

Significance
Acute myeloid leukemia (AML) is a hematologic malignancy with poor survival. Current treatment with chemotherapy does not target the leukemic cells specifically and is associated with severe side effects. Here we demonstrate that antibodies directed at the cell surface molecule IL-1 receptor accessory protein (IL1RAP), expressed on immature AML cells, show strong antileukemic effects in mice transplanted with human AML cells and that the mechanism behind the cell killing is through recruitment of effector cells. Using antibodies against IL1RAP also capable of blocking IL-1 signaling, we show that the proliferation of human AML cells can be inhibited, providing a second mode of action of IL1RAP antibodies. These results provide critical evidence in support of a rapid clinical development of an antibody-based anti-IL1RAP therapy in AML.
Keywords: IL1RAP, AML, antibody, leukemia, immunotherapy
Abstract
Acute myeloid leukemia (AML) is associated with a poor survival rate, and there is an urgent need for novel and more efficient therapies, ideally targeting AML stem cells that are essential for maintaining the disease. The interleukin 1 receptor accessory protein (IL1RAP; IL1R3) is expressed on candidate leukemic stem cells in the majority of AML patients, but not on normal hematopoietic stem cells. We show here that monoclonal antibodies targeting IL1RAP have strong antileukemic effects in xenograft models of human AML. We demonstrate that effector-cell–mediated killing is essential for the observed therapeutic effects and that natural killer cells constitute a critical human effector cell type. Because IL-1 signaling is important for the growth of AML cells, we generated an IL1RAP-targeting antibody capable of blocking IL-1 signaling and show that this antibody suppresses the proliferation of primary human AML cells. Hence, IL1RAP can be efficiently targeted with an anti-IL1RAP antibody capable of both achieving antibody-dependent cellular cytotoxicity and blocking of IL-1 signaling as modes of action. Collectively, these results provide important evidence in support of IL1RAP as a target for antibody-based treatment of AML.
Acute myeloid leukemia (AML) is a genetically heterogeneous disease characterized by clonal expansion of leukemic cells. Despite an increased understanding of the underlying disease biology in AML, the standard treatment with cytotoxic chemotherapy has remained largely unchanged over the last decades and the overall 5-y survival remains poor, being <30% (1, 2). Hence, there is a pressing need for novel therapies with increased efficacy and decreased toxicity, ideally targeting the AML stem cells because these cells are believed to be critical in the pathogenesis of AML, and their inadequate eradication by standard therapy is thought to contribute to the high incidence of relapse (3, 4). Although therapeutic antibodies directed at cell-surface molecules have proven effective for the treatment of malignant disorders such as lymphomas and acute lymphoblastic leukemia, as well as solid tumors (5, 6), no antibody-based therapy is currently approved for AML.
The interleukin 1 receptor accessory protein (IL1RAP), also called IL1R3, is a coreceptor of type 1 interleukin 1 receptor (IL1R1) and is indispensable for transmission of IL-1 signaling (7). We have previously reported that IL1RAP is a biomarker for putative chronic myeloid leukemia stem cells (8). In a recent study, we showed that IL1RAP is expressed on the cell surface in ∼80% of AML patients and that candidate CD34+CD38− AML stem cells can be selectively killed in vitro by antibody-dependent cellular cytotoxicity (ADCC) (9). Furthermore, IL1RAP is up-regulated on immature cells in high-risk AML with chromosome 7 aberrations, and increased IL1RAP expression correlates with poor prognosis (10). These findings could suggest IL1RAP as a novel and specific target for an antibody-based therapy in AML; however, in vivo evidences for therapeutic effects of IL1RAP targeting are lacking. Here, we demonstrate that antibodies targeting IL1RAP in immunodeficient mice transplanted with human AML cells have strong in vivo antileukemic effects. We further show that effector-cell–mediated killing is a critical mechanism for the observed therapeutic effects and that IL1RAP antibodies capable of inhibiting IL-1 signaling suppress the proliferation of primary human AML cells, suggesting that this dual mode of action of anti-IL1RAP antibodies is an efficient and promising approach in future treatment of AML.
Results
IL1RAP-Targeting Antibodies Show Therapeutic Effects in a Xenograft Model of Human AML.
To evaluate whether an antibody-based therapy targeting IL1RAP has therapeutic effects in a preclinical in vivo model of AML, we first used human immortalized MA9Ras cord blood cells coexpressing an MLL/AF9 fusion gene and an activated NRAS gene. MA9Ras cells express IL1RAP on the cell surface (Fig. 1A) and have been shown to faithfully recapitulate human AML in a xenograft setting (11). Untreated nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice engrafted with MA9Ras cells developed acute leukemia with a latency of 50–65 d and displayed high leukemic engraftment in bone marrow (BM) and spleen (Fig. S1A). In this model, approximately one-third of the mice normally develop extramedullary myeloid sarcoma consisting of leukemic cells, tumors that are infrequent in human AML patients (12). Treating mice with the anti-IL1RAP monoclonal antibody (mAb) 81.2 resulted in a significantly reduced frequency of leukemic cells in peripheral blood (PB) 35 d after transplantation, compared with the control group receiving an isotype control antibody (Fig. 1B). Platelet counts remained stable in mAb81.2-treated mice, suggesting improvement in hematopoietic parameters as a consequence of reduced leukemic burden (Fig. 1C). At day 62, all mAb81.2-treated mice showed low frequencies of leukemic cells in the PB, in contrast to controls (Fig. 1D). The mAb81.2-treated mice displayed a significantly prolonged survival (median 75 d, range 64–76) compared with control mice (median 64 d, range 62–67) and were killed due to MA9Ras-consisting myeloid sarcoma (Fig. 1E). Notably, despite being killed on average 11 d later than controls, the mAb81.2 treatment group had significantly reduced levels of leukemic cells in the BM (Fig. 1F). Moreover, mAb81.2-treated mice showed markedly reduced levels of leukemic cells in the spleen and lower spleen weights (Fig. 1G and Fig. S1B). Hematoxylin/eosin (H&E)-stained and human CD45-stained sections of BM and spleen revealed close to normal architecture in mAb81.2-treated mice, whereas the control mice displayed substantial infiltration of leukemic cells and disrupted architecture (Fig. 1H and Fig. S1C).
Fig. 1.

Treatment with mAb81.2 has antileukemic effects and prolongs survival in the MA9Ras xenotransplantation model. (A) Expression of IL1RAP on the surface of human MA9Ras AML cells (red, isotype control; blue, anti-IL1RAP) using flow cytometry analysis. (B and C) Frequency of leukemic cells and platelet counts in PB 35 d after transplantation of MA9Ras AML cells into NOD/SCID mice treated with the IL1RAP-targeting antibody mAb81.2 or a mIgG2a isotype control antibody (means ± SD; n = 7 in both groups). (D) Leukemic cell frequency in PB 62 d after transplantation (means ± SD). (E) Overall survival rates with mAb81.2 (median 75 d) compared with isotype control (median 64 d). P value was calculated by using the Mantel Cox log rank test. (F and G) Leukemic cell frequency in BM and spleen (means ± SD). (H) H&E-stained and human CD45-immunostained BM sections from representative isotype control and mAb81.2-treated mice. (Scale bars, 300 μm.) Unless otherwise stated, P values were calculated with Student’s t test.
Fig. S1.
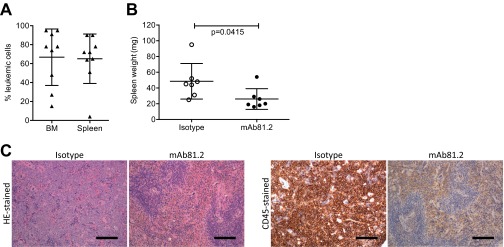
Treatment with mAb81.2 reduces the leukemic burden in the MA9Ras xenotransplantation model. (A) MA9Ras cell engraftment in BM and spleen of unconditioned NOD/SCID mice without antibody treatment (means ± SD; n = 9). (B) Spleen weight in mice engrafted with MA9Ras and treated with mAb81.2 or isotype control (means ± SD; n = 7 in both groups). (C) H&E-stained and human CD45-immunostained spleen sections from representative control and mAb81.2-treated mice. (Scale bars, 300 μm.) P values were calculated with Student’s t test.
To validate IL1RAP as the specific target for the observed in vivo therapeutic effects, a second antibody, mAb3F8, was generated that binds to a different epitope on IL1RAP than mAb81.2 and with higher affinity. Similar to mAb81.2, treatment with mAb3F8 resulted in reduced levels of leukemic cells in PB, BM, and spleen, accompanied by a significantly prolonged survival in the MA9Ras xenograft model (Fig. S2 A–G). A mAb81.2 dose titration study showed that the antibody dose could be lowered from 20 mg/kg to 0.16 mg/kg with maintained effect in PB, BM and spleen (Fig. S2 A, B, D, and E). Collectively, the investigations in the MA9Ras xenotransplantation model with two independent antibodies demonstrate that antibody-based immunotherapy against IL1RAP results in strong anti-leukemic in vivo effects.
Fig. S2.

In vivo results using mAb3F8 and mAb81.2 dose titration. (A and B) PB leukemic cell frequency (A) and platelet counts (B) at 35 d after transplantation in NOD/SCID mice engrafted with MA9Ras cells and treated with mAb81.2 or mAb3F8 or a corresponding isotype control antibody (means ± SD; n = 3–7 per group). (C) Overall survival rates after treatment with mAb81.2 (median 58 d), mAb3F8 (median 57 d), and control (median 51 d) (P < 0.0001; n = 7 for all groups). P values were calculated by using the Mantel Cox log rank test. (D and E) Leukemic cell frequency in BM (D) and spleen (E) (means ± SD). (F and G) Spleen weight (F) and estimated leukemic spleen burden (G) calculated as frequency of leukemic cells multiplied by spleen weight (means ± SD). Unless otherwise stated, P values were calculated with Student’s t test.
Murine Effector Cells Mediate the Therapeutic Effect in the MA9Ras Xenograft Model.
We next investigated the mechanistic basis for the observed antileukemic effects on MA9Ras cells. Upon addition of mAb81.2 or mAb3F8 to suspension cultures of MA9Ras cells, no effects on cellular expansion or degree of apoptosis was observed (Fig. 2 A and B), suggesting that the antibody-induced therapeutic effect observed in vivo is dependent on interactions with murine effector cells. To delineate this hypothesis, MA9Ras cells were engrafted in NOD/SCID IL-2–receptor γc-deficient (NSG) mice, which have reduced effector cell activity compared with NOD/SCID mice due to an inherent lack of natural killer (NK) cells. Following treatment with mAb81.2, only a minor reduction in BM leukemic cell frequency was observed, suggesting that NK cell-mediated cytotoxicity was involved in leukemic cell killing in the MA9Ras NOD/SCID model (Fig. 2C). However, a therapeutic effect was noted in the spleen (Fig. 2D), consistent with previous reports demonstrating that cell types other than NK cells, such as macrophages, can act as effector cells in mice (13, 14).
Fig. 2.

Antileukemic in vivo effect on MA9Ras cells depends on murine effector cells. (A) Fold expansion of MA9Ras cells in suspension cultures with addition of 10 μg/mL IL1RAP-targeting antibodies mAb81.2 and mAb3F8 or an isotype control antibody (means ± SEM from four experiments with technical duplicates). (B) Relative levels of viable, apoptotic, and necrotic MA9Ras cells after treatment with 10 μg/mL mAb81.2, mAb3F8, or an isotype control antibody. Staurosporine treatment (STP) was used as positive control for apoptosis (means ± SEM from three experiments with technical duplicates). (C and D) NSG mice were engrafted with MA9Ras cells and treated with mAb81.2 or an isotype control antibody (n = 5 in both groups). Graphs show BM (C) and spleen (D) leukemic cell frequency at death 39 d after transplantation (means ± SD). (E) IL-1 activates NF-kβ in the HEKblue IL1R1 reporter cell line. The graph shows the NF-kβ activation measured by absorbance in the presence of IL-1 upon addition of mAb81.2 or mAb3F8 or an isotype control antibody (means ± SEM from technical duplicates; one representative experiment out of three). (F) Absorbance of NF-kβ activation in the HEKblue IL1R1 reporter cell assay in the absence of IL-1 with IL1RA included as a control (one representative experiment out of three). P values were calculated with Student’s t test.
Multiple studies have shown that interference with IL-1 signaling by anti–IL-1 antibodies or an IL1R1 antagonist (IL1RA) suppress proliferation of leukemic cells from a majority of AML patients (15–23). We therefore investigated the ability of the developed antibodies to inhibit IL-1 signaling. Whereas mAb81.2 showed almost no inhibitory effect in an IL-1 reporter assay, mAb3F8 displayed potent inhibition of IL1R1 signaling in a dose-dependent manner (Fig. 2E). Neither of the antibodies affected IL1R1 signaling in the absence of IL-1, thereby excluding agonistic functions on IL-1–induced signaling (Fig. 2F). To investigate whether the MA9Ras cells were responsive to IL-1, suspension cultures were supplemented with IL-1, albeit no impact on cellular expansion or rate of apoptosis was observed (Fig. S3). Together, these results strongly suggest that the observed in vivo therapeutic effects in the MA9Ras model rely upon interactions with murine effector cells.
Fig. S3.

MA9Ras cells do not respond to IL-1 in suspension cultures. (A) Fold expansion of MA9Ras cells without IL-1 or with 0.4 or 10 ng/mL IL-1 (means ± SEM from four experiments with technical duplicates). (B) Relative levels of viable, apoptotic, and necrotic MA9Ras cells in the absence or presence of IL-1 (means ± SEM from two experiments with technical duplicates). P values were calculated with Student’s t test.
Anti-IL1RAP Treatment Shows Therapeutic Effect on Primary Human AML Cells in Vivo.
To verify that the therapeutic in vivo effects from targeting IL1RAP in the MA9Ras model could be reproduced with primary AML cells, samples obtained from AML patients were transplanted into immunodeficient mice (Table S1), followed by treatment with mAb81.2 or an isotype control antibody. Because effector cell function was found to be essential for the therapeutic effects in the MA9Ras model, we first transplanted three AML samples (AML1, AML2, and AML3) into NOD/SCID mice. Although it is challenging to obtain engraftment of primary AML cells in this strain, sample AML2 showed successful BM engraftment of leukemic cells, thereby allowing us to evaluate the therapeutic effect on primary human AML cells in vivo. Notably, the mAb81.2-treated mice displayed a significant reduction of BM leukemic cell frequency compared with controls (Fig. 3A and Fig. S4A). Increased engraftment of primary human AML cells can be obtained by using preconditioning and more severely immunocompromised mouse strains such as NSG (4). Hence, we next transplanted three samples (AML1, AML3, and AML4) into sublethally irradiated NSG or NSG SGM3 (NSGS) mice. Despite the less efficient effector-cell–mediated cell killing in these mice, mAb81.2 treatment of mice engrafted with sample AML4 resulted in significantly reduced levels of leukemic cells in BM and spleen (Fig. 3B). Treatment with mAb81.2 of samples AML1 and AML3 in vivo resulted in a similar trend (Fig. S4 B and C). Collectively, these results demonstrate that antibodies targeting IL1RAP also show in vivo therapeutic effects on primary human AML cells.
Table S1.
Characteristics of primary AML cells used in this study
| Sample | Sex/age, y | Source | FAB | Karyotype | Molecular aberrations | IL1RAP expression |
| AML1 | M/40 | PB | ND* | 46,XY | FLT3-ITD−/FLT3-TKD−, NPM1+ | High |
| AML2 | F/46 | PB | M2 | 46,XX | FLT3-ITD+/FLT3-TKD−, NMP1+ | High |
| AML3 | F/71 | BM | M5 | 46,XX | FLT3−, NPM1− | Intermediate |
| AML4 | F/65 | PB | M4/M5 | 47,XX,+8[21]/46,XX[4] | FLT3-ITD−/FLT3-TKD−, NPM1+ | High |
| AML5 | F/67 | BM | ND* | 46,XX | FLT3+ | High |
| AML6 | M/52 | BM | ND* | 46,XY,t(9;11) | FLT3−, NPM1− | Intermediate |
| AML7 | F/45 | BM | M2 | 46,XX | FLT3-ITD+/FLT3-TKD−, NPM1+ | High |
| AML8 | F/74 | BM | ND* | 46,XX | FLT3-ITD−/FLT3-TKD−, NPM1− | Intermediate |
| AML9 | F/72 | BM | M4 | 46,XX | FLT3-ITD−/FLT3-TKD+ | Intermediate |
IL1RAP expression was determined by flow cytometry as described (9). F, female; FAB, French–American–British classification; M, male; ND, not determined.
Fig. 3.

Targeting of IL1RAP results in reduced frequencies of primary human AML cells in vivo. (A) Frequency of leukemic cells in BM of NOD/SCID mice engrafted with sample AML2 and treated with mAb81.2 (n = 10) or isotype control antibody (n = 8) at death 28 d after transplantation (Left; means ± SD), and flow cytometry plots of representative isotype control (Center) and mAb81.2-treated (Right) mice. (B) Frequency of leukemic cells in BM (Left) and spleen (Right) of NSGS mice engrafted with sample AML4 and treated with mAb81.2 or isotype control antibody (n = 6 in both groups) at death 56 d after transplantation (means ± SD). P values were calculated with Mann–Whitney test.
Fig. S4.

Treatment with mAb81.2 has an antileukemic effect in mice engrafted with primary human AML cells. (A) Flow cytometry pseudocolor plots from analyzed BM of all NOD/SCID mice transplanted with sample AML2 and treated with mAb81.2 or isotype control antibody. CD45+CD33+ cells were considered human leukemic cells. Two nontransplanted and untreated mice were included in the analysis as specificity controls for the CD45 and CD33 antibodies. (B) Frequency of leukemic cells in BM (Upper) and spleen (Lower) of NSGS mice transplanted with sample AML1 at death 57 d after transplantation. In BM, three of five mAb81.2-treated mice were below the median of the control (n = 5 in both groups), and in spleen, all five were (medians ± interquartile range). (C) Frequency of leukemic cells of NSG mice transplanted with sample AML3 at death 35 d after transplantation. In BM (Upper) and spleen (Lower), all six mAb81.2-treated mice were below the median of the control (n = 6 in both groups; medians ± interquartile range). P values were calculated with the Mann–Whitney test.
IL1RAP-Targeting Antibodies Mediate ADCC with Human NK Cells.
To investigate possible antibody-mediated effector-dependent mechanisms for cell killing in a human setting, chimeric mAb81.2 and mAb3F8 of human IgG1 subtype were analyzed for ADCC, antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) activity, by using human effector cells or complement. In agreement with previous results using primary human AML cells (9), MA9Ras cells were specifically killed by ADCC using human NK cells with both antibodies (Fig. 4A). By contrast, neither of the antibodies directed human macrophages to phagocytosis over the levels of an isotype control (Fig. 4B). Nor did the IL1RAP-targeting antibodies mediate specific cell death by CDC (Fig. 4C). The ability of the IL1RAP-targeting antibodies to mediate ADCC on different human AML cells was verified by using the human AML cell lines EOL1, MonoMac6, and OCI-AML1 (Fig. S5). Given that ADCC also could have an effect on normal tissues expressing IL1RAP, we next investigated the tissue expression of IL1RAP on an array of 30 different normal human tissues using mAb3F8. We have previously shown that, among normal blood cells, IL1RAP is expressed mainly on monocytes, and that primary AML cells are more sensitive to mAb81.2-mediated ADCC than corresponding normal BM cells (8, 9). We now show that IL1RAP staining was observed in only five tissues: BM, esophagus, lymph node, placenta, and spleen (Fig. S6 and Table S2). All together, these results suggest that antibody-based targeting of IL1RAP in humans would be associated with few side effects, although toxicity studies will be needed to confirm this finding.
Fig. 4.

IL1RAP-targeting antibodies mediate ADCC with human NK cells, and mAb3F8 blocks proliferation of AML cells. (A) Frequency of dead MA9Ras cells with human NK cells in an ADCC assay (means ± SEM from technical duplicates; one representative experiment out of three). (B) Relative amount of human phagocytic macrophages in an ADCP assay with MA9Ras target cells. Data were normalized to 1.0 at conditions without antibody addition (means ± SEM from four experiments with technical duplicates in which the typical frequency of MA9Ras+ macrophages were 20–50% without the addition of antibody). (C) Frequency of dead MA9Ras cells in a CDC assay using human normal serum (NS) or heat-inactivated serum (HIS) (means ± SEM from three experiments with technical duplicates). (D) Proliferation rate of three primary human AML samples with IL-1, as measured by incorporation of EdU. Data normalized to proliferation without IL-1 set to 1 (means ± SEM from one experiment with technical duplicates or triplicates). (E) Proliferation rate of samples AML5 and AML6 in the presence of IL-1 and mAb3F8 or isotype control antibody measured by EdU incorporation. Data were normalized to conditions without IL-1 set to 1 as indicated by the dotted line (means ± SEM from one experiment with technical duplicates or triplicates). (F) Total expansion of sample AML6 after 6 d of culture, with the dotted line indicating the number of cells in conditions without IL-1 (means ± SEM from one experiment with technical triplicates). (G) The effect of mAb3F8 or isotype control antibody on the IL-1–induced proliferation of primary human AML cells as measured by expression of the proliferation marker Ki-67. Data were normalized to conditions with IL-1 only set to 1 (means ± SEM from four AML patients with technical triplicates). P values were calculated with Student’s t test.
Fig. S5.

IL1RAP-targeting antibodies mAb81.2 and mAb3F8 mediate ADCC with human NK cells on human AML cell lines. Frequency of dead target cells using MonoMac6 (A), EOL1 (B), and OCI-AML1 cells (C) (means ± SEM from technical duplicates; one representative experiment out of two).
Fig. S6.

Immunostaining with mAb3F8 on 30 normal tissues reveals staining in only five. Displayed are the tissues that showed some degree of staining with 0.1 μg/mL mAb3F8 in at least one individual out of three analyzed. (Scale bars, 100 μm.) A mIgG1 antibody at a concentration of 0.1 μg/mL was used as isotype control. IL1RAP high-expressing SKMEL5 cells were used as positive control, and IL1RAP low-expressing KG1 cells were used as negative control.
Table S2.
IL1RAP-immunostaining of normal tissues
| Tissue | Anti-IL1RAP mAb3F8 mIgG1 | mIgG1 isotype control |
| Adrenal 1 | 1+ diffuse in stromal parts | 0 |
| Adrenal 2 | 1+ diffuse in stromal parts | 0 |
| Adrenal 3 | 1+ diffuse in stromal parts | 0 |
| BM 1 | 2–3+ in undefined cells | 1–2+ in undefined cells |
| BM 2 | 2–3+ in undefined cells | 1–2+ in undefined cells |
| BM 3 | 2–3+ in undefined cells | 1–2+ in undefined cells |
| Breast, 1 | 0 | Local in mucin |
| Breast, 2 | 0 | 0 |
| Breast, 3 | 0 | Local in mucin |
| Brain, cerebellum, 1 | 0 | 0 |
| Brain, cerebellum, 2 | 0 | 0 |
| Brain, cerebellum, 3 | 0 | 0 |
| Brain, cortex, 1 | 0 | 0 |
| Brain, cortex, 2 | 0 | 0 |
| Brain, cortex, 3 | 0 | 0 |
| Brain, pituitary, 1 | 0 | 0 |
| Brain, pituitary, 2 | 0 | 0 |
| Brain, pituitary, 3 | 0 | 0 |
| Colon, 1 | 0 | 0 |
| Colon, 2 | 2+ diffuse in lamina propria and epithelial cells | 1–2+ diffuse in lamina propria and epithelial cells |
| Colon, 3 | 0 | 0 |
| Aorta,1 | 0 | 0 |
| Aorta,2 | 0 | 0 |
| Aorta,3 | 0 | Some unspecific at edge of section |
| Esophagus, 1 | 1+ in epithelial cells | 0 |
| Esophagus, 2 | 1+ in epithelial cells | 0 |
| Esophagus, 3 | 1+ in epithelial cells | 0 |
| Fallopian tube, 1 | 0 | 0 |
| Fallopian tube, 2 | 1+ diffuse local | 1+ diffuse local |
| Fallopian tube, 3 | 1+ diffuse local and some endogenous peroxidase | 1+ diffuse local and some endogenous peroxidase |
| Heart, 1 | 0 | 0 |
| Heart, 2 | 0 | 0 |
| Heart, 3 | 0 | 0 |
| Kidney, 1 | 0 | 0 |
| Kidney, 2 | 0 | 0 |
| Kidney, 3 | 0 | 0 |
| Liver, 1 | 1+ diffuse | Some very weak diffuse |
| Liver, 2 | 1+ diffuse | Some very weak diffuse |
| Liver, 3 | 1+ diffuse | Some very weak diffuse |
| Lung, 1 | 2–3+ in undefined cells | 2–3+ in undefined cells |
| Lung, 2 | 1–2+ mostly local | 1–2+ local |
| Lung, 3 | 1–2+ mostly local | 1–2+ local |
| Lymph node, 1 | 1+, diffuse and some endogenous peroxidase | Endogenous peroxidase |
| Lymph node, 2 | 1+ diffuse | Endogenous peroxidase |
| Lymph node, 3 | 1+ diffuse | Endogenous peroxidase |
| Ovary, 1 | 0 | 0 |
| Ovary, 2 | 0 | 0 |
| Ovary, 3 | 0 | 0 |
| Pancreas, 1 | Local 1–2+ in mucin | Local 1–2+ in mucin |
| Pancreas, 2 | Local 1–2+ in mucin | Local 1–2+ in mucin |
| Pancreas, 3 | Local 1–2+ in mucin | Local 1–2+ in mucin |
| Placenta, 1 | 2–3+ local staining | Endogenous peroxidase |
| Placenta, 2 | 2–3+ local staining | Some local endogenous peroxidase |
| Placenta, 3 | 2–3+ local staining | Some local endogenous peroxidase |
| Prostate, 1 | 0 | 0 |
| Prostate, 2 | 0 | 0 |
| Prostate, 3 | 0 | 0 |
| Skin, 1 | 0 | 0 |
| Skin, 2 | 0 | 0 |
| Skin, 3 | 0 | 0 |
| Spinal cord, 1 | 0 | 0 |
| Spinal cord, 2 | 0 | 0 |
| Spinal cord, 3 | 0 | 0 |
| Spleen, 1 | 1–2+ in red pulp | Endogenous peroxidase |
| Spleen, 2 | 1+ in red pulp | Endogenous peroxidase and some 1+ diffuse |
| Spleen, 3 | 1+ in red pulp | Endogenous peroxidase and some 1+ diffuse |
| Striated muscle, 1 | 0 | 0 |
| Striated muscle, 2 | 0 | 0 |
| Striated muscle, 3 | 0 | 0 |
| Stomach, 1 | 0 | Some endogenous peroxidase |
| Stomach, 2 | 2+, in mucin | 2+, in mucin |
| Stomach, 3 | 2+, in mucin | 2+, in mucin |
| Testis, 1 | 0 | Some local endogenous peroxidase |
| Testis, 2 | 0 | Some local endogenous peroxidase |
| Testis, 3 | 0 | Some local endogenous peroxidase |
| Thymus, 1 | 0 | 0 |
| Thymus, 2 | 0 | 0 |
| Thymus, 3 | 0 | 0 |
| Thyroid, 1 | 0 | 0 |
| Thyroid, 2 | 0 | 0 |
| Thyroid, 3 | 0 | 0 |
| Ureter, 1 | 0 | 0 |
| Ureter, 2 | 0 | Some local endogenous peroxidase |
| Ureter, 3 | 0 | Some local endogenous peroxidase |
| Uterus, endometrium, 1 | 0 | 0 |
| Uterus, endometrium, 2 | 0 | 0 |
| Uterus, endometrium, 3 | 2–3+ local in mucin | 2–3+ local in mucin |
| Uterus, cervix, 1 | 0 | 0 |
| Uterus, cervix, 2 | 0 | 0 |
| Uterus, cervix, 3 | 2–3+ local in mucin | 2–3+ local in mucin |
The staining intensity was determined as negative (0), weak staining (1+), moderate staining (2+), or strong staining (3+).
Anti-IL1RAP Antibody Suppresses Proliferation of IL-1–Dependent Human AML Cells.
Because mAb3F8 was found to block IL1R1 signaling in the IL-1 reporter assay in addition to mediating ADCC, we next explored whether this IL1RAP-targeting antibody could inhibit IL-1–induced proliferation of AML cells. Because MA9Ras cells are not responsive to IL-1, we used primary human AML cells. The response of human cells to murine IL-1 was found to be weak (Fig. S7), precluding in vivo assessments of IL-1 signaling inhibition. We therefore identified IL-1–responsive AML samples by culturing AML cells (samples AML5, AML6, and AML7; Table S1) in serum-free medium with or without IL-1 followed by determination of the proliferation rate by incorporation of 5-ethnynyl-2′-deoxyuridine (EdU). Samples AML5 and AML6 displayed increased proliferation after a 24-h stimulation with IL-1 (Fig. 4D). Treatment with mAb3F8 reduced the IL-1–induced cell proliferation by 25% (AML5) and 84% (AML6), compared with an isotype control antibody (Fig. 4E). In addition, in sample AML6, mAb3F8 inhibited the IL-1–induced cellular expansion by 55%, following a prolonged culture of 6 d (Fig. 4F). We also analyzed the expression of the proliferation marker Ki-67 on four IL-1–responsive primary human AML samples (AML5, AML6, AML8, and AML9; Table S1). Treatment with mAb3F8 inhibited the proliferative effect of IL-1 by 50%, as measured by expression of Ki-67 (Fig. 4G). These results demonstrate that an IL1RAP-targeting antibody can reduce proliferation of primary human AML cells by interfering with IL1R1-signaling.
Fig. S7.

Weak IL-1 signaling of human cells in response to murine IL-1. The graph shows the NF-kβ activation in the HEKblue IL1R1 reporter cell line measured by absorbance in presence of increasing concentrations of murine or human IL-1.
Discussion
Here, we provide in vivo evidence that antibodies targeting IL1RAP show therapeutic effects on human AML cells. The mechanism underlying the in vivo therapeutic effects was found to depend on interactions with effector cells. In IL-1–responsive primary AML samples, we identified antibody-induced suppression of IL-1 signaling as an additional mode of action.
Currently, only a few cell-surface markers have been shown to display preferential expression on AML stem cells compared with normal hematopoietic stem cells and with antibodies targeting these molecules demonstrating therapeutic efficacy in xenotransplantation models (24–27). However, apart from CD123, which showed limited efficacy in early clinical trials (28), to our knowledge, none has progressed to clinical evaluation in AML. IL1RAP is expressed on candidate AML stem cells, but not on corresponding normal cells (9). By using MA9Ras cells engrafted into immunodeficient mice, a strong antileukemic effect of the two independent IL1RAP-targeting antibodies mAb81.2 and mAb3F8 was observed in all hematopoietic compartments, including PB, BM, and spleen. Because the mice in the anti-IL1RAP treatment group were killed due to myeloid sarcoma formation, rarely seen in AML patients, the survival data poorly reflect the strong antileukemic effects observed in the BM, spleen, and PB. Importantly, the results were validated by using a xenograft model of primary human AML cells, where treatment with an anti-IL1RAP antibody significantly reduced the leukemic burden.
Consistent with the mechanisms of action for antibodies used to target cell-surface molecules in B-cell malignancies and in solid tumors (29–32), ADCC mediated by NK cells was found to be an important mode of action for killing of human AML cells. In contrast, neither of the anti-IL1RAP antibodies mediated specific cell death by ADCP or CDC of MA9Ras cells in vitro, suggesting that these mechanisms may be less critical for the observed effects. NK cells are considered the most important effector cell type in humans (30, 33, 34). The feasibility of an antibody-based immunotherapy depending on NK cells in AML is supported by reports showing that NK cell numbers are intact in AML patients (35) and that the levels of CD16, which activates NK cells for ADCC (36), is equal in AML patients and healthy subjects (37). Collectively, these data show that the effect of anti-IL1RAP immunotherapy in the MA9Ras xenograft model is mediated by effector cells and suggests that effector-cell–mediated mechanisms is an important and accessible mode of action in a future clinical setting.
Although MA9Ras cells share many properties with primary human AML cells, they do not respond to IL-1 stimulation and hence cannot be used for determining the impact of IL1RAP targeting antibodies on IL-1–dependent AML cell growth. In addition, because human cells respond poorly to murine IL-1, the therapeutic relevance of blocking IL-1 signaling cannot accurately be assessed in a murine xeno-transplantation setting. These features represent a limitation of our study because the effect of blocking IL-1 signaling with mAb3F8 could not be explored in vivo, and thus the relative impact of IL-1 signaling blockade on the total treatment effect could not be determined. To explore the effects of IL-1 signaling inhibition by an IL1RAP-targeting antibody, we instead performed in vitro investigations on primary cells harvested from AML patients. In agreement with previous reports using anti–IL-1 antibodies or the IL-1 receptor antagonist IL1RA (15, 17–20, 22), blocking IL-1 signaling with the IL1RAP-targeting antibody mAb3F8 significantly suppressed the proliferation of primary AML cells responsive to IL-1. The importance of IL-1 signaling in AML cells has recently obtained further support by a study that used a combined cytokine and siRNA screen against cell-surface receptors and identified IL-1 signaling inhibition as having the most dramatic effect on regulating growth of AML cells (38). Hence, based on available data, we propose that IL-1 signaling inhibition as a second mode of action in an antibody-based immunotherapy against IL1RAP should be important in the future treatment of AML.
Nonspecific binding of a therapeutic antibody may lead to serious adverse effects. IL1RAP is expressed on candidate AML stem cells, but not on corresponding normal hematopoietic stem cells (9, 10). Among normal blood cells, IL1RAP is expressed mainly on monocytes, and AML cells are more sensitive than corresponding normal BM cells to ADCC induced by IL1RAP-targeting antibodies (8, 9). Here, absent or weak IL1RAP expression was shown in a majority of normal human tissues. Collectively, these data would suggest a low risk of adverse effects of an antibody-mediated immunotherapy against IL1RAP in AML; however, toxicity studies using animal species with cross-reactivity for the IL1RAP-targeting antibodies will be needed to provide a better prediction.
In summary, we demonstrate that IL1RAP provides a therapeutic target in AML and that the dual mode of action of IL1RAP-targeting antibodies, combining effector-cell–mediated mechanisms with IL-1–signaling suppression, is predicted to be beneficial. These findings strongly support a rapid clinical development of an antibody-based IL1RAP therapy in AML.
Materials and Methods
Culturing and Isolation of Cells.
MA9Ras cells were previously generated by retroviral transduction of primitive cord blood cells with an MLL/AF9 fusion gene and an activated NRAS gene (11). Cells were grown in Iscove's modified Dulbecco's medium supplemented with 10% (vol/vol) FBS and 1% penicillin/streptomycin/glutamine and with a 1/1,000 volume of mercapto-ethanol added freshly. Cell lines EOL1, MonoMac6, and OCI-AML1 were cultured according to the supplier’s instructions (DSMZ). For primary cells, patients diagnosed with AML donated BM or PB after informed consent. The protocol was approved by the Institutional Ethics Committee and was in accordance with the Declaration of Helsinki. Mononuclear cells (MNCs) were isolated by density gradient separation.
IL1RAP-Targeting Antibodies.
The antibodies mAb81.2 and mAb3F8 were generated by standard hybridoma technique as described (9).
In Vivo Antibody Treatment.
All animal experiments were approved by The Swedish Board of Agriculture, Malmö/Lund animal ethics committee in Lund, Sweden. Unconditioned NOD/SCID (NOD.CB17-Prkdcscid/J) mice were engrafted with 107 MA9Ras cells by tail vein injection. To allow for homing of the leukemic cells, treatment initiation was delayed to 3 d after transplantation. MAb81.2, mAb3F8, or a corresponding mIgG2a isotype control was administered biweekly by i.p. injections. In the initial MA9Ras studies, 500 μg per dose was given, corresponding to ∼20 mg per kg of body weight, with the first dose as a bolus of double amount antibody. Sublethally irradiated (200 rad) NSG (NOD.Cg-PrkdcscidI/2rgtm1Wjl/SzJ) mice were engrafted with 106 MA9Ras cells and treated with 50 μg per dose of mAb81.2 or mIgG2a, first dose of 100 μg. PB was drawn from vena saphena. Mice were killed by cervical dislocation at a fixed endpoint or, for survival experiments, upon signs of severe disease as judged by hunchback, untidy fur, and decreased mobility or due to extramedullary myeloid sarcomas consisting of MA9Ras cells.
For treatment studies with primary human AML cells, 2 × 106 primary MNCs or tertiary spleen cells from NSGS (NOD.Cg-PrkdCscidI/2rgtm1WjlTg(CMV-IL3,CSF2,KITLG)1Eav/MloySzj) mice serially engrafted with human AML MNCs were transplanted into unconditioned NOD/SCID mice or sublethally irradiated (200 rad) NSG or NSGS mice. Treatment was initiated 3 d after transplantation and administered by biweekly i.p. injections with mAb81.2 or mIgG2a isotype control antibody of 100 μg per dose, corresponding to 4 mg/kg, for all doses except for the first dose, which was 500 μg. Experiments were terminated at 28 d after transplantation for NOD/SCID mice and 35–57 d after transplantation for NSG/NSGS mice.
Sample Preparation and Flow Cytometry.
BM cells from mice were isolated by flushing of one femur or by crushing femurs, tibiae, and the pelvis. Spleens were mashed through a 70-μm mesh filter. PB was prepared for flow cytometry by ammonium chloride treatment to remove red cells. The following antibodies (all anti-human) were used in flow cytometry analysis: CD45-APC (BD Biosciences), CD33-BV421, CD33-PE/Cy7, CD19-PerCp/Cy5.5, CD3-PE/Cy7 (Biolegend), and IL1RAP-PE (R&D Systems). In the in vivo experiments with primary human AML cells, CD45+CD33+ cells were considered to be leukemic. For all flow-cytometric analysis, a FACS Canto (BD) was used.
Total Cell Expansion.
MA9Ras cells were seeded in technical duplicates at a density of 10,000 cells in a volume of 100 μL in normal culture medium with or without the addition of 10 μg/mL mAb81.2, mAb3F8, or a mIgG1 isotype control antibody and incubated for 48 h. For primary AML cells, 20,000 cells were plated in 100 μL of serum-free medium in technical triplicates and cultured for 6 d. The total cell numbers were determined with CountBright Absolute Counting Beads (Molecular Probes) and flow cytometry.
Apoptosis.
For analysis of apoptotic cells, 50,000–75,000 MA9Ras cells were seeded in technical duplicates and cultured for 48 h, followed by detection of viable, apoptotic, or dead cells by staining with Annexin V and 7-amino-actinomycin D (7AAD). Samples were analyzed by flow cytometry.
Proliferation.
Primary human AML MNCs were seeded in technical duplicates or triplicates at a density of 100,000 cells per well in a volume of 100 μL of serum-free medium with or without the addition of 0.5–10 ng/mL IL-1β and 10 μg/mL mAb3F8 or a corresponding mIgG1 isotype control. Proliferation was determined by measuring the incorporation of EdU after 24 h with Click-iT Plus EdU Flow Cytometry Assay (Molecular Probes) according to manufacturer’s instructions. Expression of the proliferation marker Ki-67 was determined after 48 h of culture, by using a Ki-67–APC antibody (Miltenyi Biotec) upon fixation and permeabilization of the cells with 1.6% paraformaldehyde and ice-cold 95% ethanol.
ADCC Assay.
Target cells were labeled with PKH26 (Sigma-Aldrich) according to the manufacturer’s instructions and seeded in technical triplicates. After 30 min of preincubation with 0.01–100 μg/mL mAb81.2, mAb3F8, or a corresponding hIgG1 isotype control, human NK cells were added at a 10:1 effector-to-target cell (E/T) ratio, and plates were incubated overnight. The following day, the percentage of dead target cells (PKH26+7AAD+) was measured by flow cytometry.
ADCP Assay.
Human monocytes were isolated from PB and matured into macrophages by addition of M-CSF to the culture medium. Proper maturation was confirmed by morphology and flow cytometry analysis after staining for CD206. Mature macrophages were seeded at a density of 40,000 cells per well and allowed to adhere overnight. Target MA9Ras cells were stained with PKH26, preincubated with 10 μg/mL mAb81.2, mAb3F8, or a corresponding hIgG1 isotype control antibody for 30 min, and added at a 1:1 E/T ratio to the wells in technical duplicates. After incubation for 2 h at 37 °C, cells were detached, and the amount of PHK26-positive macrophages was determined by using flow cytometry. At this E/T ratio, there was a variation of 20–50% MA9Ras+ macrophages without antibody addition between the individual experiments. Thus, at presentation, data were normalized to 1.0 relative amount of MA9Ras+ macrophages without antibody addition.
CDC Assay.
MA9Ras cells were seeded in technical duplicates at a density of 100,000 cells per well in serum-free StemSpan medium (Stem Cell Technologies). Cells were preincubated with 10 μg/mL mAb81.2, mAb3F8, or a corresponding hIgG1 isotype control for 30 min. Native or heat-inactivated human serum was added to a final concentration of 10% or 50%. After incubation for 2 h at 37 °C, cells were stained with ToPro3-iodide and analyzed by flow cytometry.
IL-1 Signaling Reporter Cell Assay.
HEK-Blue IL-1β cells (Invivogen) were harvested and plated in technical duplicates at a density of 50,000 cells per well in a 96-well plate. MAb81.2, mAb3F8, or a corresponding mIgG1 control antibody was added to the wells in a concentration range of 0.01–10 μg/mL. After preincubation for 30 min, IL-1β was added to a final concentration of 0.5 ng/mL, and the plate was incubated overnight. To ensure assay functionality, one set of samples was pretreated with 10 ng/mL IL1RA (R&D Systems). To examine whether any of the antibodies were able to induce IL-1R activation in the absence of IL-1β, samples were incubated with 10 μg/mL antibody without addition of the ligand. The following day, substrate was added to the supernatants, and samples were analyzed for absorbance at 620 nm. For measuring the response to murine IL-1, the cells were stimulated with 0.1 pg to 10 ng of human or murine IL-1 in the absence of antibody.
Immunostaining of Normal Tissue Array.
Immunohistochemistry on normal tissue arrays was performed at MicroMorph AB (Lund). Briefly, a tissue array containing 30 different normal human tissues from three individuals was immunostained with mAb3F8 or a mIgG1 isotype control antibody. High IL1RAP-expressing SKMEL-5 cells were used as positive control and KG1 cells as negative. For detection, an HRP-conjugated anti-mouse antibody (Dako) was used. The staining intensity was determined as follows: negative (0), weak staining (1+), moderate staining (2+), or strong staining (3+).
Statistical Analyses.
GraphPad Prism software (GraphPad Software) was used for statistical analyses. Statistical differences were determined to be significant at P < 0.05 using two-tailed Student’s t test (MA9Ras xenograft data and all in vitro data), two-tailed Mann–Whitney log-rank test (xenograft data using primary AML cells), or Mantel Cox log rank test (survival). In vivo data are presented as mean ± SD or median ± interquartile range, with the value for each individual mouse represented as a symbol. In vitro data are presented as mean ± SEM.
Acknowledgments
We thank Drs. Anki Malmborg Hager and Karin von Wachenfeld for fruitful discussions and Cantargia AB for contributing antibodies used in the study. This work was supported by grants from the Swedish Cancer Society, the Swedish Children’s Cancer Foundation, the Medical Faculty of Lund University, the Swedish Research Council and BioCARE strategic research grants and a project grant (to T.F.), a contract research grant from Cantargia AB (to M.J. and T.F.), and NIH Center of Excellence in Molecular Hematology P30 Award DK090971 and Grant CA168369 (to J.C.M.). J.C.M. is a Leukemia and Lymphoma Society Scholar.
Footnotes
Conflict of interest statement: K.S., M.J., and T.F. are cofounders of Cantargia AB (Medicon Village, Lund), formed together with Lund University Bioscience AB. Cantargia AB is the owner of the intellectual property rights for agents targeting IL1RAP for use in the treatment and diagnosis of neoplastic hematologic disorders. J.R. has stock options in Cantargia.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422749112/-/DCSupplemental.
References
- 1.Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 3.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5(7):738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa F, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 5.Hoelzer D. Targeted therapy with monoclonal antibodies in acute lymphoblastic leukemia. Curr Opin Oncol. 2013;25(6):701–706. doi: 10.1097/CCO.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SE, Chester JD. Personalised cancer medicine. Int J Cancer. 2015;137(2):262–266. doi: 10.1002/ijc.28940. [DOI] [PubMed] [Google Scholar]
- 7.Subramaniam S, Stansberg C, Cunningham C. The interleukin 1 receptor family. Dev Comp Immunol. 2004;28(5):415–428. doi: 10.1016/j.dci.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Järås M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci USA. 2010;107(37):16280–16285. doi: 10.1073/pnas.1004408107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Askmyr M, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood. 2013;121(18):3709–3713. doi: 10.1182/blood-2012-09-458935. [DOI] [PubMed] [Google Scholar]
- 10.Barreyro L, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120(6):1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wunderlich M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24(10):1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohanian M, et al. Is acute myeloid leukemia a liquid tumor? Int J Cancer. 2013;133(3):534–543. doi: 10.1002/ijc.28012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theocharides APA, et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209(10):1883–1899. doi: 10.1084/jem.20120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wunderlich M, et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121(12):e90–e97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakai K, et al. Autocrine stimulation of interleukin 1 beta in acute myelogenous leukemia cells. J Exp Med. 1987;166(5):1597–1602. doi: 10.1084/jem.166.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cozzolino F, et al. Interleukin-1 and interleukin-2 control granulocyte- and granulocyte-macrophage colony-stimulating factor gene expression and cell proliferation in cultured acute myeloblastic leukemia. Int J Cancer. 1990;46(5):902–907. doi: 10.1002/ijc.2910460525. [DOI] [PubMed] [Google Scholar]
- 17.Cozzolino F, et al. Interleukin 1 as an autocrine growth factor for acute myeloid leukemia cells. Proc Natl Acad Sci USA. 1989;86(7):2369–2373. doi: 10.1073/pnas.86.7.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delwel R, et al. Interleukin-1 stimulates proliferation of acute myeloblastic leukemia cells by induction of granulocyte-macrophage colony-stimulating factor release. Blood. 1989;74(2):586–593. [PubMed] [Google Scholar]
- 19.Bradbury D, Bowen G, Kozlowski R, Reilly I, Russell N. Endogenous interleukin-1 can regulate the autonomous growth of the blast cells of acute myeloblastic leukemia by inducing autocrine secretion of GM-CSF. Leukemia. 1990;4(1):44–47. [PubMed] [Google Scholar]
- 20.Rambaldi A, et al. Modulation of cell proliferation and cytokine production in acute myeloblastic leukemia by interleukin-1 receptor antagonist and lack of its expression by leukemic cells. Blood. 1991;78(12):3248–3253. [PubMed] [Google Scholar]
- 21.Rambaldi A, Torcia M, Dinarello CA, Barbui T, Cozzolino F. Modulation of cell proliferation and cytokine production in AML by recombinant interleukin-1 receptor antagonist. Leukemia. 1993;7(Suppl 2):S10–S12. [PubMed] [Google Scholar]
- 22.Estrov Z, et al. Inhibition of acute myelogenous leukemia blast proliferation by interleukin-1 (IL-1) receptor antagonist and soluble IL-1 receptors. Blood. 1992;79(8):1938–1945. [PubMed] [Google Scholar]
- 23.Stosić-Grujicić S, Basara N, Milenković P, Dinarello CA. Modulation of acute myeloblastic leukemia (AML) cell proliferation and blast colony formation by antisense oligomer for IL-1 beta converting enzyme (ICE) and IL-1 receptor antagonist (IL-1ra) J Chemother. 1995;7(1):67–70. doi: 10.1179/joc.1995.7.1.67. [DOI] [PubMed] [Google Scholar]
- 24.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 25.Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Kikushige Y, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–717. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 28.He SZ, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma. 2015;56(5):1406–1415. doi: 10.3109/10428194.2014.956316. [DOI] [PubMed] [Google Scholar]
- 29.Le Jeune C, Thomas X. Antibody-based therapies in B-cell lineage acute lymphoblastic leukaemia. Eur J Haematol. 2015;94(2):99–108. doi: 10.1111/ejh.12408. [DOI] [PubMed] [Google Scholar]
- 30.Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Musolino A, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 32.Bibeau F, et al. Impact of FcgammaRIIa-FcgammaRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–1129. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 33.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 34.Cartron G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 35.Vidriales MB, et al. Lymphoid subsets in acute myeloid leukemias: Increased number of cells with NK phenotype and normal T-cell distribution. Ann Hematol. 1993;67(5):217–222. doi: 10.1007/BF01715050. [DOI] [PubMed] [Google Scholar]
- 36.Bryceson YT, March ME, Ljunggren H-G, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costello RT, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood. 2002;99(10):3661–3667. doi: 10.1182/blood.v99.10.3661. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal A, et al. Targeted suppression of interleukin-1 signaling inhibits growth of primary human acute myeloid leukemia cells. Cancer Res. 2014;74(19 Suppl):abstr 5248. [Google Scholar]