ZRSR2, have been implicated in MDS, and the mutations typically are mutually exclusive, strongly suggesting that a splicing-related function must be involved in the pathogenesis of MDS. Second, we now know that mutation of Pro-95 in SRSF2 alters RNA binding in a way consistent with observed alterations in splicing, again implicating a splicing-related process. A second hypothesis to explain the link between SRSF2 mutations and MDS involves distinguishing between “driver” splicing events—i.e., splice variations that cause disease—and “passenger” splicing events—i.e., splice variations that result from the mutations but do not play an active role in the disease. We might hypothesize that a few crucial driver splice variations are responsible for the disease, whereas the passengers have no effect. However, because of the small magnitude of the splicing changes detected by us and by Kim et al. (7), this scenario also appears unlikely. It is difficult to imagine that the alterations of a mere 10–30% of a disease-crucial isoform could produce such a large effect. A more likely possibility, we believe, is that multiple splicing changes contribute to disease, leading to “death by a thousand cuts.” This idea is consistent with the nature of the splicing alterations detected, but if it is correct it will be challenging to unravel the combination of deregulated splicing events that bring about MDS. The small magnitude of the splicing changes also is consistent with the view that the pathogenesis of MDS is a gradual process (2), so that the small effects of altered splicing on the cellular proteome only manifest themselves with time.
What are the potentially important splicing targets? One is suggested by Kim et al. (7), who provided evidence that the chromatin remodeler Enhancer of zeste homolog 2 (EZH2) may be a significant target. Although this possibility is supported by their data, we note that in our RNA-seq analysis the inclusion of the EZH2 cassette exon in mutSRSF2 cells was increased by only 4.6% (below our initial 10% cutoff), and RT-PCR analysis likewise suggested a minimal change (Fig. S3F). A number of the targets we validated also are potentially of disease relevance. For example, by mutually exclusive splicing of exons 7A and 7B, the tyrosine kinase FYN can produce two isoforms, FYN-B and FYN-T, which differ in the exon 7-encoded SH2-kinase linker region (41). FYN-T is expressed primarily in the hematopoietic lineage and regulates cell survival. Alternative splicing leads to production of FYN-B, typically expressed in brain but up-regulated by mutSRSF2, which auto-inhibits kinase activity because of its altered SH2-linker region (41). Another example is ARMC10, which is a putative tumor-suppressor gene located in chromosome 7q22, a region frequently deleted and/or translocated in MDS and AML (42, 43). A specific isoform of ARMC10 including exon 5 and exon 6, which is the isoform elevated by mutSRSF2, is up-regulated in hepatocellular carcinomas (44) and accelerates growth rate and confers tumorigenicity by binding to p53 (44, 45).
In conclusion, our studies have shown how an MDS-associated hotspot mutation in the SR protein splicing factor SRSF2 subtly alters RNA-binding affinity either positively or negatively, depending on the sequence, to sites in target transcripts, and how this alteration can lead in turn to correspondingly subtle increases or decreases in exon inclusion. Among the hundreds of splicing changes we detected, several are of potential disease relevance, but additional studies will be required to determine the significance of these, or other, misregulated splicing events to MDS.
Materials and Methods
Expression Constructs.
The human SRSF2 coding sequence (followed by an HA tag and two stop codons) was cloned into the vector p3xFLAG-CMV-14 (Sigma) using the restriction sites HindIII and BamHI. Because there were two stop codons following the HA tag, the 3xFLAG tag in the vector was not included. Site-directed mutagenesis was used to generate the P95H mutant construct. The eGFP with an HA tag and two stop codons also was cloned into p3xFLAG-CMV-14 using the same restriction sites.
Antibodies and Immunoblotting.
We used the primary antibodies mAb104 (CRL-2067; ATCC), anti-HA tag (G166; ABM), anti-ACTIN (A2066; Sigma), anti-SF3B1 (A300-996A; Bethyl Laboratories), anti-U2AF1 (ab86305; Abcam), and anti-snRNP70 (sc-9571; Santa Cruz). Immunoblotting was performed either with LI-COR secondary antibodies, donkey anti-rabbit IgG (926-68073; LI-COR) and goat anti-mouse IgG (926-32210; LI-COR) or with HRP secondary antibody, donkey anti-goat IgG-HRP (sc-2020; Santa Cruz).
CRISPR Guide RNA Vector Construction and Clone Isolation.
The human SRSF2 CRISPR guide RNA (gRNA) (5′-GGCGCGCTACGGCCGCCCCCcgg-3′) was selected using a CRISPR design tool (crispr.mit.edu) and cloned into the pSpCas9(BB)-2A-GFP (PX458; Addgene) vector. SRSF2 gRNA vector along with either WT (5′-ATGGACGGGGCCGTGCTGGACGGCCGCGAGCTGCGGGTGCAAATGGCGCGCTACGGCCGgCCCCCaGACTCACACCACAGCCGCCGGGGACCGCCACCCCGCAGGTACGGGGGCGGTGGCTAC-3′) or P95H mutant (5′-ATGGACGGGGCCGTGCTGGACGGCCGCGAGCTGCGGGTGCAAATGGCGCGCTACGGgCGCCaCCCaGACTCACACCACAGCCGCCGGGGACCGCCACCCCGCAGGTACGGGGGCGGTGGCTAC-3′) single-stranded oligodeoxynucleotide (ssODN) (Integrated DNA Technologies) was electroporated into K562 cells using the NEON transfection system (Invitrogen). After 48–72 h, cells were sorted for expression of GFP using the BD FACSAria cell sorter. After confirmation of gene knockin via PCR amplification (forward primer: 5′-TCCCGCGGCTTCGCCTTCGTTC-3′; reverse primer: 5′-CCGCCTCCCGCGGTCCCCTCAG-3′) and sequencing, the rest of the sorted cells were plated on 96-well plates for single-clone isolation. Positive single clones were selected and confirmed following PCR amplification and DNA sequencing.
RNA Sequencing, Gene Expression Levels, and Exon Inclusion Measurements.
Total RNA was isolated from K562 and CRISPR cells using the miRNeasy Mini Kit (Qiagen). Poly(A)+ RNAs were selected, and Illumina libraries were prepared using TruSeq RNA Sample Prep Kit. RNA sequencing was performed on the Illumina HiSeq 2000 system with 60 million paired-end 2 × 101 bp reads per sample (Columbia University Genome Center). Reads were mapped to the National Center for Biotechnology Information human genome reference build 37.2 using TopHat v2.0.4 with the arguments “--max-multihits 10 --no-coverage-search --mate-inner-dist 100” (46). The number of read fragments mapping to each gene was counted using HTSeq 0.6.1p1 (47) and then was normalized by gene length, which was obtained by merging all exons annotated to each gene. To obtain gene-expression levels, an additional normalization by sequencing depth was performed using DESeq2 (48). Alternative 3′ and 5′ splice sites, skipped exons, mutually exclusive exons, and retained introns from the TopHat alignments were quantified using rMATS v3.0.8 with Ensembl annotation GRCh37.75 (16). The difference in inclusion level of each candidate splicing event was calculated using reads mapping to the body of exons as well as splice junctions from four mutant samples and four WT samples. Differentially spliced events were required to have an absolute difference in inclusion level greater than 10% and a false-discovery rate less than 10%.
Motif Identification and K-mer Enrichment Analysis.
MEME was used to identify conserved sequence motifs in the published SRSF2-binding sites (37). A site distribution of zero or one occurrence per sequence and a 10-nt width were used in MEME. A position-dependent scoring matrix was generated from the identified motif, and FIMO was used to search for sequence matches in the validated splicing targets (37). Enriched motifs in the regulated cassette exons were identified by computationally scanning all the exons nucleotide by nucleotide with a window of 4 nt (4-mers) or 5 nt (5-mers), and the occurrences of all 4-mers and 5-mers were counted. The frequency of occurrence of a 4-mer or 5-mer was defined by the occurrence of the 4-mer or 5-mer divided by the total occurrences of all 4-mers or 5-mers in the exons. The frequency of occurrence in random sequence was calculated by multiplying the probabilities of all relevant bases in a 4-mer or 5-mer based on the base composition in all scanned 4-mers or 5-mers. Enriched 4-mers and 5-mers were ranked by the observed frequency in the cassette exons relative to the frequency in random sequence.
RT-PCR.
Total RNA was extracted from CRISPR cells, K562 cells, and 293T cells transfected with 500 ng plasmid expressing HA-tagged WT or mutSRSF2 using TRIzol (Life Technologies). Reverse transcription was carried out with 2 μg total RNA and 0.3 μL of Maxima Reverse Transcriptase (Thermo Scientific) using 50 pmol oligo-dT primer. The synthesized cDNA library was used as template in 10-μL PCR reactions, each containing 0.6 μCi [α-32P] dCTP (PerkinElmer). PCR products were resolved by 6% nondenaturing PAGE and then were visualized by autoradiography and quantified using ImageQuant (Molecular Dynamics). In the case of FYN, PCR products were digested with PstI before gel electrophoresis. Primers used in the PCR reactions were ATF2 forward, 5′-TTCTATGTACTGCGCCTGGA-3′; ATF2 reverse, 5′-GGTGTTGCAAGAGGGGATAA-3′; MELK forward, 5′-ATGATCACCTCACGGCTACC-3′; MELK reverse, 5′-TGCAGGTGTTCTGCATAAGG-3′; WDR45 forward, 5′-CAGGTGTGCGCATCTACAAC-3′; WDR45 reverse, 5′-ACAGAAAGCACTGGCTTGGT-3′; DGUOK forward, 5′-CTTCGAGCACCCTTCAGTTC-3′; DGUOK reverse, 5′-GGGCTCCAGCTGTACTTTCA-3′; ARMC10 forward, 5′-AACCTGAGTGTGAATGTTGAAAA-3′; ARMC10 reverse, 5′-GGGCACATTCTTCTCCATGT-3′; CDK5 regulatory subunit-associated protein 2 (CDK5RAP2) forward, 5′-GCAGCTGCTCTCACAGAATG-3′; CDK5RAP2 reverse, 5′-CCTGGGAGGAATCAAACAGA-3′; FYN forward, 5′-TCCGTGATTGGGATGATATG-3′; FYN reverse, 5′-CACCACTGCATAGAGCTGGA-3′; Abl-interactor 1 (ABI1) forward, 5′-TGGAGGAAGTGGAAGTCGAG-3′; ABI1 reverse, 5′-GGGAGGTGGAGAGTCATCAA-3′; Carnitine acetyltransferase (CRAT) forward, 5′-AGCGAAGATGTTAGCCTTCG-3′; CRAT reverse, 5′-GCCTTCAGGTAGTGGTCCAG-3′; SLC25A26 forward, 5′-CCGGAAGTTCAAGACAGACC-3′; SLC25A26 reverse, 5′-GAATCAGCATGCAAAAACCA-3′; PFKM forward, 5′-CATCATCATTGTGGCTGAGG-3′; PFKM reverse, 5′-CACCTGGACACATTCCATGA-3′; Cell division cycle 45 (CDC45) forward, 5′-TTCCCGCCTATGAAGACATC-3′; CDC45 reverse, 5′-AAGCCAGCTCAAACATCACC-3′; EZH2 forward, 5′-TTTCATGCAACACCCAACACT-3′ (7); EZH2 reverse, 5′-CCCTGCTTCCCTATCACTGT-3′ (7).
Purification of Recombinant Proteins.
Coding sequences of WT and mutSRSF2 (amino acids 1–101) followed by a His6 tag and two stop codons were cloned into the pET-26b(+) vector (Addgene) using the restriction sites NdeI and EcoRI. Protein expression was induced in Escherichia coli Rosetta cells at 20 °C overnight by 1 mM isopropyl β-D-1-thiogalactopyranoside. Proteins were purified with nickel affinity chromatography using the protocol described (15), except that we performed an additional series of washes (with 50 mM Na2HPO4, pH 5.5, containing decreasing concentrations of NaCl from 900–0 mM in 100-mM decrements) before elution with buffer containing 50 mM Na2HPO4 (pH 5.5) and 500 mM imidazole. Proteins were dialyzed overnight at 4 °C and then were concentrated using Amicon filters (EMD Millipore). Protein concentrations were determined by measuring their optical density at 280 nm.
RNA Gel Shift Assays.
RNA oligonucleotides (Integrated DNA Technologies) were radiolabeled at the 5′-hydroxyl terminus with [γ-32P] ATP using T4 polynucleotide kinase (New England Biolabs). RNA gel shift assays were performed with 20 fmol of radiolabeled RNA oligonucleotide, 100-fold molar excess tRNA, 5 U RNasin, 20 mM NaH2PO4 buffer (pH 5.5), 100 mM NaCl, and 10% glycerol. Increasing concentrations of purified His6-tagged WT or mutSRSF2 (amino acids 1–101) were added to the reaction mixtures, as indicated in the figure legends. Samples were incubated for 30 min at 37 °C and then were resolved by electrophoresis in 1× Tris-borate-EDTA buffer at 4 °C on an 8% nondenaturing polyacrylamide gel containing 15% triethylene glycol. Free and bound RNA oligonucleotides were visualized by autoradiography and quantified using ImageQuant (Molecular Dynamics). The percentage of bound RNA was plotted against the protein concentration using SigmaPlot (Systat). A sigmoidal curve fitting was used to estimate the apparent dissociation constant (Kd), i.e., the concentration of free protein at which 50% of the RNA oligonucleotides were bound.
Minigene Reporter Assays.
A minigene containing MELK (NM_014791.3) exons 12, 13, 14, and 15 and truncated introns 12, 13, and 14 was cloned into the pcDNA3 vector (Invitrogen). Another minigene containing ATF2 (NM_001880.3) exons 4, 5, and 6, part of exon 7, and truncated introns 4 and 5 also was cloned into pcDNA3. Mutations of the native SRSF2 sites to different 10-nt SRSF2 sites were introduced into these two minigenes by PCR-based site-directed mutagenesis (49). HEK293T cells were transfected with 100 ng minigene and 500 ng plasmid expressing HA-tagged WT or mutSRSF2 using Lipofectamine 2000 (Life Technologies). After ∼48 h, total RNA was isolated using TRIzol (Life Technologies). Reverse transcription was carried out with 2 μg total RNA (DNase-treated) using 50 pmol oligo-dT primer and 0.2 pmol vector-specific reverse primer (5′-TAGAAGGCACAGTCGAGG-3′). The synthesized cDNA library was used as a template in PCR reactions containing [α-32P] dCTP (PerkinElmer). PCR products were resolved by 6% nondenaturing PAGE, visualized by autoradiography, and quantified using ImageQuant (Molecular Dynamics). Primers used in the PCR reactions were vector-specific forward primer, 5′-TAATACGACTCACTATAGGGAG-3′; ATF2 reverse, 5′-GGTGTTGCAAGAGGGGATAA-3′; MELK reverse, 5′-TGCAGGTGTTCTGCATAAGG-3′.
Coimmunoprecipitation Assay.
HEK293T cells were transfected with 2 μg plasmid expressing HA-tagged WT or mutSRSF2 using the calcium phosphate precipitation method. A transfection with 2 μg plasmid expressing HA-tagged eGFP and a mock transfection without plasmid were performed as negative controls. After ∼48 h, cells were lysed using lysis buffer containing 50 mM Tris⋅Cl (pH 7.4), 150 mM NaCl, 2 mM EDTA (pH 8.0), 0.5% Nonidet P-40, and protease inhibitor mixture (Roche) in the presence of 100 μg RNase A. After incubation at 4 °C for 15 min and centrifugation at 21,130 × g for 15 min, cell extracts were incubated with 5 μL anti-HA rabbit polyclonal antibody for 30 min at 4 °C before protein G beads were added. After incubation for 2–3 h at 4 °C, the beads were washed three times with lysis buffer. Proteins were eluted from the beads with SDS buffer before immunoblot analysis.
Statistics.
In the 32P RT-PCR results (Fig. 1 and Fig. S3), Student’s t test was performed to compare the exon inclusion/exclusion levels between WT and mutSRSF2 cells. *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank Inder Verma and Tushar Menon (Salk Institute) for help with CRISPR/Cas9 technology and Ritam Neupane for technical help during the early stages of this work. This work was supported in part by NIH Grant R01 GM048259 (to J.L.M.) and by the Partnership for Cures (A.R.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE71299).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1514105112/-/DCSupplemental.
References
- 1.Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340(21):1649–1660. doi: 10.1056/NEJM199905273402107. [DOI] [PubMed] [Google Scholar]
- 2.Corey SJ, et al. Myelodysplastic syndromes: The complexity of stem-cell diseases. Nat Rev Cancer. 2007;7(2):118–129. doi: 10.1038/nrc2047. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 4.Haferlach T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thol F, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119(15):3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 6.Wu SJ, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120(15):3106–3111. doi: 10.1182/blood-2012-02-412296. [DOI] [PubMed] [Google Scholar]
- 7.Kim E, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27(5):617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010;24(21):2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3(11):1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long JC, Caceres JF. The SR protein family of splicing factors: Master regulators of gene expression. Biochem J. 2009;417(1):15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 11.Tacke R, Manley JL. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14(14):3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75(6):1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 13.Kohtz JD, et al. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368(6467):119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 14.Papaemmanuil E, et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627, quiz 3699. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daubner GM, Cléry A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012;31(1):162–174. doi: 10.1038/emboj.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen S, et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA. 2014;111(51):E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Bergami P, Lau E, Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10(1):65–76. doi: 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandel H, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29(3):337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 19.Bieber LL. Carnitine. Annu Rev Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, et al. CDK5RAP2 is required for spindle checkpoint function. Cell Cycle. 2009;8(8):1206–1216. doi: 10.4161/cc.8.8.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scita G, et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401(6750):290–293. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- 22.Hopwood B, Dalton S. Cdc45p assembles into a complex with Cdc46p/Mcm5p, is required for minichromosome maintenance, and is essential for chromosomal DNA replication. Proc Natl Acad Sci USA. 1996;93(22):12309–12314. doi: 10.1073/pnas.93.22.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang P, Zhang D. Maternal embryonic leucine zipper kinase (MELK): A novel regulator in cell cycle control, embryonic development, and cancer. Int J Mol Sci. 2013;14(11):21551–21560. doi: 10.3390/ijms141121551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haack TB, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91(6):1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahsan H, et al. Familial Breast Cancer Study A genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidemiol Biomarkers Prev. 2014;23(4):658–669. doi: 10.1158/1055-9965.EPI-13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrimi G, et al. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J. 2004;379(Pt 1):183–190. doi: 10.1042/BJ20031664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavaloc Y, Bourgeois CF, Kister L, Stévenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5(3):468–483. doi: 10.1017/s1355838299981967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaal TD, Maniatis T. Selection and characterization of pre-mRNA splicing enhancers: Identification of novel SR protein-specific enhancer sequences. Mol Cell Biol. 1999;19(3):1705–1719. doi: 10.1128/mcb.19.3.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dreumont N, et al. Antagonistic factors control the unproductive splicing of SC35 terminal intron. Nucleic Acids Res. 2010;38(4):1353–1366. doi: 10.1093/nar/gkp1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arrisi-Mercado P, Romano M, Muro AF, Baralle FE. An exonic splicing enhancer offsets the atypical GU-rich 3′ splice site of human apolipoprotein A-II exon 3. J Biol Chem. 2004;279(38):39331–39339. doi: 10.1074/jbc.M405566200. [DOI] [PubMed] [Google Scholar]
- 31.Zahler AM, Damgaard CK, Kjems J, Caputi M. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J Biol Chem. 2004;279(11):10077–10084. doi: 10.1074/jbc.M312743200. [DOI] [PubMed] [Google Scholar]
- 32.Hallay H, et al. Biochemical and NMR study on the competition between proteins SC35, SRp40, and heterogeneous nuclear ribonucleoprotein A1 at the HIV-1 Tat exon 2 splicing site. J Biol Chem. 2006;281(48):37159–37174. doi: 10.1074/jbc.M603864200. [DOI] [PubMed] [Google Scholar]
- 33.Caputi M, Zahler AM. SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J. 2002;21(4):845–855. doi: 10.1093/emboj/21.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian W, et al. Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011;39(14):6161–6171. doi: 10.1093/nar/gkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaal TD, Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol. 1999;19(1):261–273. doi: 10.1128/mcb.19.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crovato TE, Egebjerg J. ASF/SF2 and SC35 regulate the glutamate receptor subunit 2 alternative flip/flop splicing. FEBS Lett. 2005;579(19):4138–4144. doi: 10.1016/j.febslet.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 37.Bailey TL, et al. 2009. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res 37(Web Server issue):W202–208.
- 38.Gozani O, Potashkin J, Reed R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol. 1998;18(8):4752–4760. doi: 10.1128/mcb.18.8.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lander ES, et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. and corrections (2001) 411:720 and (2001) 412:565. [DOI] [PubMed] [Google Scholar]
- 40.Shen S, et al. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40(8):e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brignatz C, et al. Alternative splicing modulates autoinhibition and SH3 accessibility in the Src kinase Fyn. Mol Cell Biol. 2009;29(24):6438–6448. doi: 10.1128/MCB.00398-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Beau MM, et al. Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood. 1996;88(6):1930–1935. [PubMed] [Google Scholar]
- 43.Fischer K, et al. Molecular cytogenetic delineation of deletions and translocations involving chromosome band 7q22 in myeloid leukemias. Blood. 1997;89(6):2036–2041. [PubMed] [Google Scholar]
- 44.Huang R, et al. A specific splicing variant of SVH, a novel human armadillo repeat protein, is up-regulated in hepatocellular carcinomas. Cancer Res. 2003;63(13):3775–3782. [PubMed] [Google Scholar]
- 45.Zhou X, Yang G, Huang R, Chen X, Hu G. SVH-B interacts directly with p53 and suppresses the transcriptional activity of p53. FEBS Lett. 2007;581(25):4943–4948. doi: 10.1016/j.febslet.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M, David CJ, Manley JL. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat Struct Mol Biol. 2012;19(3):346–354. doi: 10.1038/nsmb.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.