

Significance
Lipid homeostasis is a fundamental process for understanding antimicrobial susceptibility. Modification of the polar head group of phosphatidylglycerol into the respective aminoacyl-ester of phosphatidylglycerol is a widely used strategy to mediate bacterial resistance. Here we present the structures of the catalytic domains of aminoacyl-phosphatidylglycerol synthases from Pseudomonas aeruginosa and Bacillus licheniformis. These prototypical enzymes specifically catalyze the tRNA-dependent synthesis of alanyl-phosphatidylglycerol and lysyl-phosphatidylglycerol, respectively. A central tunnel architecture facilitates binding of the polar aminoacyl-tRNA molecule opposite the hydrophobic lipid substrate as a fundamental principle for the catalysis at the water–lipid interface. Specific inhibition of aminoacyl-phosphatidylglycerol synthases might be a promising strategy to render Gram-positive and Gram-negative pathogenic bacteria more susceptible to antimicrobial treatment.
Keywords: A-PGS, L-PGS, MprF, structure, tRNA
Abstract
The cytoplasmic membrane is probably the most important physical barrier between microbes and the surrounding habitat. Aminoacylation of the polar head group of the phospholipid phosphatidylglycerol (PG) catalyzed by Ala-tRNAAla–dependent alanyl-phosphatidylglycerol synthase (A-PGS) or by Lys-tRNALys–dependent lysyl-phosphatidylglycerol synthase (L-PGS) enables bacteria to cope with cationic peptides that are harmful to the integrity of the cell membrane. Accordingly, these synthases also have been designated as multiple peptide resistance factors (MprF). They consist of a separable C-terminal catalytic domain and an N-terminal transmembrane flippase domain. Here we present the X-ray crystallographic structure of the catalytic domain of A-PGS from the opportunistic human pathogen Pseudomonas aeruginosa. In parallel, the structure of the related lysyl-phosphatidylglycerol–specific L-PGS domain from Bacillus licheniformis in complex with the substrate analog L-lysine amide is presented. Both proteins reveal a continuous tunnel that allows the hydrophobic lipid substrate PG and the polar aminoacyl-tRNA substrate to access the catalytic site from opposite directions. Substrate recognition of A-PGS versus L-PGS was investigated using misacylated tRNA variants. The structural work presented here in combination with biochemical experiments using artificial tRNA or artificial lipid substrates reveals the tRNA acceptor stem, the aminoacyl moiety, and the polar head group of PG as the main determinants for substrate recognition. A mutagenesis approach yielded the complementary amino acid determinants of tRNA interaction. These results have broad implications for the design of L-PGS and A-PGS inhibitors that could render microbial pathogens more susceptible to antimicrobial compounds.
Bacteria can adapt rapidly to changing environmental conditions by controlling the physical properties of biological membranes (1). One important strategy is the tRNA-dependent aminoacylation of the polar head group of phosphatidylglycerol (PG) by aminoacyl-phosphatidylglycerol synthases (aa-PGSs). The resulting products alanyl-phosphatidylglycerol (A-PG) or lysyl-phosphatidylglycerol (L-PG) (Fig. 1) reduce the overall net negative charge of the membrane, making it less susceptible to cationic antimicrobial peptides (CAMPs). Such CAMPs often are synthesized as innate immunity host-defense peptides in response to bacterial infections (2–5). Fundamental work with the Gram-positive pathogen Staphylococcus aureus indicated aa-PGS–mediated nonsusceptibility to vancomycin (an antibiotic of last resort), resistance to host antimicrobial peptides (defensins), and protection against neutrophil killing (6–9). Accordingly, aa-PGS enzymes can be considered virulence factors and thus have been termed “multiple peptide resistance factors.” In Pseudomonas aeruginosa, aminoacylation of PG also was found in response to acidic environmental conditions (2, 5), inter alia. P. aeruginosa is the dominant pathogen that colonizes the lung of patients suffering from cystic fibrosis. Because of the underlying defect in bicarbonate ion transport, acidification of the airway surface liquid contributes to cystic fibrosis pathogenesis (10). Furthermore, during inflammatory response local acidification by the production of acids was observed (11).
Fig. 1.

tRNA-dependent aminoacylation of phosphatidylglycerol with alanine or lysine. A-PGS and L-PGS (also named “MprF”) catalyzed the formation of A-PG (A) and L-PG (B), respectively.
Aa-PGS catalysis is one of the rare instances in which a ribosomal aminoacyl-tRNA is used other than in protein biosynthesis. Aa-PGS enzymes are classified as (i) A-PGS, found, for example, in Gram-negative P. aeruginosa; (ii) L-PGS, found in Gram-positive S. aureus, Bacillus anthracis, and Bacillus licheniformis, inter alia; and (iii) aa-PGS enzymes with a broadened substrate specificity (synthesis mainly of L-PG together with A-PG) as described for Bacillus subtilis and Enterococcus faecium (2–5, 12). Recent results demonstrate that precise tuning of cellular A-PG and/or L-PG concentrations is fundamental for bacterial resistance (2). Furthermore, regulatory circuits including specific aminoacyl-phosphatidylglycerol (aa-PG) hydrolases have been described for P. aeruginosa and E. faecium (13, 14).
The aa-PGS enzymes consist of a separable, water-soluble C-terminal domain showing full enzymatic activity (4, 15). This domain contains all elements for the specific recognition of the ∼26 kDa water-soluble aminoacyl-tRNA (Ala-tRNAAla or Lys-tRNALys) and for binding of the hydrophobic PG substrate (15). The N-terminal transmembrane domain anchors aa-PGS proteins in the cytoplasmic membrane and harbors an additional translocase activity responsible for flipping the newly synthesized lipid into the outer leaflet of the cytoplasmic membrane (16).
This study presents the first, to our knowledge, structures of the catalytic A-PGS domain from P. aeruginosa together with the related L-PGS structure from B. licheniformis in the presence of a small substrate analog. Structural biology in combination with biochemical experiments using a series of artificial aminoacyl-tRNA or artificial lipid substrates allows the molecular understanding of aa-PGS substrate recognition as a basis for the future exploitation of this class of previously unidentified antimicrobial targets.
Results and Discussion
The determination of the crystal structures of the catalytic A-PGS and L-PGS domains from P. aeruginosa and B. licheniformis is summarized in Table S1 (effective resolution, 2.4 Å for each). The structural superposition of both structures with an rmsd of 1.1 Å is depicted in Fig. 2A (34.1% amino acid sequence identity; compare with alignment in Fig. S1). Despite this high degree of structural conservation, a marked specificity for the synthesis of A-PG or L-PG was observed for A-PGS and L-PGS in enzymatic assays using Ala-tRNAAla or Lys-tRNALys as substrates, respectively (Fig. S2).
Table S1.
Data collection and refinement statistics
| Dataset | A-PGS SeMet derivative | A-PGS native | L-PGS |
| Temperature, K | 100 | 100 | 100 |
| Wavelength, Å | 0.9797 | 0.9184 | 1.0332 |
| Δϕ, ° | 0.7 | 0.5 | 0.2 |
| Space group | P4122 | P4122 | P1 |
| Unit cell parameters | a = 94.0 Å | a = 94.0 Å | a = 46.9 Å |
| b = 94.0 Å | b = 94.0 Å | b = 66.3 Å | |
| c = 166.5 Å | c = 166.5 Å | c = 71.2 Å | |
| α = γ = 90° | α = γ = 90° | α = 112.4° | |
| β = 90° | β = 90° | β = 94.1° | |
| γ = 90° | γ = 90° | γ = 98.9° | |
| Resolution range dmax-dmin, Å | 50.0–3.1 (3.2–3.1) | 20.0–2.3 (2.4–2.3) | 40–2.1 (2.2–2.1) |
| No. of observed reflections | 315,054 (23,659) | 262,982 (26,038) | 128,799 (2,501) |
| Completeness | 0.999 (0.999) | 0.995 (0.991) | 0.806 (0.191) |
| Multiplicity | 12.3 (12.2) | 7.8 (7.9) | 3.5 (3.5) |
| Rmerge* | 0.307 (1.557) | 0.159 (1.343) | 0.095 (0.596) |
| Average I/σ,I | 12.9 (2.1) | 12.3 (2.0) | 8.7 (2.0) |
| CC(1/2)† | 0.994 (0.853) | 0.997 (0.729) | 0.994 (0.720) |
| Effective dmin‡, Å | 3.1 | 2.4 | 2.4 |
| Wilson B, Å2 | 56.1 | 37.4 | 27.5 |
| Solvent content | 0.74 | 0.74 | 0.52 |
| Refinement | |||
| Rwork§ | 0.182 | 0.175 | 0.197 |
| Rfree¶ | 0.213 | 0.199 | 0.248 |
| No. of nonhydrogen atoms per asymmetric unit | |||
| Total | 2,598 | 2,728 | 5,468 |
| Protein | 2,585 | 2,557 | 5,234 |
| No. of molecules per asymmetric unit | |||
| Protein | 1 | 1 | 2 |
| Water | — | 111 | 192 |
| Ligands | 5 | 16 | 3 |
| Atomic displacement factors B, Å2 | |||
| Overall | 50.6 | 48.7 | 36.0 |
| Protein | 50.4 | 48.4 | 36.0 |
| Water | — | 46.2 | 34.4 |
| Ligands | 99.8 | 69.4 | 36.5 |
| Rmsd from ideal | |||
| Bond lengths, Å | 0.002 | 0.008 | 0.002 |
| Bond angles, ° | 0.64 | 1.11 | 0.54 |
| Ramachandran plot | |||
| Favored, % | 96.3 | 95.6 | 95.7 |
| Allowed, % | 2.5 | 3.8 | 3.0 |
Values in parentheses are for the highest-resolution shell.
Rmerge = Σhkl,i(Σi|Ihkl,i-〈Ihkl〉|/Σhkl,i〈Ihkl〉.
Fraction of correlation between intensities from random half-datasets (48).
Resolution beyond which data completeness falls below the arbitrary threshold of 0.9 or the I/σ(I) average falls below 2.
Rwork = (Σ||Fo|-|Fc||/Σ|Fo|); |Fo|, structure factor magnitudes observed; |Fc|, structure factors calculated.
Rfree is computed as Rwork but using 5% randomly assigned reflections excluded from refinement.
Fig. 2.

X-ray structures of the catalytic domain of A-PGS from P. aeruginosa and L-PGS from B. licheniformis. (A) Structural superposition of A-PGS (light blue, blue, purple, and gray) and L-PGS (light gray) in a wall-eye stereo view. (B) Topology diagram of the tandem repeated GNAT fold of A-PGS and L-PGS. GNAT domains 1 and 2 (light blue and blue) share strands E and K (purple). The detailed sequence assignment for A-PGS and L-PGS is depicted in Fig. S1. (C, Center) A longitudinal section of the A-PGS structure from P. aeruginosa. (Left and Right) The resulting halves reveal a tunnel which contains the upper tRNA-binding pocket and the lower PG-binding tunnel. The key catalytic residues of A-PGS (yellow sticks) and residues located at the constriction of the tunnel (light gray sticks) and the C terminus (semitransparent gray surface) are located above the respective sectional plane. The protein is represented as a van der Waals surface and is colored according to its surface electrostatics [−20 (red) to +20 (blue) KbT/ec]. (D, Center) Longitudinal section of L-PGS in complex with the competitive inhibitor L-lysine amide. (Left and Right) The resulting halves reveal a tunnel that contains the L-lysine amide (LYN, green ball-and-stick representation)-binding site in the upper tRNA-binding pocket (Left) and the lower PG-binding cavity. Important amino acid residues are highlighted and colored as in C. The enlarged view in the left panel shows the polar interactions of key catalytic residues (yellow sticks) with the L-lysine amide, which is an analog of the aminoacyl moiety of the substrate Lys-tRNALys. The position of the structurally related A-PGS amino acid residues is superimposed (light gray sticks). (E) Superposition of the FemX/tRNA structure (20) with the L-PGS/L-lysine amide complex reveals a plausible binding mode of the aminoacylated tRNA acceptor arm. A structural overlay of L-PGS/L-lysine amide with the FemX/tRNA complex localizes the terminal tRNA nucleotides (CCA) in a defined cavity of L-PGS. The 3′ hydroxyl oxygen atom of the terminal ribose is placed only 1.0 Å away from the cocrystallized L-lysine amide (LYN, offset indicated by asterisk). (F) Model for the interaction of A-PGS and tRNA. A representative tRNA molecule (PDB ID code 1TN1) was positioned onto the structure of A-PGS. The aminoacyl-binding pocket and the CCA-binding pocket were used as reference points. Mutational analysis for amino acid residues Lys676, Arg684, Arg687, and Asn683 located on helix 5 resulted in impaired A-PGS activity. The terminal base pairings G2-C71, G3-U70, and G4-C69 have been identified previously as tRNA-recognition elements (15). The theoretical position of the U70 base is indicated by an asterisk. (G) L-lysine amide (LYN) bound within the aminoacyl-binding pocket of L-PGS (yellow bonds). Hydrogen bonds as shown as dashed lines with distances indicated in Ångstroms. Generated with LIGPLOT (43). (H) Wall-eye stereo view of the lipid-binding tunnel of A-PGS. Enlarged view of the longitudinal section of Fig. 2C, Right indicating conserved amino acid residues (gray and blue). The functional role of amino acid residues Gln636, Glu658, and Ser763 (gray) was demonstrated by mutagenesis (see also Table S2). Docking calculations [using Autodock Vina (41)] revealed the theoretical binding mode of the PG lipid substrate (green sticks, representing PG C5:0/C8:0).
Fig. S1.

Structure-based sequence alignment of the catalytic domain of A-PGS from P. aeruginosa, L-PGS from B. licheniformis, and FemX from Weissela viridescens (20, 21). Alignments were calculated using University of California, San Francisco Chimera (38, 39). Clustal Omega (44) was used for primary sequence analysis. Conserved and partly conserved residues are indicated by an asterisk (*) and colon (:) or period (.), respectively, and are named Cons1 for conservation between A-PGS sequences, Cons2 for L-PGS sequences, and Cons3 for the overall alignment of A-PGS, L-PGS, and FemX. Helices (boxes) and β-strands (arrows) are marked. GNAT domain 1 is colored in light blue, GNAT domain 2 in blue, secondary structure elements of A-PGS and L-PGS that are part of both GNAT domains are highlighted in purple, and inserted elements are colored in gray. Underlined fonts indicate L-PGS residues that are involved in L-lysine amide interaction. Green fonts indicate conserved residues of the lipid tunnel. Red fonts mark residues of FemX (and A-PGS and L-PGS), which are involved (might be involved) in the interaction with the CCA end of tRNA. Orange fonts highlight residues proposed for tRNA acceptor stem interaction. Blue fonts indicate FemX residues that are involved in pentapeptide interaction (20).
Fig. S2.

Incorporation of radioactively labeled 14C-lysine and 14C-alanine into the organic lipid phase by the enzymatic activities of the catalytic domain of A-PGS and L-PGS. The crude cellular extract of E. coli cells overproducing A-PGS543–881 and L-PGS519–850, respectively, were supplemented with radioactively labeled 14C-lysine (A) or 14C-alanine (B) and an ATP regenerating system as described (5). Samples were subjected to lipid extraction, and synthesis of radioactively labeled lipids was analyzed by liquid scintillation counting.
Both aa-PGS structures share a tandem repeated GNAT (GCN5-related N-acetyltransferase) fold (17). The topology diagram in Fig. 2B highlights GNAT domains 1 and 2 (colored light blue and blue, respectively) partially arranged with internal “pseudo-twofold symmetry” (rhombus), which share strands E and K (purple), respectively. Internal superposition of the central elements of GNAT domains 1 and 2 resulted in rmsds of 0.99 Å for A-PGS and 0.96 Å for L-PGS. Domain 2 shares the specific sequential arrangement of the GNAT fold (17, 18) with an insertion of two α-helices (8a and 9), whereas domain 1 lacks the most N-terminal β-strand of the superfamily.
The strongest structural homology (19) of both aa-PGS structures was found to the alanyl transferase FemX [Protein Data Bank (PDB) code 4II9], which is another Ala-tRNAAla–dependent enzyme involved in peptidoglycan interpeptide bridge formation. FemX catalyzes the transfer of L-Ala to the side chain of the ε-amino group of L-Lys of the peptidoglycan precursor UDP-MurNAc-pentapeptide (20). The core of the FemX protein also is composed of two GNAT domains that are related by pseudo-twofold symmetry (rmsd for internal superposition, 2.0 Å) (21). Structural domains 1 and 2 are separated by an extended cleft of 20 Å that is 15 Å deep to accommodate both the UDP-MurNAc-pentapeptide on GNAT domain 1 and the unpaired CCA acceptor arm of the charged tRNA on GNAT domain 2 (20). The superposition of GNAT domains 2 from A-PGS and FemX revealed an rmsd of 1.39 Å (GNAT domains 2 from L-PGS and FemX, 1.36 Å).
Notably, FemX was cocrystallized with a stable aminoacyl-tRNA analog. The peptidyl-RNA conjugate used mimics the peptide substrate in parallel with the instable Ala-tRNAAla cosubstrate, resulting in a structure that resolves the three terminal CCA nucleotides of the unpaired tRNA acceptor arm (20).
Structural superposition of the L-PGS (or A-PGS) structure with the FemX/peptidyl-RNA complex clearly localizes the terminal tRNA nucleotides in a defined cavity located on GNAT domain 2 of L-PGS (see Fig. 2E) (or A-PGS). This cavity is delineated mainly by secondary structure elements comprising a series of fully conserved amino acid residues [sequence excerpt; fully conserved residues are in bold, and residues in van der Waals distance are underscored: helix 6 and following loop region (SDAWL713 A-PGS/SDEWL688 L-PGS), strand I and following loop region (DLMRVHPDAPKLTM778 A-PGS/DLMRYSKKAPKGIM742 L-PGS), and helix 10 (LRRFK840 A-PGS/FSGLRSFK814 L-PGS)].
Notably, Phe839 and Lys840 of A-PGS (or Phe813 and Lys814 of L-PGS) are located in a spatial position identical to that of the catalytically relevant residues Phe304 and Lys305 of FemX. Therefore, the proposed CCA-binding mode was substantiated further by mutagenesis of related A-PGS residues. Mutational analyses revealed complete A-PGS inactivation with the mutagenesis of Lys840 and Phe839, as is consistent with related results for FemX mutagenesis (20). Therefore, we concluded that Lys840 and Phe839 have a central role in tRNA substrate interaction (compare Fig. S1 and Table S2).
Table S2.
Comparison of conserved amino acid residues of A-PGS, L-PGS, and FemX indicating related enzymatic activities of mutant proteins and the inhibition of L-PGS in the presence of L-lysine amide
| A-PGS residue | Related L-PGS residue (FemX residue) | Mutation | In vivo activity | Relative in vitro activity ± SD, % |
| Wild type | +++ | 100 | ||
| Coordination of aminoacyl moiety of the tRNA substrate | ||||
| S709 | S684 | S709A* | +++ | 5 ± 1 |
| S709N* | +++ | 17 ± 3 | ||
| E720 | E693 | E720Q* | +++ | 10 ± 3 |
| Y732 | Y705 | Y732A* | + | 3 ± 1 |
| D765 | D739 | D765A* | — | 0 |
| D765N* | — | 0 | ||
| R768 | R742 (Y199)† | R768S* | — | 0 |
| R768Q* | — | 0 | ||
| Coordination of 3′ CCA end of tRNA | ||||
| W712 | W687 | |||
| V769 | Y733 (S259)† | |||
| A773 | A737 (T264)† | |||
| K775 | K739 | |||
| M778 | M752 | M778A* | +++ | 50 ± 2 |
| L836 | S808 (L301)† | |||
| F839 | F813 (F304; F304A: 6%; F304L: 3%)† | F839A | — | 0 |
| F839L | + | 6 ± 1 | ||
| K840 | K814 (K305; K305A: <0.01%; K305M: 0.5%; K305R: 0.1%)† | K840S* | — | 0 |
| K840Q* | — | 0 | ||
| Coordination of the acceptor stem | ||||
| K676 | A649 (K169)† | K676S | ++ | 29 ± 8 |
| K676E | — | 0 | ||
| N683 | N656 | N683D | ++ | 11 ± 1 |
| N683S | +++ | 71 ± 2 | ||
| R684 | R657 (R176)† | R684S | ++ | 21 ± 1 |
| R684E | — | 0 | ||
| R687 | R660 (R179)† | R687S | ++ | 13 ± 1 |
| Conserved residues of the PG tunnel | ||||
| D606 | D581 | |||
| G609 | G584 | |||
| Q636 | Q611 | Q636R | + | 13 ± 3 |
| Q636W | + | 10 ± 3 | ||
| E640 | E615 | |||
| E658 | E633 | E658R | + | 6 ± 3 |
| E658W | +++ | 23 ± 4 | ||
| S763 | S737 | S763N* | +++ | 11 ± 1 |
| G800 | G774 | |||
| M801 | M775 | |||
| R850 | K824 | |||
| L-PGS | ||||
| Wild type | 100 | |||
| Wild type, 0.1 mM L-lysine amide | 80 | |||
| Wild type, 1 mM L-lysine amide | 75 | |||
| Wild type, 10 mM L-lysine amide | 20 | |||
Standard in vivo and in vitro A-PGS assays have been described in ref. 15. The specific activity observed in the in vitro activity assay for wild-type A-PGS was set as 100%, and all other values of mutant proteins were related to this value. A-PG formation in the in vivo assay was classified as: +++, activity comparable with wild-type enzyme; ++, slightly decreased activity compared with wild-type enzyme; +, decreased activity compared with wild-type enzyme; —, no detectable A-PGS activity. For L-PGS inhibition studies 10 µM of purified enzyme were supplemented with L-lysine amide, and L-PG synthesis was analyzed analogously (15).
Data from Hebecker et al., 2011 (15).
Data from Fonvielle et al., 2013 (20).
In addition, residues Asp765 and Arg768 were identified as key catalytic A-PGS residues (yellow sticks in Fig. 2C). Remarkably, the L-PGS/FemX structural superposition depicted in Fig. 2E places the 3′ hydroxyl group of the terminal ribose moiety only 1.0 Å away from the cocrystallized L-lysine amide molecule (offset to the amide nitrogen of LYN, indicated by an asterisk in Fig. 2E). This compound (electron density depicted in Fig. S3) is a weak competitive inhibitor of L-PG synthesis (see Table S2). These structural and mutational analyses suggest a conserved binding mode for the unpaired CCA of the acceptor arm in aa-PGS and FemX proteins (compare Fig. 2E). The lower part of the L-lysine amide inhibitor-binding pocket is lined with a series of highly conserved amino acid residues, which are involved in an identical 3D network of polar interactions in the A-PGS and L-PGS structure (A-PGS/L-PGS: Ser709/Ser684, Glu720/Glu693, Tyr732/Tyr705, Asp765/Asp739, and Arg768/Arg742) (compare enlarged views in Fig. 2 D and G). Functional relevance for all these polar residues has been confirmed by mutagenesis as summarized in Table S2 (15). Combined interaction of Tyr732/Tyr705 and Asp765/Asp739 with the substrate α-amino group suggests an important role in the recognition of the substrate aminoacyl linkage. With regard to the catalyzed transesterification, residues Glu720/Glu693 and Ser709/Ser684 play a fundamental role in positioning Arg768/Arg742, which interacts directly with the α-carbonyl group of the cocrystallized inhibitor (compare Fig. 2 D and G). Structural and biochemical data suggest a nucleophilic attack of the 3′ hydroxyl group of PG on the Arg768/Arg742-activated α-carbonyl carbon of aminoacyl-tRNA.
Fig. S3.
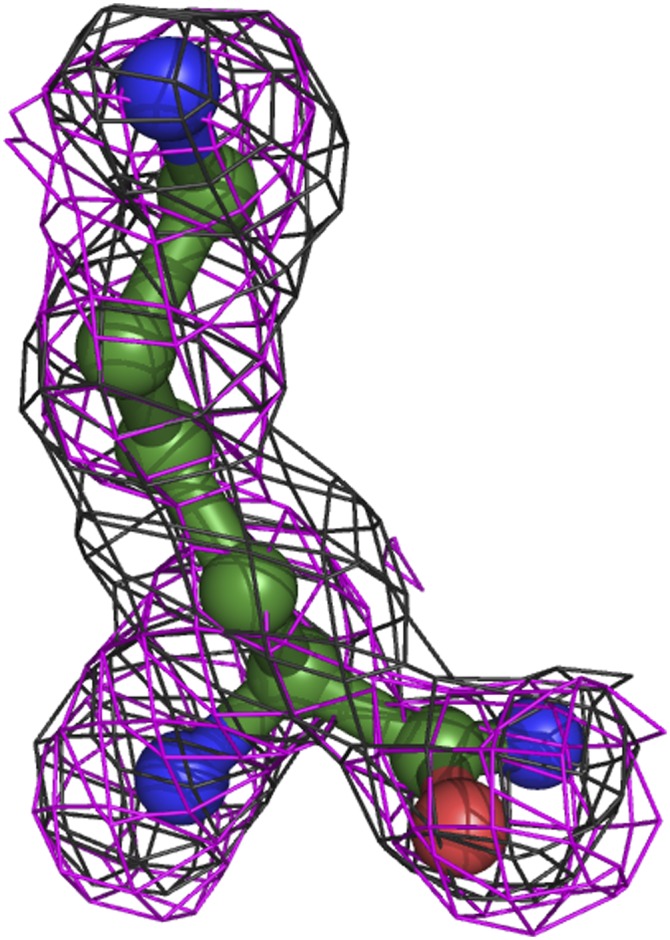
Electron density map of L-lysine amide in the L-PGS structure. A feature-enhanced electron density map (45) at 1.5 σ is shown as black mesh. An Fo-Fc omit density map at 2.1 σ is shown in magenta.
Specific synthesis of A-PG or L-PG requires accurate recognition of Ala-tRNAAla versus Lys-tRNALys. Inspection of the upper part of the aminoacyl-binding cavity of the L-PGS and A-PGS structure did not reveal any conserved or supplemental amino acid residues as direct determinants for the specific recognition (or steric exclusion) of the lysyl- versus the much smaller alanyl-substrate moiety. The four-aminobutyl side chain of the cocrystallized L-lysine amide is solely in contact with main-chain L-PGS atoms, which were found in an almost identical position in the related A-PGS structure (compare Fig. 2 D and G). One might argue that the sole amino acid moiety of the tRNA substrate does not account for the overall specificity of aa-PGS enzymes. Accordingly, we investigated whether the tRNA portion was a main aa-PGS determinant by using tRNALys that was mischarged with the smaller amino acid alanine.
The highly specific A-PGS or L-PGS enzyme was analyzed in the presence of tRNALys that was mischarged with the amino acid alanine (Ala-tRNALysC70U; Fig. 3). The specific requirement for the C70U mutation within the synthesis of Ala-tRNALysC70U is illustrated in the sequence comparison of the tRNA species we used, depicted in Fig. S4. This artificial Ala-tRNALysC70U substrate was not accepted by L-PGS (compare Fig. 3 and Fig. S5). In contrast, the A-PGS enzyme revealed a relative activity of 80% compared with the natural Ala-tRNAAla substrate (Fig. 3). These experiments might indicate that the tRNA moiety in combination with the amino acid moiety of Ala-tRNAAla is relevant for A-PGS substrate recognition. These findings are supported further by earlier experiments (using artificial tRNA microhelices), which revealed the five terminal base pairings as important elements of A-PGS substrate recognition (15).
Fig. 3.

A-PG synthesis of A-PGS and L-PGS using mischarged 14C-Ala-tRNALysC70U. (A) 14C-Ala-tRNAAla or 14C-Ala-tRNALysC70U was used as substrate of A-PGS (0.2 µM) and L-PGS (1 µM) in combination with PG. Control reactions with purified L-PGS in the presence of the Lys-tRNALys substrate revealed efficient L-PG formation (compare with Fig. S5). Synthesis of aa-PG was analyzed by lipid extraction and liquid scintillation analysis. (B) Sequence of Ala-tRNAAla from B. licheniformis. (C) Sequence of Ala-tRNALysC70U from P. aeruginosa. The mutated base is indicated by an asterisk.
Fig. S4.

Comparison of the tRNA acceptor stem of Ala-tRNAAla, Lys-tRNALys, and misacylated Ala-tRNALysC70U, identity elements of aminoacyl-tRNA synthetases and substrate recognition of A-PGS and L-PGS. The acceptor stems of Ala-tRNAAla from B. licheniformis and Lys-tRNALys from P. aeruginosa and from misacylated Ala-tRNALysC70U (point mutation indicted by asterisk) are shown. Enzymatic misacylation of tRNAAla or tRNALys requires the respective tRNA identity elements (highlighted in gray) of alanyl-tRNA synthetase [AlaRS; A73 discriminator base, G3-U70 (37, 46)] or lysyl-tRNA synthetase [LysRS; A73 discriminator base, G3-C70, UUU34-36 anticodon (36, 47)], respectively. Sequence differences for tRNAAla and tRNALys are highlighted by blue boxes. Substrate recognition of aa-PGS was analyzed using the artificial substrate Ala-tRNALysC70U. Synthesis of this misacylated tRNA by AlaRS requires a single base mutation (36). Ala-tRNALysC70U was not accepted by L-PGS but was efficiently converted by A-PGS. Notice that tRNALysC70U is not accepted as a substrate of LysRS because of the C70U mutation (36).
Fig. S5.

L-PG synthesis of L-PGS and A-PGS using 14C-Lys-tRNALys as substrate. Isolated 14C-Lys-tRNALys was used as substrate in the presence of purified A-PGS (1 µM) or L-PGS (1 µM) in the presence of PG. L-PG synthesis was analyzed by lipid extraction and liquid scintillation counting.
The theoretical comparison of the tRNAAla and the tRNALys acceptor stem sequences shown in Fig. S4 reveals only base pairings G4–C69/U4–A69, C6–G67/G6–C67, and U70/C70 for the discrimination of tRNAAla/tRNALys. However, the observed A-PGS activity in the presence of tRNALysC70U indicates that G4, C69, C6, and G67 are nondiscriminating bases of A-PGS substrate recognition. Therefore, we concluded that U70 has a relevant role in A-PGS substrate recognition. In addition, specific substrate discrimination also might include dynamic interaction among aa-PGS and the overall tRNA substrate as exemplified for other transient protein/tRNA complexes (22, 23). Future cocrystallization experiments in the presence of sophisticated nonhydrolyzable aminoacyl-tRNA analogs might reveal such conformational rearrangements with relevance for the specific recognition of Ala-tRNAAla or Lys-tRNALys.
Inspection of the protein surface of A-PGS and L-PGS revealed a series of conserved amino acid residues located on helix 5 (Lys676, Arg684, Arg687, and Asn683) as candidates for additional tRNA interaction. Mutagenesis of these residues into small polar residues always resulted in moderately retained activities (K676S, 29%; R684S, 21%; R687S, 13%; N683S, 71%; and N683D, 11%). In contrast, the individual charge reversal caused by glutamate insertion resulted in a complete loss of A-PGS activity, indicating a potential tRNA interaction. Subsequently, a representative tRNA structure from the Protein Data Bank (PDB ID code 1TN1) was positioned manually onto the structure of A-PGS and L-PGS using the aminoacyl-binding pocket and the CCA-binding pocket as points of reference. This experiment revealed the mutagenized amino acid residues of helix 5 (Lys676, Arg684, Arg687) in van der Waals distance from the previously determined recognition elements of the tRNAAla acceptor stem (base pairings G2–C71, G3–U70, and G4–C69). Fig. 2F shows an enlarged view of this A-PGS/tRNA acceptor stem interaction, and the overall models of tRNA/A-PGS and tRNA/L-PGS interaction are depicted in Fig. S6.
Fig. S6.

Model for L-PGS and A-PGS tRNA interaction. A tRNA molecule (PDB ID code 1TN1) was manually positioned onto the structure of L-PGS (A) and A-PGS (B) using the aminoacyl-binding pocket and the CCA-binding pocket as reference points. Substitution of amino acid residues Lys676, Arg684, and Arg687 located on helix 5 results in impaired A-PGS activity. The terminal base pairings G2–C71, G3–U70, and G4–C69 have been identified previously as tRNA recognition elements (compare Table S2 and ref. 15). The theoretical position of the C70 or U70 base in the model of L-PGS or A-PGS is indicated by an asterisk.
Aa-PGS catalysis requires the recruitment of the hydrophobic lipid substrate, whereas FemX binds the polar UDP-MurNAc-pentapeptide in an extended cleft located on GNAT domain 1 (20). Accordingly, we propose a completely unrelated aa-PGS substrate-binding mode with respect to PG. A comparison of the overall structure of aa-PGS and FemX reveals that the core secondary structural elements of GNAT domain 1 are tilted by up to 6 Å toward the domain interface, thereby closing the UDP-MurNAc-pentapeptide–binding cavity of FemX by virtue of helix 1 (PDGGLALT577) and loop region ARRGRSMI602 and also by an insertion (loop region EKGFSLGR727) and the extension (LIAGGLTGL878) located at the C terminus of A-PGS.
In Fig. 2 C and D an intersection of both aa-PGS proteins reveals a 23-Å tunnel with a diameter of 4–8 Å, which is connected directly to the aminoacyl-tRNA– binding cavity of A-PGS and L-PGS, respectively. This theoretical “backdoor entrance” is delineated by conserved residues of GNAT domains 1 and 2 (see Fig. S1) and provides an elegant solution for the aa-PGS catalysis at the water–lipid interface: The water-insoluble lipid substrate was placed into the lower part of the A-PGS tunnel by running a molecular docking simulation (Autodock Vina software in combination with PG C5:0/C8:0; PDB ligand ID code AGA). The relative positioning of the PG molecule used in the postulated lipid-binding site of A-PGS is depicted in Fig. 2H. The 3′ hydroxyl of the polar lipid head group protrudes toward the aminoacyl-binding site, whereas the branched diacylglycerol moiety appears to have a high degree of flexibility concerning the recognition of the respective fatty acid moieties of PG [C16:0/C19:0 cis 9, 10 cyclopropane predominant in P. aeruginosa (5, 24)]. This theoretical docking mode is in agreement with earlier experiments investigating A-PGS activity in the presence of artificial PG substrates (15). Modification of the respective fatty acid alkyl chains by (i) variation of saturation, (ii) fourfold methylation, (iii) using a monoacylated PG derivative, or (iv) using a short-chain C6 fatty acid PG variant did not hamper the activity of A-PGS. However, the alteration of the polar head group into an ethylene glycol moiety or by using diphosphatidylglycerol with a symmetrically branched glycerol moiety did not result in any detectable A-PGS activity, indicating that the polar head group of PG is the dominant determinant of lipid substrate recognition (15).
The proposed PG substrate-binding mode was analyzed experimentally by site-directed mutagenesis of solvent-exposed amino acid residues located at the bottleneck that forms the connection to the aminoacyl-tRNA–binding site of A-PGS (see Fig. 2C and the close-up view depicted in Fig. 2H). Replacing residues Gln636, Glu658, and Ser763 (highlighted in light gray in Fig. 2H) with significantly bulkier side chains (tryptophane, arginine, or asparagine) resulted in only moderately retained activities of 6% for E658R, 13% for Q636R, 10% for Q636W, 23% for E658W, and 11% for S763N (Table S2). These biochemical and structural findings suggest the binding of the polar aminoacyl-tRNA molecule opposite the hydrophobic lipid substrate as a fundamental principle for the aa-PGS catalysis at the water–lipid interface. The elucidated modes of substrate recognition provide a framework for the future development of aa-PGS inhibitors as a new strategy to render pathogenic bacteria more susceptible to established antibiotics and also to the wide range of naturally occurring antimicrobial defense molecules of the human host.
Materials and Methods
Production and Purification of P. aeruginosa A-PGS and B. licheniformis L-PGS.
Base pairs 1627–2643 of ORF PA0920 from P. aeruginosa PAO1 and base pairs 1555–2550 of ORF yfiX from B. licheniformis DSM13 were PCR-amplified using oligonucleotide pairs 1 and 2 and 3 and 4 (Table S3), respectively, and were cloned into the XmaI and SacI sites of pET52b(+) (Novagen) for expression with a cleavable N-terminal Strep-II-tag. The SerP (surface entropy reduction) server (25) was used to identify mutations that may facilitate optimized crystallization of A-PGS (KGKE674 to AGAA674). To exchange amino acid residues of the catalytic domain of A-PGS, the QuikChange kit (Agilent) was used according to the manufacturer’s instructions in combination with oligonucleotide pairs 5 and 6 to 31 and 32.
Table S3.
Oligonucleotides used in this study
| Primer | 5′ – 3′ sequence |
| 1 | TTAACCCGGGCGCGCGGCACC |
| 2 | GAGTGAGCTCTCAGCGTTTCACCAATC |
| 3 | GCGCCCCGGGAACAGGAAAACGAAAGAGATC |
| 4 | GCGCGAGCTCTCATTTCGTTCTCCTTCC |
| 5 | CCGGTGTTCTACTGGGTGCGTGCCGAGAAC |
| 6 | GTTCTCGGCACGCACCCAGTAGAACACCGG |
| 7 | CCGGTGTTCTACCGTGTGCGTGCCGAGAAC |
| 8 | GTTCTCGGCACGCACACGGTAGAACACCGG |
| 9 | CAAGCTCGGCGAACGTGCGCGAGTC |
| 10 | GACTCGCGCACGTTCGCCGAGCTTG |
| 11 | CCTCAAGCTCGGCGAATGGGCGCGAGTCGAC |
| 12 | GTCGACTCGCGCCCATTCGCCGAGCTTGAGG |
| 13 | GGGCAAGGAAATGGAAGACCTGCGCTACAC |
| 14 | GTGTAGCGCAGGTCTTCCATTTCCTTGCCC |
| 15 | GAACAAGGGCAAGGAAATGTCTGACCTGCGCTACACCTG |
| 16 | CAGGTGTAGCGCAGGTCAGACATTTCCTTGCCCTTGTTC |
| 17 | GCTACACCTGGGACCGCGGCCAGC |
| 18 | GCTGGCCGCGGTCCCAGGTGTAGC |
| 19 | CTGCGCTACACCTGGTCTCGCGGCCAGCGCG |
| 20 | CGCGCTGGCCGCGAGACCAGGTGTAGCGCAG |
| 21 | GCGCTACACCTGGAACGAAGGCCAGCGCGACG |
| 22 | CGTCGCGCTGGCCTTCGTTCCAGGTGTAGCGC |
| 23 | GCGCTACACCTGGAACTCTGGCCAGCGCGACG |
| 24 | CGTCGCGCTGGCCAGAGTTCCAGGTGTAGCGC |
| 25 | GGAACCGCGGCCAGTCTGACGGCCTGGCC |
| 26 | GGCCAGGCCGTCAGACTGGCCGCGGTTCC |
| 27 | CAGGGGCTGCGACGCGCTAAGGACAAGTTCCAG |
| 28 | CTGGAACTTGTCCTTAGCGCGTCGCAGCCCCTG |
| 29 | CAGGGGCTGCGACGCCTGAAGGACAAGTTCCAG |
| 30 | CTGGAACTTGTCCTTCAGGCGTCGCAGCCCCTG |
| 31 | GCTGCGCTTCGACCTGGAGAACGCTGGCGCTGCTATGAAAGACCTGCGCTACACCTG |
| 32 | CCAGGTGTAGCGCAGGTCTTTCATAGCAGCGCCAGCGTTCTCCAGGTCGAAGCGCAC |
| 33 | AATTCTAATACGACTCACTATAGGGGCCTTAGCTCAGCTGGGAGAGCGCCTGCTTTGCACGCAGGAGGTCAGCGGTTCGATCCCGCTAGGCTCCACCAG |
| 34 | GATCCTGGTGGAGCCTAGCGGGATCGAACCGCTGACCTCCTGCGTGCAAAGCAGGCGCTCTCCCAGCTGAGCTAAGGCCCCTATAGTGAGTCGTATTAG |
| 35 | AATTCTAATACGACTCACTATAGGGTCGTTAGCTCAGTCGGTAGAGCAGTTGGCTTTTAACCAATTGGTCGTAGGTTCGAATCCTACACGATCCACCAG |
| 36 | GATCCTGGTGGATCGTGTAGGATTCGAACCTACGACCAATTGGTTAAAAGCCAACTGCTCTACCGACTGAGCTAACGACCCTATAGTGAGTCGTATTAG |
| 37 | AATTCTAATACGACTCACTATAGGGTCGTTAGCTCAGTCGGTAGAGCAGTTGGCTTTTAACCAATTGGTCGTAGGTTCGAATCCTACACGACCCACCAG |
| 38 | GATCCTGGTGGGTCGTGTAGGATTCGAACCTACGACCAATTGGTTAAAAGCCAACTGCTCTACCGACTGAGCTAACGACCCTATAGTGAGTCGTATTAG |
Restriction sites are shown in italics. Bold fonts indicate exchanged nucleotides for mutagenesis of the catalytic domain of A-PGS from P. aeruginosa.
A-PGS and L-PGS genes were expressed, and recombinant proteins were purified to apparent homogeneity as follows: Transformed Escherichia coli BL 21(DE3) cells for the production of A-PGS543–881 and Tuner (DE3) cells for production of L-PGS519–850 were cultivated at 37 °C in LB medium supplemented with 100 µg/mL ampicillin. At an OD578 of 0.5, protein production was induced with 50 µM of isopropyl β-d-1-thiogalactopyranoside, and cells were shifted to 17 °C for 18 h. Selenomethionine-labeled surface mutant A-PGS543–881 AGAA with 90% SeMet occupancy was produced as described elsewhere (26).
Cells were harvested by centrifugation and disrupted by a French press at 19,200 psi in lysis buffer [100 mM Tris⋅HCl (pH 7.5), 400 mM NaCl, 20 mM MgCl2, 5% (wt/vol) glycerol, 2 mM DTT].
After ultracentrifugation for 1 h at 110,000 × g at 4 °C, the supernatant was applied to 1 mL of Strep-Tactin Superflow resin (IBA), which was equilibrated with lysis buffer. Following washing with 10 mL of lysis buffer and 10 mL of elution buffer 1 [20 mM Tris⋅HCl (pH 7.5), 200 mM NaCl, 0.5 mM MgCl2, 5% (wt/vol) glycerol, 2 mM DTT] for A-PGS543–881 AGAA and A-PGS543–881, or alternatively elution buffer 2 [20 mM Tris⋅HCl (pH 7.5), 300 mM NaCl, 5 mM MgCl2, 5% (wt/vol) glycerol, 2 mM DTT] for L-PGS519–850, the proteins were liberated and eluted from the resin by cleavage of the Strep-II-tag via PreScission protease treatment (GE Healthcare). The GST-tagged protease was removed using Glutathione Sepharose 4FF (GE Healthcare). The elution fractions containing A-PGS and L-PGS proteins, respectively, were concentrated to ∼10 mg/mL using Vivaspin 15 centrifugal concentrators with a 10-kDa cutoff (Sartorius).
Protein Crystallization.
Crystals were obtained in hanging-drop vapor-diffusion experiments at 4 °C by mixing 2 µL of protein with 2 µL of reservoir solution. Crystals of selenomethionine-labeled A-PGS543–881 AGAA grew from a solution containing 7.5 mM CoCl2, 85 mM Mes (pH 5.7), 1.53 M (NH4)2SO4, and 15% (vol/vol) glycerol. Crystals of native A-PGS543–881 AGAA grew from 85 mM Na-acetate (pH 6.37), 1.6 M (NH4)2SO4, and 20% (vol/vol) glycerol. Crystals of L-PGS519–850 were obtained from a solution of 0.2 M NaCl, 0.1 M phosphate-citrate (pH 4.2), and 10% (wt/vol) PEG3000 supplemented with 0.5 mM L-lysine amide. Needle-shaped crystals grew within 1–2 wk. Crystals were shock-cooled in liquid nitrogen. L-PGS519–850 crystals were cryoprotected with 30% (vol/vol) glycerol in reservoir solution before cooling in liquid nitrogen.
Data Collection, Structure Determination, and Refinement.
Diffraction data of A-PGS and L-PGS crystals were collected on beamline 14.2 (27) of the Berlin Electron Storage Ring Society for Synchrotron Radiation (BESSY) II electron storage ring (Berlin-Adlershof) and on beamline P11 (28) of the Positron-Electron Tandem Ring Accelerator (Petra) III at DESY (Hamburg, Germany). Integration and space group assignment were carried out with XDS (29). A crystal of selenomethionine-derivatized A-PGS was used for data collection at the absorption edge of selenium. The resulting anomalous signal was sufficient to obtain experimental phases in a single-wavelength anomalous dispersion experiment and to compute initial electron density with phenix.autosol (30). A first structural model was built by phenix.autobuild (30), which subsequently was improved by manual rebuilding in COOT (31) and refinement in phenix.refine (30). An improved A-PGS model was obtained through refinement against high-resolution data of a native A-PGS crystal. Diffraction data of L-PGS crystals showed significant anisotropy and were subjected to anisotropy correction using the anisotropy correction server (32). The phase problem was solved by molecular replacement with phaser (33) using a pruned A-PGS monomer as search model. Phaser placed two monomers in the asymmetric unit with reasonable confidence. However, the resulting electron density was poor. Hence, model and density were improved by alternate rebuilding and relaxation cycles with phenix.mr_rosetta (34) and then finalized through manual rebuilding and refinement. The complete data collection and refinement statistics are shown in Table S1.
A-PGS/L-PGS Enzyme Assay.
The in vivo A-PGS and L-PGS activities were determined as described before (15). To validate the purified (mutant) proteins, our well-established in vitro activity assay was performed (15). In brief, an E. coli strain overproducing either alanyl-tRNA synthetase (15) or lysyl-tRNA synthetase (35) provided the substrate molecules PG and aminoacylated tRNAAla or tRNALys. The formation of A-PG or L-PG, respectively, was determined by liquid scintillation counting using radioactively labeled [1-14C]-L-alanine (51 mCi/mmol; Moravek Biochemicals) or [U-14C]-L-lysine (288 mCi/mmol; Moravek Biochemicals) (15).
Preparation, Purification, and Aminoacylation of RNA Transcripts.
The tRNAAla gene from B. licheniformis, the tRNALys gene from P. aeruginosa, and the sequence for tRNALysC70U (carrying identity elements for misacylation with alanine by alanyl-tRNA synthetase; G3-C70 base pair mutated to G3-U70) were cloned into the pUC18 vector (using oligonucleotides 33–38). In vitro-transcribed tRNAs were purified via MonoQ chromatography, folded, and acylated with [1-14C]-L-alanine (for tRNAAla) or [U-14C]-L-lysine (for tRNALys), as described elsewhere (15, 36).
Misacylation of tRNALys Using Alanyl-tRNA Synthetase.
Substrate recognition of alanyl-tRNA synthetase is one of the rare instances in which only a single base pair of the acceptor stem acts as a major identity element of tRNA recognition (37). Therefore, native tRNALys from P. aeruginosa functions as a substrate of alanyl-tRNA synthetase because of the sole mutation of the base pairing G3-C70 into G3-U70 (36). Hence, in vitro synthesis of the misacylated Ala-tRNALysC70U offers an alternative methodology to investigate aa-PGS substrate recognition. In vitro-transcribed tRNALysC70U was misacylated efficiently in the presence of 0.42 µM of 14C-Ala and 1 µM E. coli alanyl-tRNA synthetase (15). Synthesis of a related Lys-tRNALysC70U is strongly hampered because a lysyl-tRNA synthetase revealed a loss of activity by a factor of >1,000 as the result of a G3-U70 mutation (36).
A-PGS/L-PGS Activity Assays in the Presence of Ala-tRNAAla and Misacylated Ala-tRNALysC70U.
We used 0.42 µM of 14C-Ala-tRNAAla or 14C-Ala-tRNALysC70U as substrate for A-PGS (0.2 µM) and L-PGS (0.1, 1 µM, and 10 µM) in the presence of 2 mg/mL PG (Sigma-Aldrich) supplemented with 1.76 mg/mL Triton X-100 in the respective elution buffers. Synthesis of A-PG was analyzed by lipid extraction and liquid scintillation analysis. Control experiments using 14C-Lys-tRNALys were performed to demonstrate L-PGS activity. This experimental setup was used to elucidate the overall contribution of the tRNA substrate moiety.
Structure-Based Sequence Analysis.
Structure-based sequence analyses were calculated by the MatchMaker and MatchAlign subroutines of UCSF Chimera (38, 39). Figures were prepared with Pymol (40) and UCSF Chimera. Rmsds were calculated in Pymol.
Molecular Docking of PG.
Docking calculations were performed by means of Autodock Vina (41) as part of the MGLTools package (mgltools.scripps.edu/) to investigate the binding mode of the lipid substrate. The PG ligand (PG C5:0/C8:0, PDB ligand ID AGA, from structure 1q16; ref. 42) was extracted from PDB, charges and rotational bonds were assigned, and no flexibility was allowed for side-chain residues for the A-PGS structure. The search volume was assigned first to the whole A-PGS and then stepwise, limited to the lower part of the substrate tunnel to exclude docking to the outer shell of the molecule. All conformers showing diametrically opposed fatty acid moieties were considered biologically irrelevant. The theoretical lipid-binding mode depicted in Fig. 2H shows the best result, having the lowest binding energy of the remaining list.
Acknowledgments
We thank the staffs of beamline P11 of Petra III and beamline 14.2 of BESSY II for their support. This work was supported by Deutsche Forschungsgemeinschaft Grants MO 1749/1-2 and HE 1852/14-2. Parts of this research were carried out at the light source Petra III at the German Electron Synchrotron, Hamburg, and at BESSY II, Berlin, both members of the Helmholtz Association.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (accession nos. 4V34, 4V35, and 4V36).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511167112/-/DCSupplemental.
References
- 1.Zhang YM, Rock CO. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol. 2008;6(3):222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 2.Arendt W, Hebecker S, Jäger S, Nimtz M, Moser J. Resistance phenotypes mediated by aminoacyl-phosphatidylglycerol synthases. J Bacteriol. 2012;194(6):1401–1416. doi: 10.1128/JB.06576-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peschel A, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193(9):1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy H, Ibba M. Broad range amino acid specificity of RNA-dependent lipid remodeling by multiple peptide resistance factors. J Biol Chem. 2009;284(43):29677–29683. doi: 10.1074/jbc.M109.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein S, et al. Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol Microbiol. 2009;71(3):551–565. doi: 10.1111/j.1365-2958.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 6.Pillai SK, et al. Daptomycin nonsusceptibility in Staphylococcus aureus with reduced vancomycin susceptibility is independent of alterations in MprF. Antimicrob Agents Chemother. 2007;51(6):2223–2225. doi: 10.1128/AAC.00202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillai SK, et al. Development of reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus. Clin Infect Dis. 2009;49(8):1169–1174. doi: 10.1086/605636. [DOI] [PubMed] [Google Scholar]
- 8.Peschel A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002;10(4):179–186. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 9.Kristian SA, Dürr M, Van Strijp JA, Neumeister B, Peschel A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect Immun. 2003;71(1):546–549. doi: 10.1128/IAI.71.1.546-549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coakley RD, et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA. 2003;100(26):16083–16088. doi: 10.1073/pnas.2634339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmen HP, Battaglia H, Giovanoli P, Blaser J. Analysis of pH, pO2 and pCO2 in drainage fluid allows for rapid detection of infectious complications during the follow-up period after abdominal surgery. Infection. 1994;22(6):386–389. doi: 10.1007/BF01715494. [DOI] [PubMed] [Google Scholar]
- 12.Samant S, Hsu FF, Neyfakh AA, Lee H. The Bacillus anthracis protein MprF is required for synthesis of lysylphosphatidylglycerols and for resistance to cationic antimicrobial peptides. J Bacteriol. 2009;191(4):1311–1319. doi: 10.1128/JB.01345-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arendt W, Groenewold MK, Hebecker S, Dickschat JS, Moser J. Identification and characterization of a periplasmic aminoacyl-phosphatidylglycerol hydrolase responsible for Pseudomonas aeruginosa lipid homeostasis. J Biol Chem. 2013;288(34):24717–24730. doi: 10.1074/jbc.M113.482935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith AM, Harrison JS, Sprague KM, Roy H. A conserved hydrolase responsible for the cleavage of aminoacylphosphatidylglycerol in the membrane of Enterococcus faecium. J Biol Chem. 2013;288(31):22768–22776. doi: 10.1074/jbc.M113.484402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebecker S, et al. Alanyl-phosphatidylglycerol synthase: Mechanism of substrate recognition during tRNA-dependent lipid modification in Pseudomonas aeruginosa. Mol Microbiol. 2011;80(4):935–950. doi: 10.1111/j.1365-2958.2011.07621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst CM, et al. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009;5(11):e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: A structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vetting MW, et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433(1):212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Holm L, Rosenstrom P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fonvielle M, et al. The structure of FemX(Wv) in complex with a peptidyl-RNA conjugate: Mechanism of aminoacyl transfer from Ala-tRNA(Ala) to peptidoglycan precursors. Angew Chem Int Ed Engl. 2013;52(28):7278–7281. doi: 10.1002/anie.201301411. [DOI] [PubMed] [Google Scholar]
- 21.Biarrotte-Sorin S, et al. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: Insights into FemABX family substrates recognition. Structure. 2004;12(2):257–267. doi: 10.1016/j.str.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Francklyn C, Perona JJ, Puetz J, Hou YM. Aminoacyl-tRNA synthetases: Versatile players in the changing theater of translation. RNA. 2002;8(11):1363–1372. doi: 10.1017/s1355838202021180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francklyn CS. DNA polymerases and aminoacyl-tRNA synthetases: Shared mechanisms for ensuring the fidelity of gene expression. Biochemistry. 2008;47(45):11695–11703. doi: 10.1021/bi801500z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hancock IC, Meadow PM. The extractable lipids of Pseudomonas aeruginosa. Biochim Biophys Acta. 1969;187(3):366–379. doi: 10.1016/0005-2760(69)90010-1. [DOI] [PubMed] [Google Scholar]
- 25.Goldschmidt L, Cooper DR, Derewenda ZS, Eisenberg D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007;16(8):1569–1576. doi: 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Nimtz M, Rinas U. Optimized procedure to generate heavy isotope and selenomethionine-labeled proteins for structure determination using Escherichia coli-based expression systems. Appl Microbiol Biotechnol. 2011;92(4):823–833. doi: 10.1007/s00253-011-3603-x. [DOI] [PubMed] [Google Scholar]
- 27.Mueller U, et al. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J Synchrotron Radiat. 2012;19(Pt 3):442–449. doi: 10.1107/S0909049512006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meents A, et al. Development of an in-vacuum x-ray microscope with cryogenic sample cooling for beamline P11 at PETRA III. Proc SPIE. 2013;8851:88510K (abstr). [Google Scholar]
- 29.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strong M, et al. Toward the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103(21):8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terwilliger TC, et al. phenix.mr_rosetta: Molecular replacement and model rebuilding with Phenix and Rosetta. J Struct Funct Genomics. 2012;13(2):81–90. doi: 10.1007/s10969-012-9129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibba M, Bono JL, Rosa PA, Söll D. Archaeal-type lysyl-tRNA synthetase in the Lyme disease spirochete Borrelia burgdorferi. Proc Natl Acad Sci USA. 1997;94(26):14383–14388. doi: 10.1073/pnas.94.26.14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambrogelly A, Frugier M, Ibba M, Söll D, Giegé R. Transfer RNA recognition by class I lysyl-tRNA synthetase from the Lyme disease pathogen Borrelia burgdorferi. FEBS Lett. 2005;579(12):2629–2634. doi: 10.1016/j.febslet.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Hou YM, Schimmel P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988;333(6169):140–145. doi: 10.1038/333140a0. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Yang Z, et al. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J Struct Biol. 2012;179(3):269–278. doi: 10.1016/j.jsb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrödinger L. (2010) The PyMOL molecular graphics system (Schrödinger, LLC, New York), Version 1.5.0.4.
- 41.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertero MG, et al. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Biol. 2003;10(9):681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 43.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 44.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Afonine PV, Headd JJ, Terwilliger TC, Adams PD. New tool: Phenix.real_space_refine. Computational Crystallography Newsletter. 2013;4:51–58. [Google Scholar]
- 46.Francklyn C, Schimmel P. Aminoacylation of RNA minihelices with alanine. Nature. 1989;337(6206):478–481. doi: 10.1038/337478a0. [DOI] [PubMed] [Google Scholar]
- 47.Tamura K, Himeno H, Asahara H, Hasegawa T, Shimizu M. In vitro study of E. coli tRNA(Arg) and tRNA(Lys) identity elements. Nucleic Acids Res. 1992;20(9):2335–2339. doi: 10.1093/nar/20.9.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]