

Abstract
Experience suggests that African Americans may express autoimmune disease differently than other racial groups. In the context of systemic sclerosis (scleroderma), we sought to determine whether race was related to a more adverse expression of disease. Between January 1, 1990, and December 31, 2009, a total of 409 African American and 1808 white patients with scleroderma were evaluated at a single university medical center. While the distribution by sex was virtually identical in both groups, at 82% female, African American patients presented to the center at a younger mean age than white patients (47 vs. 53 yr; p < 0.001). Two-thirds of white patients manifested the limited cutaneous subset of disease, whereas the majority of African American patients manifested the diffuse cutaneous subset (p < 0.001). The proportion seropositive for anticentromere antibody was nearly 3-fold greater among white patients, at 34%, compared to African American patients (12%; p < 0.001). Nearly a third of African American (31%) patients had autoantibodies to topoisomerase, compared to 19% of white patients (p = 0.001). Notably, African American patients experienced an increase in prevalence of cardiac (adjusted odds ratio [OR], 1.6; 95% confidence interval [CI], 1.3–2.2), renal (OR, 1.6; 95% CI, 1.2–2.1), digital ischemia (OR, 1.5; 95% CI, 1.4–2.2), muscle (OR, 1.7; 95% CI, 1.3–2.3), and restrictive lung (OR, 6.9; 95% CI, 5.1–9.4) disease. Overall, 700 (32%) patients died (159 African American; 541 white). The cumulative incidence of mortality at 10 years was 43% among African American patients compared to 35% among white patients (log-rank p = 0.0011). Compared to white patients, African American patients experienced an 80% increase in risk of mortality (relative risk [RR], 1.8; 95% CI, 1.4–2.2), after adjustment for age at disease onset and disease duration. Further adjustment by sex, disease subtype, and scleroderma-specific autoantibody status, and for the socioeconomic measures of educational attainment and health insurance status, diminished these risk estimates (RR, 1.3; 95% CI, 1.0–1.6). The heightened risk of mortality persisted in strata defined by age at disease onset, diffuse cutaneous disease, anticentromere seropositivity, decade of care at the center, and among women. These findings support the notion that race is related to a distinct phenotypic profile in scleroderma, and a more unfavorable prognosis among African Americans, warranting heightened diagnostic evaluation and vigilant care of these patients. Further, we provide a chronologic review of the literature regarding race, organ system involvement, and mortality in scleroderma; we furnish synopses of relevant reports, and summarize findings.
INTRODUCTION
Experience suggests that African Americans may express autoimmune disease differently than other racial groups. For example, among patients with autoimmune hepatitis, histologic evidence of cirrhosis was more prevalent among African American patients than white patients, occurring in the majority of the former and the minority of the latter.19 African American patients presented with more advanced liver disease than white patients, among patients with known primary biliary cirrhosis being screened in a multicenter clinical trial.33 Regarding myasthenia gravis, a prototypic autoimmune neurologic disease, serologic status has been found to differ according to race; acetylcholine receptor antibodies were more prevalent in white than African American patients, whereas among the seronegative generalized myasthenia stratum, the prevalence of seropositivity to muscle-specific tyrosine kinase was higher among African Americans.30 Further, the annual incidence of myasthenia gravis in a large United States cohort was highest among black women compared to white women and men.1 In contrast, the incidence of autoimmune thyroiditis and juvenile-onset, insulin-dependent diabetes mellitus is diminished among African Americans.14,20,31,43 Thyroid autoantibodies are less commonly observed among African American than white children with type I diabetes mellitus.5 Yet, in Allegheny County, Pennsylvania, the risk of mortality was greater among African Americans than whites with insulin-dependent diabetes mellitus.14
Similarly, in the context of the autoimmune rheumatologic disorders, experience intimates a differential expression of disease according to race. Most notably, young African American women are at greatest risk to develop severe disease manifestations of systemic lupus erythematosus, particularly a greater risk of progression to end-stage renal disease and dialysis dependence, than other sex-race groups.7 The prevalence of lupus is particularly high, at 1 in 250 persons, among African American women aged 15–64 years.8 Further, African Americans who previously smoked or continued to smoke cigarettes were found to be at heightened risk to develop both autoantibody-positive and autoantibody-negative rheumatoid arthritis.27
Systemic sclerosis (scleroderma) is no exception to this impression. Previous investigations have reported a younger age at disease onset, greater incidence and prevalence of disease, a predilection to more severe disease manifestations, a distinct serologic profile, and a worse prognosis among African American patients compared to white patients.3,9,13,22–24,28,29,37 However, the number of African American patients examined in prior reports has often been relatively small, precluding reliable estimates. It remains important to ascertain definitively whether race is independently related to disease severity and mortality in scleroderma, above and beyond the known contribution of the aforementioned key demographic, clinical, socioeconomic, and serologic predictors to survival. Moreover, the independent relation of race to survival within well-defined strata defined by cutaneous disease subset and by serologic status has not been clearly defined.
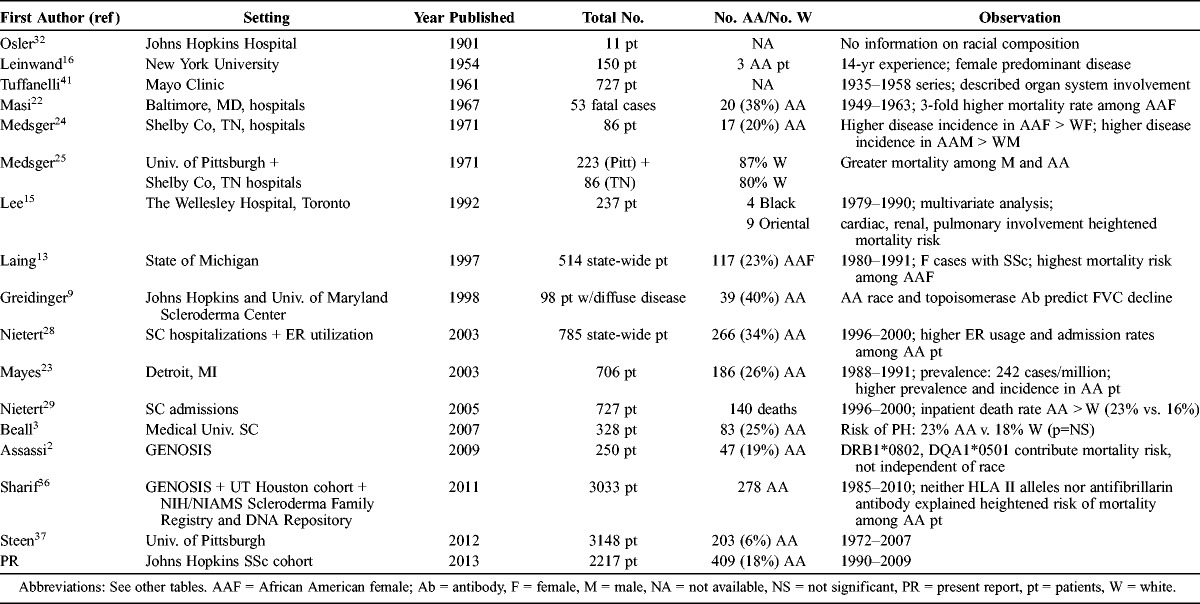
In this context, the goals of the present analyses were 2-fold. First, we sought to examine the independent association of race with the development of key organ system manifestations of scleroderma and with survival. The relatively unique aspect of this effort is the opportunity to examine an exceptionally large cohort assembled at a single university medical center over 20 years, in which 18% of the cohort was African American. Second, because relevant demographic, clinical, and serologic measures of disease have been frequently ascertained, we sought to furnish an informative chronologic review of the published literature that addresses the association of African American race with disease susceptibility, phenotypic features, and prognosis in scleroderma. Finally, we sought to consider the findings culled from the current study of 2217 consecutive African American and white patients, evaluated from 1990 to 2009 at the Johns Hopkins Scleroderma Center, within the context of the presented literature review.
METHODS
Over the 20 years from January 1, 1990, to December 31, 2009, consecutive patients evaluated at the Johns Hopkins Scleroderma Center were enrolled in a research database. Demographic, serologic, and clinical (for example, spirometry and echocardiography) data were ascertained. These parameters were collected at the initial evaluation, and then prospectively re-assessed at 6-month intervals among those cohort participants receiving follow-up care at the Scleroderma Center. The protocol was reviewed and approved by the Johns Hopkins Office of Human Subjects Research.
Similarly, each patient was queried regarding the presence of digital cold sensitivity and observed color changes—pallor, cyanosis, and hyperemia—upon cold exposure. Age at onset of Raynaud phenomenon, at onset of first non-Raynaud symptom of scleroderma, and at diagnosis of scleroderma were uniformly recorded, as were sex and reported race. Date of disease onset was defined as the date of the first non-Raynaud symptom of scleroderma. Serologic status for antinuclear antibody (ANA), anticentromere antibody, antitopoisomerase antibody, and antiribonucleoprotein (anti-RNP) antibody was ascertained on initial presentation to the center and thereafter updated if and when new serologic information became available; a smaller subset was examined for serologic status to RNA polymerase III.
Using a standard protocol at the initial evaluation, each patient was characterized based on examination findings as having the limited or the diffuse cutaneous subset of disease.17 Cohort eligibility required each patient to satisfy 1 of the following 3 criteria for a diagnosis of scleroderma: A) the American College of Rheumatology (ACR) criteria for scleroderma,35 B) 3 or more of the 5 features of the CREST (calcinosis, Raynaud, esophageal dysmotility, sclerodactyly, telangiectasia) syndrome,17,42 or C) the combination of definite Raynaud phenomenon, abnormal nail fold capillaries, and a scleroderma-specific (centromere, topoisomerase, or RNA polymerase III) antibody.18
A total of 2481 patients with scleroderma were enrolled in the database during this 20-year period, among which 2394 had a recorded date of disease onset and comprehensive assessments. The cohort participants were categorized into mutually exclusive, self-reported racial groups. The demographic distribution of these patients included 1821 white, 411 African American, 51 Asian, 17 Middle Eastern/Arabian, 16 from the Indian subcontinent, and 4 native Hawaiian/Pacific Islander; there were 74 patients whose racial identity was Other/Unknown. We considered only the white and African American patients, given the relatively small number of patients in the other racial groups. Comprehensive assessments were available for 2217 of these patients (409 African American and 1808 white), who form the basis for the present analyses.
Organ System Involvement
The clinical manifestations of scleroderma were recorded at cohort entry and as frequent as every 6 months thereafter during follow-up appointments. Measurement of lung volumes (forced vital capacity [FVC], forced expiratory volume in 1 second [FEV1], total lung capacity [TLC]) and diffusing capacity (DLCO) by pulmonary function testing (PFT) was standardized by sex and age10,12 and reported as percent predicted. Visceral involvement was defined in the following manner. Restrictive lung disease was denoted when the lowest recorded FVC was <70% predicted, provided the FEV1/FVC ratio ≥70% indicated a nonobstructive pattern. Using Doppler echocardiography, pulmonary hypertension was denoted for all patients with an estimated right ventricular systolic pressure of ≥40 mm Hg, as previously reported.21 For the subset of patients whose physician pursued a right heart catheterization evaluation, pulmonary arterial hypertension was defined by a resting mean pulmonary artery pressure ≥25 mm Hg in the setting of a pulmonary capillary wedge pressure <15 mm Hg.
In addition, cardiac, renal, and gastrointestinal organ system involvement was assigned according to the modified severity scales of Medsger (MSS).26 In this manner, renal impairment was documented for a renal severity score ≥2, corresponding to a serum creatinine ≥1.7 mg/dL, >3+ proteinuria, or dialysis dependence. Cardiac disease involvement corresponded to a heart severity score ≥1, with electrocardiographic evidence of a conduction defect, or of impaired left ventricular function with an ejection fraction <50%. Gastrointestinal involvement was noted for a gastrointestinal severity score ≥2; designating high-dose medication for gastroesophageal reflux, an abnormal small bowel series, malabsorption, episodes of pseudo-obstruction, or upon requirement for parenteral nutrition. Further, digital ischemia was defined by Raynaud phenomenon with a severity score ≥2, which corresponds to digital pitting, ulcers, or gangrene and indicates the presence of tissue damage. Muscle weakness was defined by muscle MSS ≥1, which equates to motor strength of <5/5 in the upper or lower extremity proximal muscle groups. Modified Rodnan skin scores were also determined at clinical appointments.
Given that disease features may vary with time, we characterized the involvement of each major organ system by its most extreme value at any recorded visit. Similarly, for each individual patient evaluated at the center, the minimum recorded value for each lung function parameter and heart ejection fraction was used to define the greatest extent of pulmonary and cardiac disease, respectively. For those patients who underwent a right-sided cardiac catheterization, the maximum recorded values for mean pulmonary arterial and pulmonary capillary wedge pressures were noted. For the remaining patients, the severity of pulmonary hypertension was assessed by the maximum estimated right ventricular systolic pressure at echocardiography.
Statistical Analysis
We first examined the demographic and clinical profile of the scleroderma cohort during this 20-year time span. Continuously distributed and categorical variables were summarized as means (± standard deviation) and proportions, respectively. Next, we assessed the proportion of patients who returned for follow-up care at the center; the number of visits per patient at the center was calculated. We determined the duration of care as the difference in time between the final and the first visit to the Scleroderma Center. Next, we assessed the association of demographic, serologic, and clinical parameters with race, characterized as African American versus white, using the Student t test for continuous variables and the chi-square test for categorical variables.
Logistic Regression Analysis
Using univariate logistic regression analysis, odds ratios (ORs) and 95% confidence intervals (CIs) were calculated to estimate the strength of association between race and the various organ system manifestations of scleroderma. Next, the independent contribution of race to organ system involvement was examined in multivariate regression models, with adjustment for potential confounding due to age at diagnosis, sex, the limited versus diffuse cutaneous subset of disease, and for disease duration at presentation to the Scleroderma Center. Specifically, the analyses of race with prevalent restrictive lung disease are based on spirometric measurement of FVC and are not race-adjusted. We undertook stratified analyses based on disease subtype to determine whether the association of race with visceral disease manifestations varied in those with examination evidence of the limited compared to the diffuse cutaneous subset of disease. In addition, we examined the association of race with the various disease manifestations in strata defined by seropositivity (versus seronegativity) to anticentromere and antitopoisomerase antibodies.
Survival Analysis
We ascertained vital status in 2 ways. First, among both the African American and white patients evaluated at the center, approximately two-thirds returned for follow-up medical care. In this subset of the cohort with longitudinal data, those who died and their date of death were recorded in the center database. Second, we examined the vital status of all 2217 patients in the cohort using the Social Security Death Index. The cumulative incidence of mortality following entry into the Johns Hopkins Scleroderma Center database was estimated using Kaplan-Meier analysis;11 the log-rank test was employed to examine whether the incidence of death differed according to racial group.34 Cox proportional hazards analysis was used to examine the independent contribution of race to mortality, with adjustment for age at disease onset, sex, disease subtype, disease duration, and topoisomerase (Scl-70) serologic status.6 We further adjusted for 2 measures of socioeconomic status, namely educational attainment and health insurance status. Lastly, stratified analyses were performed according to serologic status, cutaneous disease subtype, age stratum at disease onset, and sex. We examined for possible secular trends by stratifying the cohort into those patients who first presented to the center from 1990 through 1999, versus those who presented from 2000 through 2009. All analyses were performed using Stata IC 10.0; reported p values are 2 sided with α = 0.05.
RESULTS
Among the study population of 2217 patients, there were 409 African American and 1808 white patients with scleroderma (Table 1). Whereas the distribution by sex was virtually identical in both racial groups at 82% female, African American patients presented to the center at a younger mean age than white patients, at aged 47 compared to 53 years, respectively (p < 0.001). Similarly, African American patients, on average, developed their first non-Raynaud symptom of disease earlier in life than white patients (42 vs. 47 yr; p < 0.001) and experienced a shorter time before physician-diagnosis of disease (1.6 vs. 2.2 yr; p = 0.07). Moreover, the interval from first non-Raynaud symptom of disease (or a physician diagnosis, whichever came sooner) before the first evaluation at the Johns Hopkins Scleroderma Center was briefer among African American, at 4.9 years, compared to 6.3 years among white patients (p = 0.001).
TABLE 1.
Demographic, Disease Duration, and Serologic Profile Among 2217 White and African American Patients With SSc Evaluated From 1990 to 2009 at the Johns Hopkins Scleroderma Center
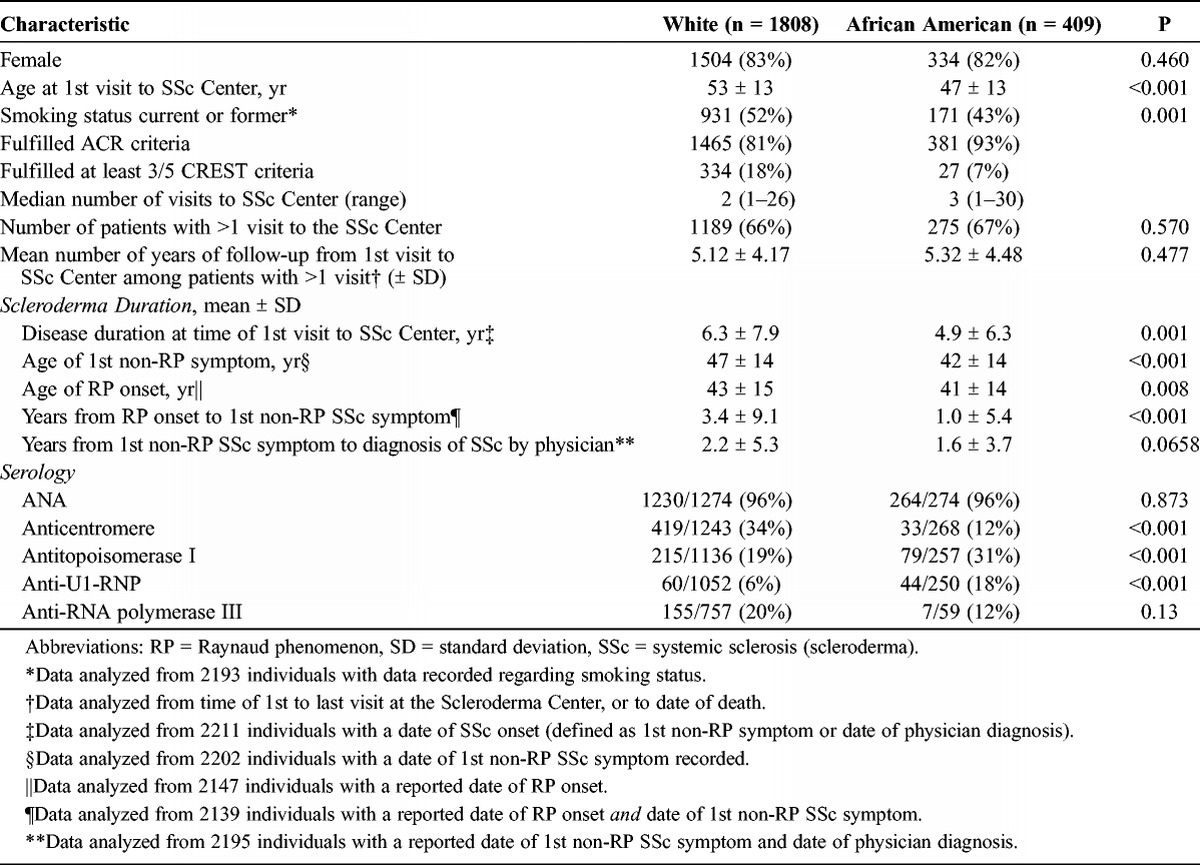
A higher proportion (93%) of African American patients satisfied the ACR criteria for scleroderma compared to 81% of white patients. In contrast, more than twice as many of the white (18%) compared to the African American (7%) patients satisfied 3 or more criteria of the CREST syndrome. Most notably, whereas two-thirds of white patients manifested the limited cutaneous subset of disease, this phenotype of scleroderma was present among only 44% of African American patients (p < 0.001). The majority of African American patients manifested the diffuse cutaneous subset of disease.
The serologic profile of the cohort was related to its racial distribution. Whereas the vast majority of all patients were seropositive for ANA, present in 96% of the cohort, other autoantibodies varied. Specifically, the proportion seropositive for anticentromere antibody was nearly 3-fold greater among white patients, at 34%, compared to African American patients (12%; p ≤ 0.001). Similarly, 31% of African American patients had autoantibodies to topoisomerase compared to only 19% of white patients (p = 0.001). Anti-U1-RNP antibody was 3-fold more prevalent: found in 18% of African American compared to 6% of white patients (p < 0.001). The frequency of RNA polymerase III autoantibody was assayed in only 37% of the cohort; the seropositive proportion was higher at 20% among white, compared to 12% among African American patients, although this difference was not statistically different (p = 0.13).
Overall, 1464 (66%) of the patients in the cohort returned to the Scleroderma Center for follow-up rheumatology care, a proportion that was indistinguishable between the 2 groups (p = 0.57). The median number of visits to the center was 2 among white and 3 among African American patients with scleroderma. We note that the mean number of years with longitudinal care at the center was quite comparable between the 2 groups: 5.3 among African American and 5.1 among white patients (p = 0.48).
Prevalence of Organ Involvement
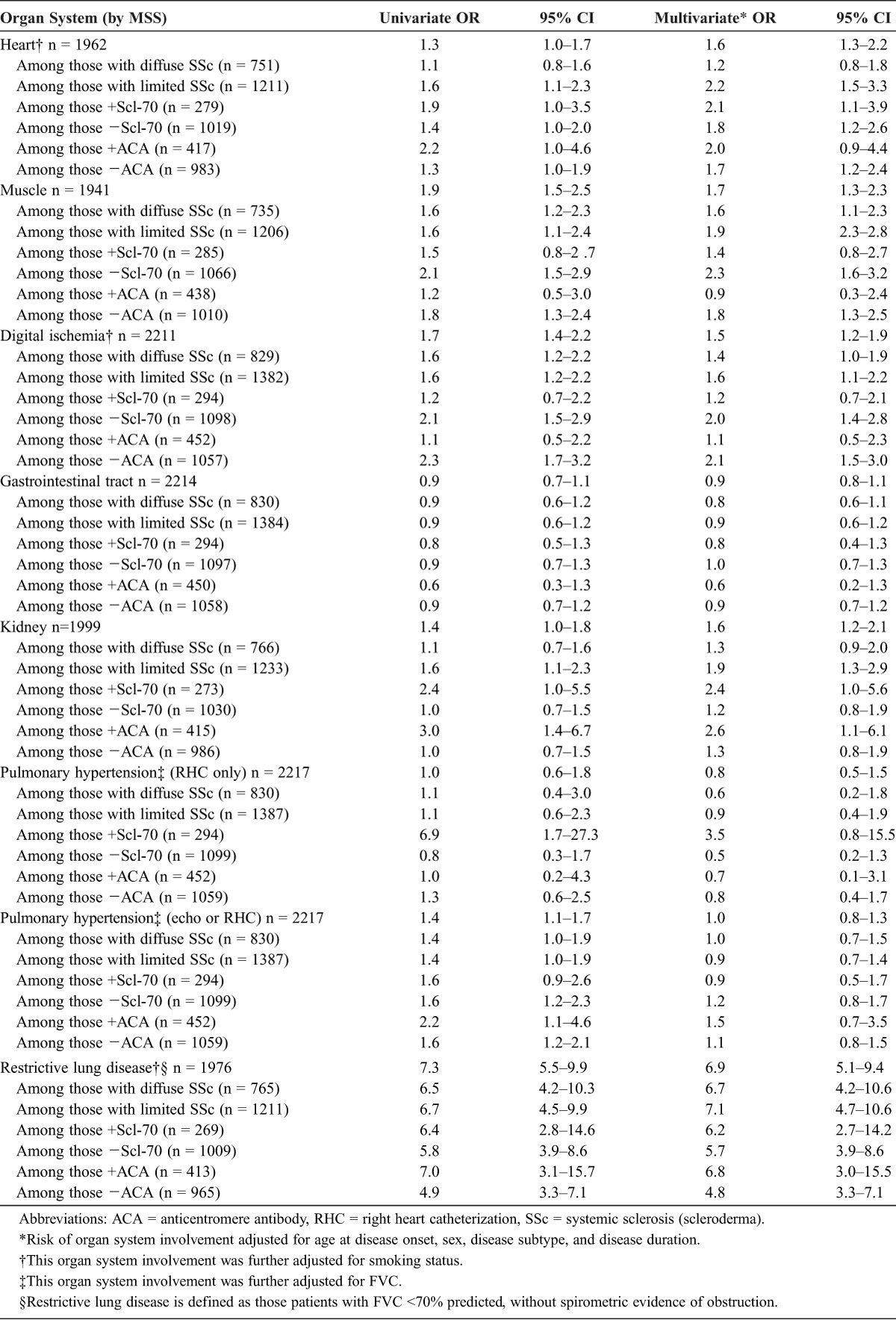
As noted, just over half, or 56%, of African American patients manifested the diffuse cutaneous subset of disease compared to substantially fewer, only 33%, of white patients (p < 0.0001). In univariate analysis, African American patients experienced a higher prevalence of cardiac, muscle, digital ischemia, renal, and restrictive lung disease (Table 2). Cardiac and renal organ system involvement, however, was of only borderline statistical significance. In contrast, the frequency of restrictive lung disease was markedly elevated in the African American patients compared to their white counterparts (OR, 7.3; 95% CI, 5.5–9.9). The association with pulmonary hypertension varied substantially in relation to the mode of ascertainment. When the definition of pulmonary arterial hypertension was restricted to those patients undergoing right heart catheterization, a relatively small proportion of the overall cohort, no difference in frequency was observed between the 2 groups. In contrast, when a diagnosis of pulmonary hypertension was met by either catheterization hemodynamics or by Doppler echocardiography, African American patients were found to experience a 40% greater frequency of pulmonary hypertension (OR, 1.4; 95% CI, 1.1–1.7). Finally, in these univariate analyses, the association of race with gastrointestinal organ system involvement was null (OR, 0.9; 95% CI, 0.7–1.1).
TABLE 2.
Association of African American Compared With White Race With Major Organ System Involvement in Scleroderma Among 2217 African American and White Patients at the Johns Hopkins Scleroderma Center, 1990–2009
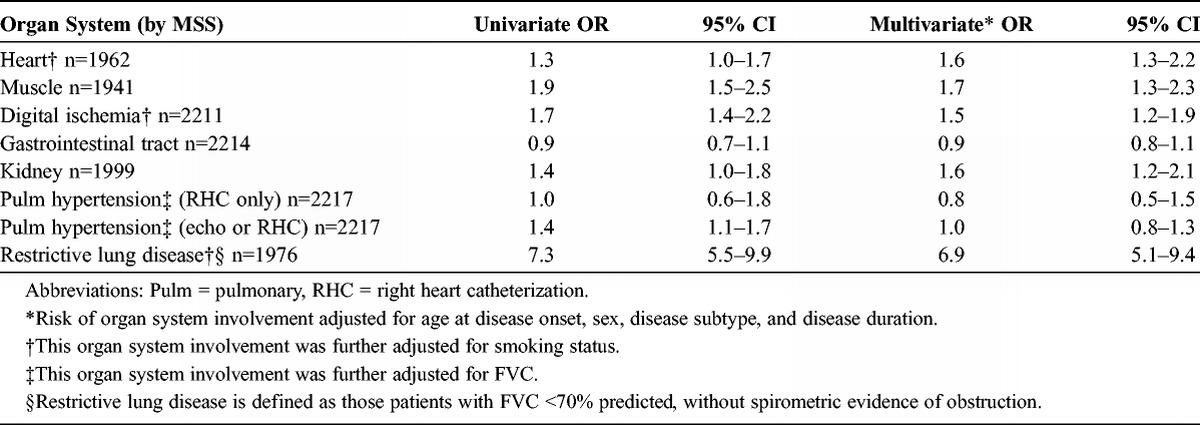
Multivariate analysis was informative in several ways (see Table 2). First, after adjustment for diffuse versus limited disease subtype, sex, age at disease onset and antitopoisomerase autoantibody status, African American patients continued to demonstrate a greater prevalence of cardiac disease involvement (OR, 1.6; 95% CI, 1.3–2.2), digital ischemia (OR, 1.6; 95% CI, 1.4–2.2), renal impairment (OR, 1.6; 95% CI, 1.2–2.1) and muscle involvement (OR, 1.7; 95% CI, 1.3–2.3). Moreover, the greater prevalence of cardiac and renal organ system involvement was strengthened and was clearly statistically significant. In contrast, gastrointestinal organ system involvement remained unrelated to race (OR, 1.0; 95% CI, 0.6–1.4). The presence of pulmonary hypertension, as assessed by echocardiography alone, or with right heart catheterization data when available, was not related to racial status. However, in the multivariate analysis, African American patients experienced a substantially heightened prevalence of restrictive lung disease (OR, 6.9; 95% CI, 5.1–9.4).
Lastly, we sought to ascertain whether the association of prevalent organ system involvement varied in strata defined by disease subtype or serologic status (see Appendix 1). In this manner, cardiac involvement was twice as prevalent among African American patients with limited cutaneous disease (OR, 2.2; 95% CI, 1.5–3.3), but was unrelated to racial status among patients with diffuse cutaneous disease (OR, 1.2; 95% CI, 0.8–1.8). Muscle disease was more prevalent in the African American patients who were seronegative for topoisomerase and centromere antibodies (that is, centromere seronegative OR, 1.8; 95% CI, 1.3–2.5) but not in the seropositive strata (centromere seropositive OR, 0.9; 95% CI, 0.3–2.4). The association of digital ischemia with African American status similarly was operative in the seronegative, but not the seropositive, strata. In contrast, a heightened prevalence of renal organ involvement was notable, and more than twice as common, in the topoisomerase and centromere seropositive, but not in the seronegative, strata. Finally, the relation of restrictive lung disease to African American race was so great and so widespread, that this association was evident in patients with both limited and diffuse disease subtypes, as well as among seropositive and seronegative strata.
Survival Analysis
During the period of follow-up, a total of 700 (159 African American and 541 white) patients died. The cumulative incidence of mortality at 10 years following the initial evaluation at the center was higher, at 43% versus 35%, in African American compared to white patients with scleroderma (Figure 1; log-rank p = 0.0011). Given the unequal distribution of age in these 2 groups, including the younger age at onset of Raynaud phenomenon, at the onset of the first non-Raynaud symptom of disease, and at first presentation to the Johns Hopkins Scleroderma Center among African Americans patients, we next examined the age-adjusted risk of mortality. After controlling for age in Cox proportional hazards analysis, African American patients experienced a 60% increase in risk of mortality compared to white patients (Table 3; relative risk [RR], 1.6; 95% CI, 1.3–2.0). The relative risk of mortality rose modestly higher, with further adjustment for disease duration at presentation to the Scleroderma Center (RR, 1.8; 95% CI, 1.4–2.2).
FIGURE 1.
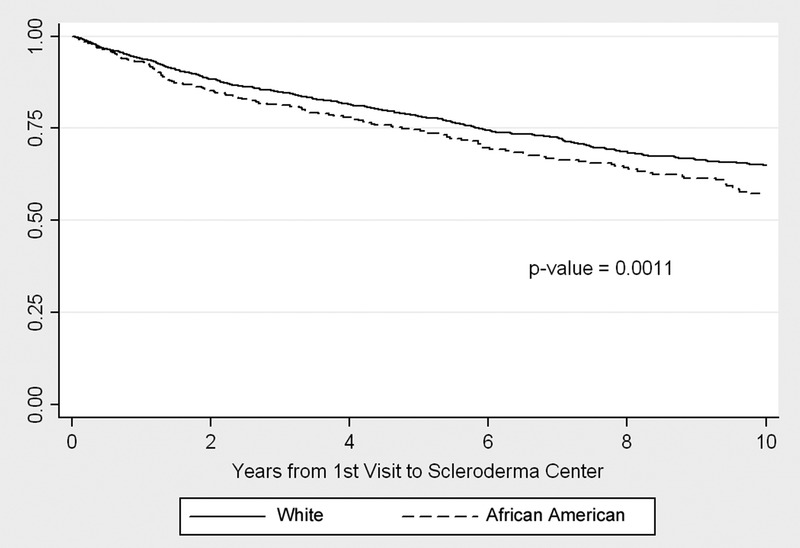
Kaplan-Meier cumulative survival by race.
TABLE 3.
Risk of Mortality Associated With Race Among 409 African American and 1808 White Patients at the Johns Hopkins Scleroderma Center, 1990–2009
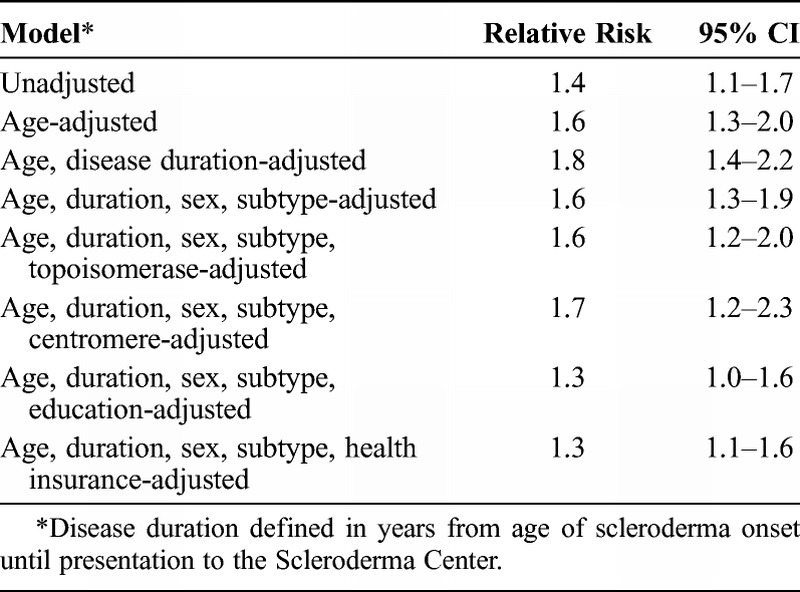
Additional known host factors related to mortality risk, including scleroderma-specific autoantibody status and the presence of diffuse cutaneous disease, were unequally distributed between the 2 racial groups. When these parameters were incorporated into the multivariate models, the risk of mortality among African American patients became somewhat attenuated, after taking age, sex, diffuse cutaneous disease subtype, and seropositivity to antitopoisomerase antibody into account (RR, 1.6; 95% CI 1.2–2.0). Results were essentially the same when seropositivity to anticentromere was substituted for antitopoisomerase antibody in these analyses (RR, 1.7; 95% CI, 1.2–2.0).
To examine the possibility that socioeconomic factors associated with racial status might have an impact on the patients’ mortality profile, we incorporated, separately, measures of educational attainment and health-insurance status into these models (see Table 3). Notably, compared to white patients, the risk of mortality associated with African American race was reduced after adjustment for education (RR, 1.3; 95% CI, 1.0–1.6) and for health insurance (RR, 1.3; 95% CI, 1.1–1.6).
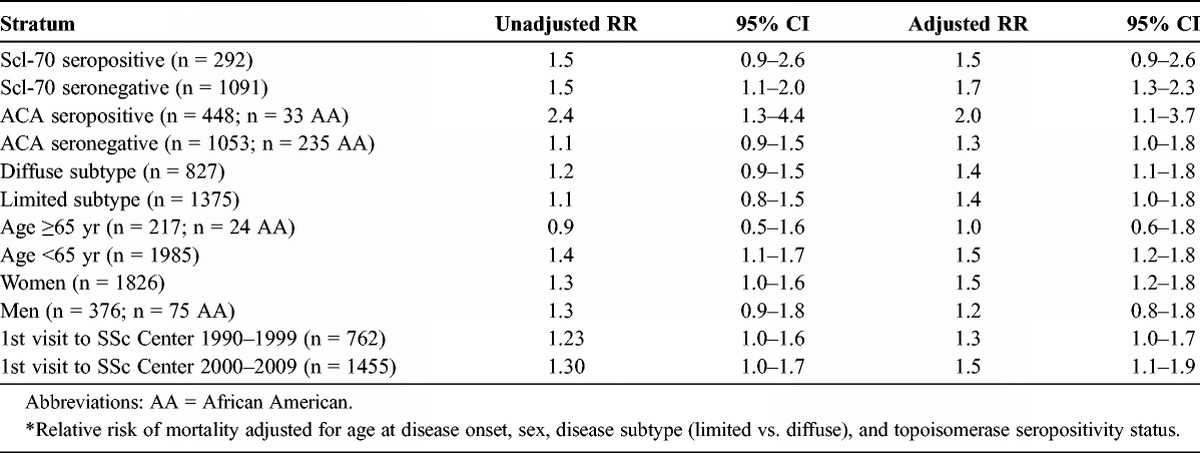
Lastly, we examined the impact of race on survival in stratified analyses according to several key clinical, demographic, and serologic parameters (Table 4). African Americans remained at heightened risk for mortality in both the limited (RR, 1.4; 95% CI, 1.0–1.8) and the diffuse (RR, 1.4; 95% CI, 1.1–1.8) cutaneous subsets of disease. We note that the stratified analyses according to age and sex clearly had an impact on the mortality risk profile. In the patients aged older than 65 years at disease onset, the fully adjusted risk of mortality was no different among African American compared to white patients (RR, 1.0; 95% CI, 0.6–1.8). In contrast, in the stratum of younger patients, aged less than 65 years, African Americans experienced a 50% increase in adjusted risk of mortality (RR, 1.5; 95% CI, 1.2–1.8). Similarly, the heightened risk of mortality for African Americans remained evident in the female (RR, 1.5; 95% CI, 1.2–1.8) but not in the male (RR, 1.2; 95% CI, 0.8–1.8) strata. In relation to serologic status, in both the Scl-70 seropositive (RR, 1.5; 95% CI, 0.9–2.6) and seronegative (RR, 1.7; 95% CI, 1.3–2.3) strata, African Americans experienced a heightened risk of mortality, although this risk was statistically significant only in the seronegative stratum. In contrast, in the anticentromere antibody-positive stratum, African American race remained related to a 2-fold greater risk of mortality (RR, 2.0; 95% CI, 1.1–3.7), whereas in the seronegative stratum, this risk was more modest and of borderline statistical significance (RR, 1.3; 95% CI, 1.0–1.8).
TABLE 4.
Risk of Mortality Associated With Race, Stratified by Various Serologic, Clinical, and Demographic Parameters, Among 409 African American and 1808 White Patients at the Johns Hopkins Scleroderma Center, 1990–2009*
CHRONOLOGIC REVIEW OF THE LITERATURE
Osler, The Principles and Practice of Medicine, 1901
At the turn of the 19th century, Sir William Osler32 reported his experience at Johns Hopkins Hospital with 11 patients diagnosed with scleroderma. In the 4th edition of The Principles and Practice of Medicine, he described key features of the disease process, which continue to characterize this fibrotic disorder to the present. Osler described cutaneous subtypes “circumscribed” and “diffuse,” and the quality of affected skin as being “brawny, hard, and inelastic,” the result of which is “a sense of stiffness or tension in making accustomed movements.” In addition, “the hands may become fixed and the fingers immobile, on account of the extreme induration of the skin over the joints.” He also described the female predominance of the disease, and noted that “remarkable vaso-motor disturbances are common, as extreme cyanosis of the hands and legs,” and that “patients are apt to succumb to pulmonary complaints or to nephritis.” Osler does not, however, make any reference to race in his description of affected patients or as race may relate to disease outcomes.
Leinwand et al, Annals of Internal Medicine, 1954
In 1954, Leinwand et al16 reported a comprehensive picture of the clinical and pathologic features of scleroderma, based on their experience with 150 patients amassed over 14 years at New York University. They emphasized that scleroderma varies in its course; dependent in large part on the number of organ systems involved, and in relation to the severity of fibrosis in affected tissues. The propensity to affect a large number of organs was attributed to widespread involvement of the arterial circulation throughout the body. They recognized that scleroderma in its most extreme fulminant form may be rapidly fatal, and that in their time, as remains the case today, curative therapy was lacking. In terms of demographic features, these authors reiterated the message that scleroderma is a female-predominant disease, with 108 (72%) of their patients being women. They conveyed their impression that scleroderma was “largely a disease of white-skinned people.” Specifically, this series of 150 patients included only 3 black patients.
Tuffanelli and Winkelmann, Archives of Dermatology, 1961
In 1961, Tuffanelli and Winkelmann41 reported a series of 727 patients with scleroderma, evaluated from 1935 through 1958, from the Section of Dermatology at the Mayo Clinic. During this 24-year period, those with the systemic subset of disease were dichotomized as having either acrosclerosis or diffuse scleroderma. The former subset, acrosclerosis, was the more common of the 2, occurring predominantly in women, beginning in the hands and associated with altered pigmentation, telangiectasia, ulceration, and calcinosis. In contrast, the diffuse subset portended a rapid course with an extremely poor prognosis, but accounted for only 5% of the total series. The onset of disease was frequently heralded by vasospastic phenomena, cutaneous sclerosis, or arthritis; most patients experienced anorexia, weight loss, fatigue, and weakness. The authors furnished detailed descriptions of disease and quantified the frequency of major organ involvement in their series. Overall, the female to male ratio was 2.9:1, with a mean age at disease onset of 40 years. Among those with follow-up information, 70% were alive at 5 years, whereas 59% were alive at 10 years after being diagnosed with scleroderma at the Mayo Clinic. However, even in this largest series of scleroderma from the first half of the 20th century, no information was furnished regarding the racial distribution of the cohort.
Masi and D’Angelo, Annals of Internal Medicine, 1967
In 1967, Masi and D’Angelo22 reported what is to our knowledge the first population-based study of scleroderma. They focused on the most severe end of the disease spectrum, identifying all residents of Baltimore, Maryland, who died after being diagnosed with scleroderma from 1949 through 1963. The authors reviewed medical records and death certificate data from all 20 Baltimore area hospitals during this 15-year period. Each case had both cutaneous evidence of scleroderma and clinical or pathologic evidence of at least 1 characteristically involved internal organ. Age-, race-, and sex-specific mortality rates were calculated for July 1956, the midpoint of the study, using United States Census data from the 1950 and 1960 surveys to estimate the population size of Baltimore. Overall, there were 53 reported deaths among patients with scleroderma. Female cases predominated over men in a ratio of 39:14 or 2.8:1. Twenty (38%) of the patients were black. Next, population-based analyses focused exclusively on the subset of 34 deaths arising among Baltimore area residents. Whereas the majority of the Baltimore City population was white during that time period, black women outnumbered white women among the scleroderma fatalities, in a ratio of 15:11 or 1.4:1. Importantly, the crude mortality rate among black women, at 6.6 deaths per million, was 3-fold greater than among white women with scleroderma, who died at a rate of 2.2 deaths per million. Among men, however, the numbers were particularly small, with only 2 black and 6 white male deaths among Baltimore residents with scleroderma. Thus, in contrast to the women, no racial difference in mortality was observed among the men. Notably, this paper made a seminal contribution in highlighting the higher mortality rate among black compared to white women; the black patients also died, on average, at a younger age.
Medsger and Masi, Annals of Internal Medicine, 1971
In 1971, Medsger and Masi24 reported a population-based examination of the epidemiology of systemic sclerosis to ascertain disease prevalence and incidence. They focused on Shelby County, Tennessee, a geographic region encompassing the city of Memphis. During the calendar period of 1947 through 1968, a total of 86 patients were identified, hospitalized with a diagnosis of scleroderma at 1 of 19 county-area hospitals. As a whole, the peak age at diagnosis of scleroderma was in the 35–54 year range. This paper was particularly influential in furnishing community-based epidemiologic data at the sex, age, and race level, using denominators derived from the 1960 census. During this time, the Memphis metropolitan area consisted of 600,000 residents, 60% white and 40% nonwhite. Among the 86 hospitalized patients with scleroderma, just under half, or 38 (44%), were residents of the county itself. Among these Shelby County residents, there were 29 women and 9 men with scleroderma, corresponding to a female to male prevalence ratio of approximately 3:1. Over the 22 years of the study, the incidence of scleroderma rose from 0.6 cases per million to 4.5 cases per million. Most importantly, this paper demonstrated that the incidence of scleroderma was greater in black compared to white residents. Specifically, the average annual age-adjusted incidence of scleroderma was 4.3 cases per million among black women compared to 3.6 among white women; the corresponding rates were 1.6 compared to 1.2 in the men, respectively. It is notable that among the 4 sex-race categories, black women had, on average, the earliest onset of disease.
By focusing entirely on cases of scleroderma culled from hospital discharges, the report by Medsger and Masi was skewed, by design, to the severe end of the disease spectrum. In the same vein, persons who manifested sclerodactyly and Raynaud phenomenon alone were not included in the report. Thus, had autoantibody testing been available during this calendar period, the calculated incidence of scleroderma would have most certainly been higher. Nevertheless, by identifying discharge diagnoses of scleroderma from all hospitalized patients in a well-defined geographic area, the authors were able to furnish incidence data on scleroderma, for age, sex, and race strata, and to follow trends in incidence rates over a 22-year period.
Lee et al, Quarterly Journal of Medicine, 1992
At the Wellesley Hospital in Toronto, Canada, Lee et al15 evaluated mortality risk among 237 patients with systemic sclerosis, a cohort assembled from January 1979 through June 1990. Each patient satisfied classification criteria for systemic sclerosis.35 Overall, 35 (57%) of the cohort manifested restricted disease involvement (sparing the trunk), while 102 (43%) developed diffuse skin thickening, including involvement of the trunk. The frequency of heart, lung, and kidney involvement was comparable among both the restricted and diffusely affected patients. However, isolated lung involvement, and the absence of any apparent internal organ involvement, were more common in those with restricted disease. During the period of follow-up, 61 patients (one-quarter of the cohort) died. The presence of cardiac, renal, and pulmonary organ involvement was each related to a heightened risk of mortality. Most of the deaths attributed to underlying systemic sclerosis were of pulmonary etiology compared to any other single organ cause. Importantly, whereas age, as expected, was an important predictor of mortality, with a 2-fold greater risk of mortality related to disease onset at or greater than age 45 years, neither sex nor extent of skin involvement predicted survival.
Notably, the fact that cutaneous disease subset did not discriminate the survivors from the nonsurvivors may in part be related to the use of a different discriminator than that frequently used today. Specifically, the presence of restricted disease in the Wellesley cohort enabled any involvement of the extremity, as long as the trunk was spared. In contrast, the dichotomy proposed by LeRoy and Medsger18 precludes invoking limited cutaneous involvement among those with skin involvement proximal to the elbow and knees in the upper and lower extremities, respectively. This report from Canada, relatively novel in its time in the use of multivariate analysis to identify predictors of survival, did not enable examination of race as a potential risk factor. The demographic composition of the cohort was virtually entirely white, other than the 4 patients who were black and the 9 who were Asian.
Laing et al, Arthritis & Rheumatism, 1997
Laing et al13 undertook a systematic effort to identify all cases of scleroderma among women residing in the state of Michigan over a 12-year period. From January 1980 through December 1991, 514 state-wide cases were identified using a 5-pronged strategy. Specifically, all women aged 18 years and older identified from 1) a comprehensive hospital discharge database in Michigan; 2) a University of Michigan hospitals database; 3) a Wayne State University hospitals database; 4) inquires to all in-state practicing rheumatologists; and by 5) direct mailing to members of the Southeast Michigan chapter of the United Scleroderma Foundation. Each woman either satisfied the ACR criteria or was deemed to have probable scleroderma based on the presence of sclerodactyly and 1 other element of the CREST syndrome. Chart abstraction was used to identify internal organ involvement. Among a total of 533 eligible female patients with scleroderma, 514 women were identified as being either black (n = 117) or white (n = 397). It is noteworthy that 58 (49.6%) or half of the black women manifested the diffuse subset, compared to only 99 (24.9%) of the white patients (p < 0.001). In addition, black women were diagnosed with scleroderma at a younger age compared to white women (44.5 vs. 51.5 yr, p < 0.001), apparent in both the diffuse and limited subsets of disease.
In Michigan, the average annual incidence of scleroderma was 14.1 cases per million women per year. This rate was higher, at 22.5 per million among black women compared to 12.8 cases per million among white women (p < 0.001). Several clinical manifestations of disease were more common among black women, including pericarditis, pulmonary hypertension, pleural effusions, myositis/myopathy, and renal insufficiency (each p ≤ 0.05). In addition, black women were more likely to have an elevated erythrocyte sedimentation rate >40 mm/h (p < 0.001). Antibodies to RNP were found in 22.4% of black compared to 12.7% of white patients (p = 0.05). In contrast, seropositivity to Scl-70 was equally prevalent, at 18%, in both the black and white patients (p = 0.99). Overall survival at 7 years following a diagnosis of scleroderma was 76.5%, and was similar among black (72.5%) and white women (77.5%, p = 0.455). In further survival analyses, both age and the diffuse (versus limited) cutaneous subset of disease were predictors of survival. Further, in age-adjusted analyses, black women had a 2-fold greater risk of mortality (RR, 1.98; 95% CI, 1.06–3.71). Yet, after further adjustment for age and diffuse disease status, race was no longer predictive of survival (p = 0.177). At the time this epidemiologic paper was published, it contained a particularly large number of black women with scleroderma and demonstrated a higher prevalence of diffuse disease, a younger age at disease onset, and a greater severity of disease in black patients, compared to white patients, including a heightened risk of mortality.
Greidinger et al, Chest, 1998
In recognition of the increasing importance of pulmonary impairment as the leading organ-specific cause of mortality in scleroderma,15 Greidinger et al,9 investigators at the Johns Hopkins and University of Maryland Scleroderma Center, examined the contribution of autoantibody status and race in relation to spirometric measures of lung decline. In total, 191 patients with the diffuse cutaneous subset of disease, satisfying ACR criteria, were evaluated between 1990 and 1996. Among this group, 39 African American and 59 white patients had both serial PFTs and serum samples performed and available for analysis. Approximately one-third of the patients were seropositive for topoisomerase antibody, a proportion greater in African American compared to white patients (44% vs. 20%; p < 0.023). The African American patients were younger, by approximately 9 years (p < 0.0001), at first PFT assessment at the Scleroderma Center. In the linear regression analyses, both African American race and topoisomerase seropositivity each contributed, independently, to a 9% drop in the percent predicted FVC. This report helped clarify that in scleroderma, race may contribute to pulmonary compromise above and beyond autoantibody status.
Nietert and Silver, Journal of Rheumatology, 2003
Nietert and Silver28 examined health care utilization patterns among patients with scleroderma in South Carolina, focusing primarily on hospitalization rates and Emergency Room (ER) visits across the state, from January 1996 to December 2000. Their data set collects information for each ER visit and hospital admission at nonfederal and nonmilitary hospitals within the state, assigning a unique identifier for each patient. In this manner, multiple ER and/or inpatient stays are still ascribed to a single individual. During this 5-year period, a total of 785 patients with scleroderma were identified, corresponding to 4402 ER visits and 2574 hospitalizations to a South Carolina hospital.
An immediately apparent demographic difference is the significantly lower mean income level of black compared to white patients. Further, although the number of admissions per patient was identical (at 3.4) for both black and white patients, the number of admissions classified as an emergency was greater among black compared to white patients, respectively (59.2% compared to 48.5%). Black patients less often had private insurance compared to white patients (29.8% vs. 37.9%, respectively) and more often had Medicaid support (22.6% versus 3.5%, respectively). We note that the incidence of both hospital admissions and ER visits for patients with scleroderma residing in the state of South Carolina was greater for black compared to white men, as for black compared to white women; the only exception to this pattern of racial disparity was in the older age groups for women (aged 50–64 yr and ≥65 yr). Lastly, black patients were less likely than white patients to receive care at the Medical University of South Carolina, a major referral center in the state for the evaluation and management of scleroderma.
Mayes et al, Arthritis & Rheumatism, 2003
Mayes et al23 examined the prevalence and incidence of scleroderma, and related survival, in the greater Detroit, Michigan, area. During the 1988–1991 calendar period, 1596 patients were identified, 72% of whom had available medical records for review. Ultimately, 706 (61%) patients satisfied classification criteria for either definite or probable scleroderma; the latter group uniformly had a rheumatologist-assigned diagnosis of sclerodactyly and at least 2 other features of the CREST syndrome. To calculate prevalence and incidence rates, a reference population was ascertained using the 1990 census data for the Detroit metropolitan area. In this manner, the 706 confirmed cases corresponded to an overall disease prevalence of 242.0 cases per million, a prevalence that was greater in women than men, at a ratio of 4.6 to 1. In addition, the prevalence of scleroderma was greater among black (315.1 cases per million) compared to white (224.7 cases per million) residents (prevalence ratio of 1.15; 95% CI, 1.02–1.30). Mean age at diagnosis of scleroderma was 46 years, similar among both men and women. However, the mean age was younger among black (41 yr) compared to white (48 yr) patients, a finding consistent among men and women.
There were 169 incident cases of scleroderma identified during the study period, corresponding to an annual incidence of 19.3 cases per million. Disease incidence was greater among women than men, with a female to male incidence ratio of 4.2:1. The incidence of scleroderma was greater in black (23.7 cases per million) compared to white (18.3 cases per million) participants, although this difference was not statistically significant. For both black women and black men with scleroderma, the peak incidence of disease occurred between the ages of 45 and 54 years. Diffuse disease was much more common, at 60%, among black patients compared to only 27% among white patients. In contrast, anticentromere antibody was less common, only 10% among black patients compared to a prevalence of 27% among white patients (p < 0.001). Topoisomerase antibody was equally common, at approximately 20%, among both black and white patients. In this study, vital status was ascertained using the hospital discharge database, medical chart review, and the National Death Index. Overall, 215 patients died; median survival was 11 years after a diagnosis of scleroderma was made. Survival was 78% survival at 5 years, 55% at 10 years, and 27% at 20 years. Notably, survival rates were greater for women than for men. However, whereas survival was marginally worse among black compared to nonblack patients during the first 12 years of follow-up, survival did not statistically differ according to race (p = 0.42).
Nietert et al, Journal of Rheumatology, 2005
Using the aforementioned administrative database maintained by the South Carolina Office of Research and Statistics, Nietert et al29 examined racial, demographic, and clinical factors associated with inhospital mortality related to systemic sclerosis. During the 5 years from 1996 to 2000, a total of 140 deaths were observed among 727 unique patients (and 2574 related hospitalizations), corresponding to a 19% death rate among individual patients, and a 5% death rate among hospitalizations, for patients with a diagnosis of scleroderma. Overall, the 5-year incidence rate of inhospital mortality was 23.0% among black patients compared to 15.7% among white patients who resided in South Carolina. In multivariate analyses, black patients experienced a 70% increase in mortality risk (OR, 1.70; 95% CI, 1.01–2.86). This fully adjusted model included measures of educational attainment and median household income, parameters themselves associated with heightened inhospital mortality, including being transferred from another hospital, being admitted on an emergent basis, having multiple prior hospital admissions during the designated 5-years, having an admission length of stay of greater than 10 days, and having a comorbid diagnosis of congestive heart failure. It is noteworthy that having comorbid hypertension was related to a 60% reduction in odds of mortality (OR, 0.41; 95% CI, 0.23– 0.71).
Beall et al, Journal of Rheumatology, 2007
Beall et al3 addressed the risk to develop pulmonary hypertension at the Medical University of South Carolina, in Charleston, among a cohort evaluated from November 1997 to January 2004. All patients satisfied the ACR criteria and were classified further as having limited versus diffuse disease based on the criteria defined by Leroy et al.18 A total of 328 patients were prospectively evaluated to ascertain the development of pulmonary hypertension, defined as a peak right ventricular systolic pressure >40 mm Hg assessed by echocardiography, or as a mean pulmonary artery pressure >25 mm Hg, assessed at right heart catheterization. Of these 328 patients, 245 were white and 83 were African American. The African American patients with scleroderma manifested the diffuse subtype of disease to a greater extent (72% vs. 43%, p < 0.0001) than their white counterparts. Overall, echocardiography was performed on 72% and right heart catheterization on 7% of the cohort. On this basis, the authors found that 18% of the white patients compared to 23% of the African American patients developed pulmonary hypertension, a difference that was not statistically significant. However, an interesting observation was that the African American patients were a full decade younger when their pulmonary hypertension was diagnosed (49 vs. 61 yr; p < 0.001). A limitation of the study is that the serologic status of the study participants was not identified, in terms of topoisomerase, centromere autoantibodies, or otherwise. As a result, one cannot ascertain the contribution of race, above and beyond seropositivity for characteristic scleroderma-associated autoantibodies, to the development of pulmonary hypertension.
Assassi et al, Arthritis & Rheumatism, 2009
Molecular developments in recent years have facilitated high volume human leukocyte antigen (HLA) allele genotyping in clinical cohorts. These markers have been examined as potential predictors of clinical outcomes, including mortality. A prime example is the Genetics versus Environment in Scleroderma Outcomes Study (GENISOS), a prospective collaborative cohort assembled from Houston, Galveston, and San Antonio, Texas.2 The GENISOS investigators focused on the influence of ethnicity on disease features, and assembled a cohort of 250 total patients, among whom 122 patients where white (48.8%), 47 were African American (18.8%), and 71 were Hispanic (28.4%). Ten others consisted of 1 Native American and 9 Asian patients. The cohort members underwent serologic evaluation for ANA, anticentromere antibody, antitopoisomerase antibody, and anti-RNA polymerase III antibody as well as genotyping for HLA class II alleles. Vital status was ascertained using the National Death Index, and then 3 years later, in 2008, was reassessed using the Social Security Death Index.
During the follow-up period, there were a total of 52 (20.8%) deaths. Whereas in the age-adjusted models, African American and Hispanic groups did not individually portend a heightened risk of mortality, when categorized in aggregate as nonwhite versus white patients, the nonwhite group demonstrated an 87% increase in mortality risk. Notably, the HLA II alleles, DRB1*0802 and DQA1*0501, did not confer further risk in mortality profile above and beyond the contribution of ethnicity. Moreover, in the fully adjusted model, there were 7 variables that were significant predictors of mortality, including older age, reduced FVC, electrocardiogram evidence of arrhythmia, absence of anticentromere antibody, hypertension, chest radiograph evidence of pulmonary fibrosis, and body mass index below the lower limit of normal. Interestingly, when the HLA II alleles were added to this final fully adjusted model, they did, in fact, confer an increase in risk of mortality, whereas the risk conferred by the race/ethnicity categories (of African American and Hispanic groups) were no longer statistically significant. Thus, it seems that the risk associated with ethnicity operates, in large part, through the HLA II gene alleles. Stated differently, the ethnicity status and class II HLA alleles appear to operate via a common biologic mechanism, as opposed to contributing via independent mechanisms, to the mortality risk of the GENISOS scleroderma cohort.
Sharif et al, Journal of Rheumatology, 2011
Subsequently, the GENISOS investigators extended their work regarding the association of HLA alleles and serologic status by expanding the number of patients under study. A total of 3033 patients with scleroderma enrolled in 3 specific cohorts were examined in aggregate.36 Sera from the GENISOS study, the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS) Scleroderma Family Registry and DNA Repository, and the University of Texas Health Science Center at Houston cohorts underwent serotyping for antifibrillarin (or anti-U3-RNP) antibody and genotyping for HLA class II alleles. Given that fibrillarin antibodies and HLA-DRB1*08 and DQB1*03:01 alleles are differentially expressed at high levels in African American patients with scleroderma, these parameters were examined in relation to survival outcomes.
The primary analysis compared the mortality profile of 278 African American patients with scleroderma seropositive to those seronegative for antifibrillarin antibodies; a third group of 328 African American controls without scleroderma was also examined. At follow-up, 30% of the 50 antifibrillarin seropositive African American patients compared to 29.5% of the 221 seronegative patients had died. Given the similarity of these event rates, it was not surprising that in the survival analyses, antifibrillarin antibody did not confer an increase in risk of mortality (RR, 0.79; p = 0.49). In fact, none of the examined HLA class II conferred an increase in mortality risk, either. Thus, neither antifibrillarin antibody nor the studied MHC class II accounted for why some African American patients experience a heightened risk of mortality. Lastly, the survival rate of the 50 antifibrillarin seropositive patients did not differ from that of the 61 African American patients who were antitopoisomerase antibody positive.
Steen et al, Arthritis & Rheumatism, 2012
Steen et al37 presented their extended experience with 3148 consecutive patients with scleroderma evaluated at the University of Pittsburgh over 35 years, from January 1972 to December 2007; 203 patients were African American and 2945 were white. Organ system involvement was ascertained at initial and follow-up visits, upon review of managing physicians’ office records, and via annual/biannual patient-completed questionnaires and phone interviews. Each patient underwent ANA testing. The following serologic assays were performed on a subset of the cohort: anticentromere, antitopoisomerase I, anti-U1-RNP, anti-RNA polymerase III, anti-U3-RNP, Ku, and anti-Th/To antibodies. Vital status was assessed through the Social Security Death Index. A higher prevalence of antitopoisomerase antibodies was identified in African American patients compared to white patients (26% vs. 17%; p = 0.01), similarly observed for anti-U3-RNP (19% vs. 3%, respectively; p < 0.0001) and U1-RNP (18% vs. 5%, respectively; p < 0.0001) antibodies. In contrast, anticentromere, anti-RNA polymerase III, and anti Th-To antibodies were each more prevalent among white patients (p value for anticentromere antibody and RNP polymerase III each < 0.01). Of note, skeletal muscle, pulmonary parenchymal, and gastrointestinal organ system involvement were each more frequent among African American patients. In contrast, pulmonary hypertension, cardiac, and renal organ involvement did not differ by race. With regard to survival, at both 5 years and 10 years after cohort entry, there was an approximate 10% higher mortality rate among African American patients compared to white patients, corresponding to 66% and 75% survival in African American and white patients, respectively, at 5 years, and 51% and 60%, respectively, at 10 years; p = 0.0063. Moreover, in age, sex, and disease subtype-adjusted multivariate analysis, African American patients experienced a 68% increase in risk of mortality at 5 years after their initial evaluation at the University of Pittsburgh compared to their white counterparts (RR, 1.68; 95% CI, 1.30–2.16).
Among the stratum of the cohort seropositive for antitopoisomerase antibody, the proportion with pulmonary fibrosis was substantially higher, at 44%, among African American patients compared to 18% (p = 0.0001) among white patients. As such, even among patients seropositive for topoisomerase antibody, a known predictor of pulmonary parenchymal involvement, African American patients experience an excess and statistically significant increase in pulmonary fibrosis and a heightened risk of mortality. In contrast, in the strata of patients seropositive for U1-RNP and those seropositive for U3-RNP antibody, race did not confer a heightened risk of mortality.
Although the proportion of patients with a high school or higher education was higher among white than African American patients (58% vs. 52%, respectively), this result was not statistically significant. It would still be of interest to know if adjusting for socioeconomic status, via education or another measure, in the multivariate modeling might have an impact on the risk of mortality associated with race.
DISCUSSION
In the current study we review the largest cohort yet published, to our knowledge, of well-characterized African American patients at a single university center (Table 5). The study results confirm and highlight that African Americans face an increase in risk of severe scleroderma and its consequences. Across the spectrum of the autoimmune rheumatologic disorders, a new diagnosis of scleroderma too often augurs a challenging course coupled with a poor prognosis.
TABLE 5.
Chronologic Review of the Literature Regarding the Association of African American Race With Disease Susceptibility, Phenotypic Features, and Prognosis in Scleroderma
The heightened risk of mortality observed in scleroderma was emphasized in a seminal report25 consisting of 223 patients diagnosed with scleroderma at the University of Pittsburgh between 1955 and 1969, together with 86 patients discharged from a Memphis, Tennessee, area hospital from 1947 to 1968 with a diagnosis of scleroderma. During this period, cumulative survival at 7 years of follow-up was only 35%. Subsequently, among 237 patients enrolled from 1979 through 1990 and prospectively followed at the Scleroderma Clinic at The Wellesley Hospital in Toronto, the survival rates were 76%, 76%, and 61% at 3, 6, and 9 years of follow-up, respectively.15 A major interval development contributing to this favorable trend in survival was the advent of angiotensin-converting enzyme inhibitor agents to treat scleroderma renal crisis.38–40 Before the introduction of these medications, death following the onset of renal crisis was essentially universal within a year; in contrast, with the use of this vital therapy, fatality may be averted and restoration of renal function achieved in a subset of patients.40 Subsequently, among 706 verified cases of scleroderma identified in the Detroit area from 1989 to 1991, survival rates were 78% at 7 years and 55% at 10 years of follow-up.23
Several demographic and clinical variables have been examined in the epidemiologic literature and identified as important determinants of outcome in scleroderma. Such parameters include the extent of cutaneous and visceral disease involvement, as well as patient’s sex.13,15,23–25,29 Patients with the diffuse cutaneous subset of disease faced a greater risk of mortality as did those with renal, pulmonary, or cardiac organ involvement.25,41 Men who developed scleroderma had a more severe prognosis than affected women.23,25 Yet, until the middle of the 20th century, reference to the association of race with phenotypic features of scleroderma and to clinically relevant outcomes, including survival, was absent from the medical literature. Earlier studies were most informative for elucidating the characteristic cutaneous subsets of disease and for describing the manifestations of internal organ involvement, but did not, to our knowledge, address the role of race in relation to outcomes in scleroderma.16,41
In contrast, over the last 50 years, several recurrent themes have emerged regarding the role of race in the expression of disease. These include the observations that compared to white patients, African American patients experience a younger age at disease onset, greater incidence and prevalence of disease, a predilection to more severe disease manifestations, a distinct serologic profile, and a worse prognosis.13,22–24 However, the number of African American patients studied in prior reports has often been relatively small (see Table 5), precluding completely reliable estimates. It remains important to ascertain definitively whether race is independently related to disease severity and mortality in scleroderma, above and beyond the known contribution of the aforementioned key demographic, clinical, and serologic predictors.
The research infrastructure of the Johns Hopkins Scleroderma Center provides a valuable opportunity to expand upon prior reports and examine these clinically relevant questions. The relatively unique aspect of this effort is the chance to examine these associations in a large disease cohort, consisting of 2217 patients assembled at a single university medical center over a 20-year period, from January 1990 to December 2009. Importantly, 18% of the cohort was African American. To our knowledge this is the largest single-center report of African American patients with scleroderma (see Table 5), whose phenotypic profiling includes dichotomization into limited versus diffuse disease, and ascertainment of each major organ system for potential sclerodermatous infiltration, as ascertained in the context of the medical care received at the center. The high proportions of patients who underwent serologic evaluation and determination of educational attainment and health insurance status are additional major strengths, enabling determination in multivariate analysis of the independent contribution of each of these known risk factors for disease burden and outcome. The relatively large cohort size further enables analysis of organ system involvement and of mortality profile, with stratification by key demographic, clinical, and serologic subsets, including cohort members who are both seropositive and seronegative for scleroderma-specific autoantibodies.
In the current Johns Hopkins Scleroderma Center cohort, there were 409 African American and 1808 white patients with scleroderma. African American patients presented to the center at a younger mean age than white patients, developed their first non-Raynaud symptom of disease earlier in life than white patients, and experienced a briefer duration of time before establishment of a physician-diagnosis of disease. Whereas two-thirds of white patients manifested the limited cutaneous subset of disease, the majority of African Americans (56%) displayed the diffuse subtype. As expected, based on these phenotypic differences, white patients had a 3-fold higher proportion seropositive for anticentromere antibody compared to African American patients, 31% of African American patients had autoantibodies to topoisomerase compared to only 19% of white patients.
Overall, a total of 700 patients (159 African American and 541 white patients) died. The cumulative incidence of mortality during 10 years of follow-up was higher, at 43% versus 35%, in African American compared to white patients with scleroderma. After adjustment for age and disease duration, African American patients experienced an 80% increase in risk of mortality compared to white patients. Further adjustment by sex, disease subtype, and scleroderma-specific autoantibody status, and for the socioeconomic measures of educational attainment and health insurance status, diminished these risk estimates. Yet, in the fully adjusted multivariate model, relative to white race, African American race was associated with a 30% increase in mortality risk, although the risk estimate was of borderline statistical significance. Further, the excess mortality associated with race persisted in strata defined by age at disease onset, diffuse cutaneous disease, anticentromere seropositivity, decade of care at the center, and among women.
Limitations of the present report include first the relatively small number of other racial and ethnic groups represented in the cohort, including Native American, Asian, and Hispanic American patients, and the inability to perform meaningful statistical analyses in these groups. However, the large number of well-defined African American patients evaluated over the 20 years at a single center provided sufficient statistical power to conduct informative multivariate analyses, often lacking in many prior reports. Second, our experience is derived at a single center, which has a potential impact on generalizability to other regions of the country or to practice patterns at other facilities. Third, although the majority of the cohort underwent serologic testing for topoisomerase, centromere, and U1-RNP autoantibodies, which is a major strength of the study design, we were nevertheless unable to ascertain serologic status within the cohort for other scleroderma-associated autoantibodies, such as Th/To and anti-U3-RNP (antifibrillarin) antibodies. HLA class II alleles were similarly not performed, which precluded examination of the contribution of these genetic markers to clinical phenotype and outcome in our cohort. Further, alhough we systematically evaluated phenotypic features of disease and survival on all patients evaluated at our center, a first visit to the center necessitated, by definition, survival long enough to be referred for evaluation at a tertiary care facility. This referral pattern may skew the severity, or clinical composition of the cohort, in relation to studies that are community-based and are able to capture all incident cases at the population level.
We further note that the heightened risk estimates for restrictive lung disease in African Africans are based on spirometric assessment of FVC. Given that national survey data in the United States demonstrate that African Americans of particular height have, on average, lower lung function than whites, the elevated estimates of restrictive lung disease in African American patients with scleroderma must take into account the known lower lung volumes observed among African American people in the general American population.4,12 As such, our findings may overestimate the prevalence of restrictive lung disease among African American patients with scleroderma. Further, in the survival analyses, we are able to report only on vital status, rather than on organ-specific causes of death. Lastly, whereas several organ system manifestations of disease were more prevalent, and overall mortality was higher, among African American patients with scleroderma compared to white patients, the present observational cohort, notwithstanding its large size and diverse composition, precludes a definitive attribution of causality between scleroderma-dependent and scleroderma-independent mechanisms.
In summary, in this large university-based scleroderma center, African American patients experienced an increased prevalence of internal organ involvement, particularly of the heart, renal, muscular, and pulmonary systems, and a heightened risk of mortality. Given the younger age at diagnosis and at referral to the Johns Hopkins Scleroderma Center, and the briefer duration of disease relative to the white counterparts, the heightened risk estimates among African Americans were accentuated after adjustment for these parameters in the cohort. It is noteworthy that this excess risk persists, and is not fully explained, after taking into account racial differences in frequency of the diffuse cutaneous disease subset and in serologic status for topoisomerase and centromere autoantibodies. Yet, the excess in risk associated with African American race is partially attenuated by imbalance in measures of socioeconomic status, as ascertained in this cohort, specifically health insurance status and educational attainment. Our results support the notion that African Americans face a more severe prognosis when afflicted by scleroderma compared to white patients. Greater vigilance and intensified screening efforts for visceral disease manifestations ought to be considered in African American patients with scleroderma, enabling the potential for earlier and more intensive therapeutic intervention.
Abbreviations
- ACR
American College of Rheumatology
- ANA
antinuclear antibody
- CI
confidence interval
- CREST
calcinosis, Raynaud, esophageal dysmotility, sclerodactyly, telangiectasia
- ER
emergency room
- FVC
forced vital capacity
- GENISOS
Genetics versus Environment in Scleroderma Outcomes Study
- HLA
human leukocyte antigen
- MSS
Medsger severity score
- NIAMS
National Institute of Arthritis and Musculoskeletal and Skin Diseases
- NIH
National Institutes of Health
- OR
odds ratio
- PFT
pulmonary function testing
- RNP
ribonucleoprotein
- RR
relative risk
APPENDIX 1
The Association of African American Compared with White Race with Major Organ System Involvement in Scleroderma Among 2217 African American and White Patients at the Johns Hopkins Scleroderma Center, 1990-2009, Overall and According to Serologic and Disease Subtype Strata
Footnotes
Financial support and conflicts of interest: Grant support provided from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) P30 Rheumatic Diseases Research Core Center Award, NIAMS T32 AR048522, Scleroderma Research Foundation, and Maryland Chapter Arthritis Foundation Arthritis Investigator Award. The authors have no conflicts of interest to disclose.
REFERENCES
- 1. Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology. 2009; 72: 1548– 1554. [DOI] [PubMed] [Google Scholar]
- 2. Assassi S, Del JD, Sutter K, McNearney TA, Reveille JD, Karnavas A, Gourh P, Estrada YMR, Fischbach M, Arnett FC, Mayes MD. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum. 2009; 61: 1403– 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beall AD, Nietert PJ, Taylor MH, Mitchell HC, Shaftman SR, Silver RM, Smith EA, Bolster MB. Ethnic disparities among patients with pulmonary hypertension associated with systemic sclerosis. J Rheumatol. 2007; 34: 1277– 1282. [PubMed] [Google Scholar]
- 4. Berry CE, Wise RA. Interpretation of pulmonary function test: issues and controversies. Clin Rev Allergy Immunol. 2009; 37: 173– 180. [DOI] [PubMed] [Google Scholar]
- 5. Burek CL, Rose NR, Guire KE, Hoffman WH. Thyroid autoantibodies in black and in white children and adolescents with type 1 diabetes mellitus and their first degree relatives. Autoimmunity. 1990; 7: 157– 167. [DOI] [PubMed] [Google Scholar]
- 6. Cox DR. Regression models and life tables. J R Stat Soc [B]. 1972; 34: 187– 220. [Google Scholar]
- 7. Dooley MA, Hogan S, Jennette C, Falk R. Cyclophosphamide therapy for lupus nephritis: poor renal survival in black Americans. Glomerular Disease Collaborative Network. Kidney Int. 1997; 51: 1188– 1195. [DOI] [PubMed] [Google Scholar]
- 8. Fessel WJ. Systemic lupus erythematosus in the community. Incidence, prevalence, outcome, and first symptoms; the high prevalence in black women. Arch Intern Med. 1974; 134: 1027– 1035. [PubMed] [Google Scholar]
- 9. Greidinger EL, Flaherty KT, White B, Rosen A, Wigley FM, Wise RA. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest. 1998; 114: 801– 807. [DOI] [PubMed] [Google Scholar]
- 10. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999; 159: 179– 187. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Amer Statist Assn. 1958; 53: 457– 481. [Google Scholar]
- 12. Knudson RJ, Kaltenborn WT, Knudson DE, Burrows B. The single-breath carbon monoxide diffusing capacity. Reference equations derived from a healthy nonsmoking population and effects of hematocrit. Am Rev Respir Dis. 1987; 135: 805– 811. [DOI] [PubMed] [Google Scholar]
- 13. Laing TJ, Gillespie BW, Toth MB, Mayes MD, Gallavan RH, Jr, Burns CJ, Johanns JR, Cooper BC, Keroack BJ, Wasko MC, Lacey JV, Jr, Schottenfeld D. Racial differences in scleroderma among women in Michigan. Arthritis Rheum. 1997; 40: 734– 742. [DOI] [PubMed] [Google Scholar]
- 14. LaPorte RE, Tajima N, Dorman JS, Cruickshanks KJ, Eberhardt MS, Rabin BS, Atchison RW, Wagener DK, Becker DJ, Orchard TJ, et al. Differences between blacks and whites in the epidemiology of insulin-dependent diabetes mellitus in Allegheny County, Pennsylvania. Am J Epidemiol. 1986; 123: 592– 603. [DOI] [PubMed] [Google Scholar]
- 15. Lee P, Langevitz P, Alderdice CA, Aubrey M, Baer PA, Baron M, Buskila D, Dutz JP, Khostanteen I, Piper S, et al. Mortality in systemic sclerosis (scleroderma). Q J Med. 1992; 82: 139– 148. [PubMed] [Google Scholar]
- 16. Leinwand I, Duryee AW, Richter MN. Scleroderma; based on a study of over 150 cases. Ann Intern Med. 1954; 41: 1003– 1041. [DOI] [PubMed] [Google Scholar]
- 17. Leroy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, Rowell N, Wollheim F. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988; 15: 202– 205. [PubMed] [Google Scholar]
- 18. Leroy EC, Medsger TA., Jr Criteria for the classification of early systemic sclerosis. J Rheumatol. 2001; 28: 1573– 1576. [PubMed] [Google Scholar]
- 19. Lim KN, Casanova RL, Boyer TD, Bruno CJ. Autoimmune hepatitis in African Americans: presenting features and response to therapy. Am J Gastroenterol. 2001; 96: 3390– 3394. [DOI] [PubMed] [Google Scholar]
- 20. Lorenzi M, Cagliero E, Schmidt NJ. Racial differences in incidence of juvenile-onset type 1 diabetes: epidemiologic studies in southern California. Diabetologia. 1985; 28: 734– 738. [DOI] [PubMed] [Google Scholar]
- 21. Manno RL, Wigley FM, Gelber AC, Hummers LK. Late-age onset systemic sclerosis. J Rheumatol. 2011; 38: 1317– 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masi AT, D’Angelo WA. Epidemiology of fatal systemic sclerosis (diffuse scleroderma). A 15-year survey in Baltimore. Ann Intern Med. 1967; 66: 870– 883. [DOI] [PubMed] [Google Scholar]
- 23. Mayes MD, Lacey JV, Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, Schottenfeld D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003; 48: 2246– 2255. [DOI] [PubMed] [Google Scholar]
- 24. Medsger TA, Jr, Masi AT. Epidemiology of systemic sclerosis (scleroderma). Ann Intern Med. 1971; 74: 714– 721. [DOI] [PubMed] [Google Scholar]
- 25. Medsger TA, Jr, Masi AT, Rodnan GP, Benedek TG, Robinson H. Survival with systemic sclerosis (scleroderma). A life-table analysis of clinical and demographic factors in 309 patients. Ann Intern Med. 1971; 75: 369– 376. [DOI] [PubMed] [Google Scholar]
- 26. Medsger TA, Jr, Silman AJ, Steen VD, Black CM, Akesson A, Bacon PA, Harris CA, Jablonska S, Jayson MI, Jimenez SA, Krieg T, Leroy EC, Maddison PJ, Russell ML, Schachter RK, Wollheim FA, Zacharaie H. A disease severity scale for systemic sclerosis: development and testing. J Rheumatol. 1999; 26: 2159– 2167. [PubMed] [Google Scholar]
- 27. Mikuls TR, Sayles H, Yu F, Levan T, Gould KA, Thiele GM, Conn DL, Jonas BL, Callahan LF, Smith E, Brasington R, Moreland LW, Reynolds RJ, Bridges SL., Jr Associations of cigarette smoking with rheumatoid arthritis in African Americans. Arthritis Rheum. 2010; 62: 3560– 3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nietert PJ, Silver RM. Patterns of hospital admissions and emergency room visits among patients with scleroderma in South Carolina, USA. J Rheumatol. 2003; 30: 1238– 1243. [PubMed] [Google Scholar]
- 29. Nietert PJ, Silver RM, Mitchell HC, Shaftman SR, Tilley BC. Demographic and clinical factors associated with in-hospital death among patients with systemic sclerosis. J Rheumatol. 2005; 32: 1888– 1892. [PubMed] [Google Scholar]
- 30. Oh SJ, Morgan MB, Lu L, Hatanaka Y, Hemmi S, Young A, Claussen GC. Racial differences in myasthenia gravis in Alabama. Muscle Nerve. 2009; 39: 328– 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Okayasu I, Hara Y, Nakamura K, Rose NR. Racial and age-related differences in incidence and severity of focal autoimmune thyroiditis. Am J Clin Pathol. 1994; 101: 698– 702. [DOI] [PubMed] [Google Scholar]
- 32. Osler W. The Principles and Practice of Medicine. 4th ed New York: Appleton; 1901. [Google Scholar]
- 33. Peters MG, Di Bisceglie AM, Kowdley KV, Flye NL, Luketic VA, Munoz SJ, Garcia-Tsao G, Boyer TD, Lake JR, Bonacini M, Combes B. Differences between Caucasian, African American, and Hispanic patients with primary biliary cirrhosis in the United States. Hepatology. 2007; 46: 769– 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peto R, Peto J. Asymptotically efficient rank invariant procedures. J R Statist Soc [A]. 1972; 135: 185– 207. [Google Scholar]
- 35.Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980; 23: 581– 590. [DOI] [PubMed] [Google Scholar]
- 36. Sharif R, Fritzler MJ, Mayes MD, Gonzalez EB, McNearney TA, Draeger H, Baron M, Furst DE, Khanna DK, del Junco DJ, Molitor JA, Schiopu E, Phillips K, Seibold JR, Silver RM, Simms RW, Perry M, Rojo C, Charles J, Zhou X, Agarwal SK, Reveille JD, Assassi S, Arnett FC. Anti-fibrillarin antibody in African American patients with systemic sclerosis: immunogenetics, clinical features, and survival analysis. J Rheumatol. 2011; 38: 1622– 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steen V, Domsic RT, Lucas M, Fertig N, Medsger TA. A clinical and serologic comparison of African-American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012; 64: 2986– 2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steen VD, Costantino JP, Shapiro AP, Medsger TA., Jr Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990; 113: 352– 357. [DOI] [PubMed] [Google Scholar]
- 39. Steen VD, Medsger TA., Jr Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000; 133: 600– 603. [DOI] [PubMed] [Google Scholar]
- 40. Traub YM, Shapiro AP, Rodnan GP, Medsger TA, McDonald RH, Jr, Steen VD, Osial TA, Jr, Tolchin SF. Hypertension and renal failure (scleroderma renal crisis) in progressive systemic sclerosis. Review of a 25-year experience with 68 cases. Medicine (Baltimore). 1983; 62: 335– 352. [DOI] [PubMed] [Google Scholar]
- 41. Tuffanelli DL, Winkelmann RK. Systemic scleroderma, a clinical study of 727 cases. Arch Dermatol. 1961; 84: 359– 371. [DOI] [PubMed] [Google Scholar]
- 42. Velayos EE, Masi AT, Stevens MB, Shulman LE. The “CREST” syndrome. Comparison with systemic sclerosis (scleroderma). Arch Intern Med. 1979; 139: 1240– 1244. [DOI] [PubMed] [Google Scholar]
- 43. Wagenknecht LE, Roseman JM, Alexander WJ. Epidemiology of IDDM in black and white children in Jefferson County, Alabama, 1979–1985. Diabetes. 1989; 38: 629– 633. [DOI] [PubMed] [Google Scholar]