

Abstract
Diffuse eosinophilic fasciitis (Shulman disease) is a rare sclerodermiform syndrome that, in most cases, resolves spontaneously or after corticosteroid therapy. It has been associated with hematologic disorders, such as aplastic anemia. The clinical features and long-term outcomes of patients with eosinophilic fasciitis and associated aplastic anemia have been poorly described. We report the cases of 4 patients with eosinophilic fasciitis and associated severe aplastic anemia. For 3 of these patients, aplastic anemia was refractory to conventional immunosuppressive therapy with antithymocyte globulin and cyclosporine. One of the patients received rituximab as a second-line therapy with significant efficacy for both the skin and hematologic symptoms. To our knowledge, this report is the first to describe rituximab used to treat eosinophilic fasciitis with associated aplastic anemia.
In a literature review, we identified 19 additional cases of eosinophilic fasciitis and aplastic anemia. Compared to patients with isolated eosinophilic fasciitis, patients with eosinophilic fasciitis and associated aplastic anemia were more likely to be men (70%) and older (mean age, 56 yr; range, 18–71 yr). Corticosteroid-containing regimens improved skin symptoms in 5 (42%) of 12 cases but were ineffective in the treatment of associated aplastic anemia in all but 1 case. Aplastic anemia was profound in 13 cases (57%) and was the cause of death in 8 cases (35%). Only 5 patients (22%) achieved long-term remission (allogeneic hematopoietic stem cell transplantation: n = 2; cyclosporine-containing regimen: n = 2; high-dose corticosteroid-based regimen: n = 1).
INTRODUCTION
As first described by Shulman in 1974,112 eosinophilic fasciitis (EF) is a rare connective tissue disease characterized by symmetrical swelling and progressive thickening and stiffness of the subcutaneous tissue, leading to a dimpled, “peau d’orange” presentation of the skin. Myalgia, inflammatory polyarthralgia, pedal and lower extremity edema and morphea are also commonly reported.75 The hands may be affected by skin sclerosis, but facial involvement is rarely observed. Visceral involvement, Raynaud phenomenon, telangiectasia, calcinosis cutis, and nail-fold capillaroscopy abnormalities are very uncommon in EF,53 usually enabling its distinction from systemic sclerosis. In up to half of the cases, the onset of symptoms seems to follow a vigorous level of exercise to which the patient was unaccustomed. Peripheral eosinophilia and hypergammaglobulinemia are often present.75 A definitive diagnosis relies on histopathologic observation of modifications of the fascia and lower subcutis, including edema and infiltration by plasma cells, lymphocytes, histiocytes, and eosinophils; later, these changes manifest as thickening and collagenization of the fascia. These alterations can extend into the dermis and underlying muscle.8 The dermatologic prognosis after corticosteroid therapy is usually good, with complete remission in most patients, yet persistent disability resulting from residual fibrosis occurs in 29%–42% of cases.37,76
EF is sometimes associated with hematologic diseases, particularly with aplastic anemia (AA) (n = 19),2,13,15,22,24,30,33,38,57,58,73,77,89,95,111,117,128 but also with T-cell lymphoma (n = 5),27,36,65,72,83 cutaneous T-cell lymphoma (n = 1),25 Hodgkin disease (n = 3),84,90,98 myeloproliferative syndromes (n = 3),61,75,85 myelomonocytic leukemia (n = 2),75,85 chronic lymphocytic leukemia (n = 2),12,75 multiple myeloma (n = 1),68 and myeloblastic leukemia (n = 1),90 and, less commonly, with solid tumors such as breast cancer (n = 5),12,90,109,127 choroidal melanoma (n = 1),125 colorectal cancer (n = 1),94 and prostate cancer (n = 1).90 Diffuse EF has also been reported in association with autoimmune disorders, such as Hashimoto thyroiditis (n = 6),2,5,13,59,114 systemic lupus erythematosus (n = 4),6,43,45,74 Crohn disease (n = 1),82 Graves disease (n = 1),114 glomerulonephritis (n = 1),63 rheumatoid arthritis (n = 1),81 type 1 diabetes (n = 1),46 and autoimmune cytopenias, including autoimmune hemolytic anemia (n = 2),5,44 immune thrombocytopenic purpura (n = 2),5,111 amegakaryocytic thrombocytopenia (n = 2),26,48 and pure red-cell aplasia (n = 1).81
It is still uncertain whether AA associated with EF is an autoimmune disease and/or the initial manifestation of an evolving clonal myeloid disorder. Among the 19 reported patients with EF and associated AA,2,13,15,22,24,30,33,38,57,58,73,77,89,95,111,117,128 8 died of complications from AA. Although most of these deaths occurred in patients receiving corticosteroids and/or antithymocyte globulin (ATG)-based regimens (without cyclosporine A [CsA]) in the 1980s, the current conventional immunosuppressive therapy of ATG and CsA was ineffective in 3 of 6 (50%) cases. We report 4 patients with severe aplastic anemia (SAA) and EF and provide a comprehensive review of the literature, focusing on clinical presentation, therapeutic challenges, and the outcomes of AA associated with EF.
PATIENTS AND METHODS
Between 1996 and 2012, 4 patients with EF and associated SAA were analyzed retrospectively at 4 French university hospitals. All of the patients had clinical and histopathologic features of EF, together with pancytopenia and, upon bone marrow examination, marked hypocellularity, and they fulfilled the established criteria for SAA diagnosis.19 Two of these patients15,33 have been previously reported, and we provide additional information on their clinical features and long-term follow-up.
We searched the National Library of Medicine’s MEDLINE database (Bethesda, MD) for relevant literature using the keywords “fasciitis” and “Shulman syndrome” together with “aplastic anemia” and “pancytopenia.” The bibliographies of all the selected articles were reviewed for additional case reports. We selected 19 patients from 15 different articles published between 1978 and 2009 in the English, French, German, and Portuguese literature.2,13,22,24,30,38,57,58,73,77,89,95,111,117,128 Patients were selected if they displayed clinical features of EF and pancytopenia and if AA was confirmed by bone marrow examination. The diagnosis of EF was confirmed by a deep skin biopsy, including the fascia, in all but 2 cases. In 1 case, a deep skin biopsy was not performed because of the risk of bleeding due to profound refractory thrombocytopenia;24 in another case,117 the deep skin biopsy was not conclusive, but it was performed after corticosteroid and ATG administration.
The clinical characteristics of the patients in the present and in previous reports were recorded and compared to those of 86 patients with EF and without AA from the benchmark Lakhanpal clinical series (n = 52)75 and from a more recent retrospective clinical study (n = 34).76 Three major caveats should be noted regarding the interpretation: there was a lack of in-depth clinical descriptions in some cases, there was possible over-reporting of unusual clinical features in the case reports compared to the clinical series, and there was a lack of long-term follow-up data in many of the case reports. Therefore, a statistical analysis was not performed.
When available, skin and hematologic outcomes were assessed using the following criteria: remission (defined as the absence of residual clinical signs of EF or as transfusion independence), long-term remission (longer than 2 yr after the start of the treatment), improvement (without complete remission), no improvement, and AA-related death.
CASE REPORTS
Patient 1
A 65-year-old retired beautician experienced petechiae, nosebleeds, and hemorrhagic bullae of the oral mucosa. She reported a 6-month history of asthenia, weight loss, myalgia, and progressive stiffness of the skin, which the patient reported had started after a swim in a river. She had been exposed to pentachlorophenol and lindane for 20 years, which are 2 pesticides used to treat lumber beams in her house. A physical examination revealed firm induration of the subcutaneous tissue, including the digits but sparing the face. There was a peau d’orange appearance of the back and venous furrowing (groove sign) ofthe anterior part of the forearms. There was no evidence of Raynaud phenomenon, dyschromia, telangiectasia, calcinosis cutis, or gastroesophageal reflux. A complete blood count disclosed pancytopenia (Hb: 8.8 g/dL; reticulocytes: 27 g/L; leukocytes: 2 g/L: neutrophils: 0.55 g/L; and platelets: 11 g/L). A bone marrow biopsy showed global hypoplasia with T lymphocyte and eosinophil infiltration. No paroxysmal nocturnal hemoglobinuria (PNH) clone was detected by flow cytometry. The antinuclear antibody titer was 1:80, and no antibodies to extractable nuclear antigens were detected, including anti-Scl70 or anticentromere antibodies. The direct Coombs test was positive with an anti-IgG antibody reagent. When the patient was admitted to the hospital, she required daily platelet transfusions, partly due to anti-HLA class I allo-sensitization. She received 3 mg/kg ATG and 2 mg/kg prednisone for 5 days and was discharged on 340 mg of oral CsA daily, weekly subcutaneous romiplostim injections, monthly intravenous immunoglobulin infusions, and platelet transfusional support 3 times per week. Six months later, her skin abnormalities had completely resolved. After transient hematologic improvement, a relapse occurred, and she required weekly platelet transfusions for 9 months after ATG treatment. Blood marrow aspiration showed persistent severe hypoplasia with 20% cellularity.
Patient 2
After a week of snowshoeing, a 57-year-old farm worker developed asthenia, myalgia, and rapidly progressive thickening of the skin on the trunk and on all 4 limbs. A physical examination revealed diffuse scleroderma, except on his face, with peau d’orange presentation of the abdomen (Figure 1) and thighs and venous furrowing of the forearms. The eosinophil count was 2.6 g/L. Three months after the onset of these symptoms, a full-thickness skin and muscle biopsy from the arm revealed perivascular lymphoplasmacytic infiltration of the hypodermis, fascia, and perimysium, indicative of EF (Figure 2). The patient was discharged on prednisone (1 mg/kg daily). Two months later, he was readmitted with diffuse petechiae and ecchymoses of the lower limbs. His platelet count was 7 g/L, hemoglobin 9.9 g/dL, reticulocytes 55 g/L, leukocytes 1.2 g/L, and neutrophils 0.4 g/L. Bone marrow aspiration disclosed severe hypocellularity (<20%) with a complete absence of megakaryocytes (Figure 3). He received ATG for 5 days, and a daily oral dose of 360 mg CsA was started. The CsA dose was tapered to 200 mg daily 6 months later after the onset of an axonal sensory polyneuropathy of the lower limbs, presumably of toxic origin. One year later, his skin condition did not improve, and he still required weekly transfusional support related to persistent anemia (Hb: 10.4 g/dL) and thrombocytopenia (platelets: 9 g/L). A second bone marrow aspiration disclosed moderate hypocellularity and an absence of megakaryocytes. He was given rituximab at a dose of 375 mg/m2 per week for 4 weeks, followed by cytopenia regression (Hb: 12 g/dL and platelets: 97 g/L) and transfusion withdrawal within 3 months. A relapse occurred, however, 6 months after the first course, and he received 4 more infusions of rituximab using the same protocol. Once again, the cytopenias improved, and transfusional support could be withdrawn. After the second course, the skin abnormalities resolved completely. CsA was tapered to 80 mg daily, and no relapse occurred during the 6 months of additional follow-up.
FIGURE 1.
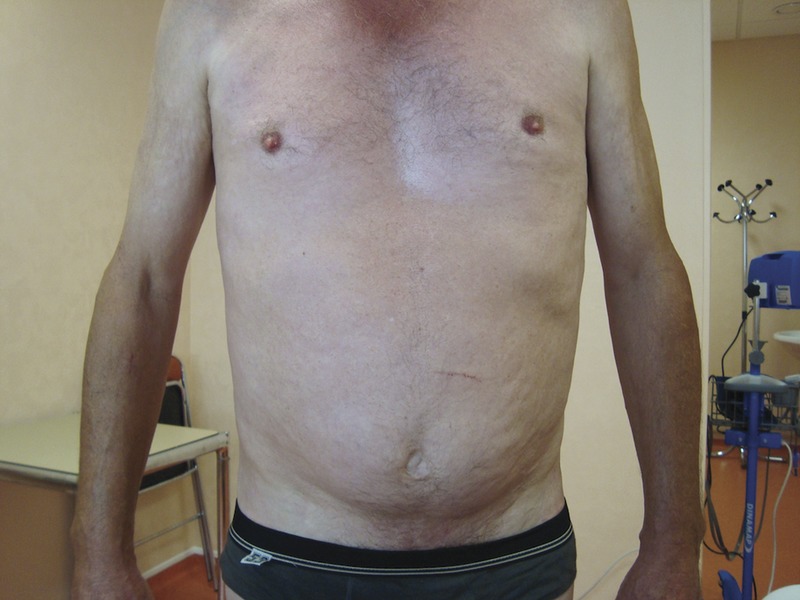
Patient 2. Dimpled, peau d’orange aspect of the abdomen. [This figure can be viewed in color online at http://www.md-journal.com].
FIGURE 2.
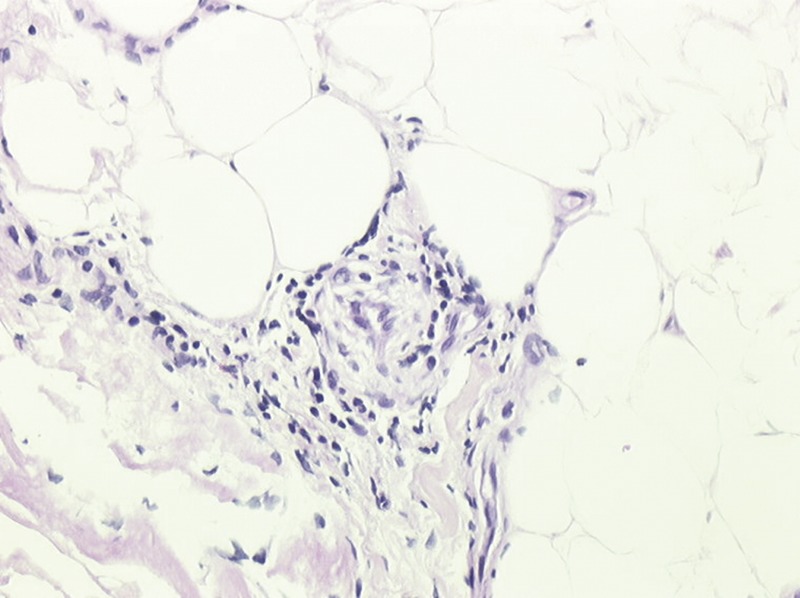
Patient 2. Full-thickness skin and muscle biopsy from the arm. Lymphoplasmocytic infiltrate of the fascia and lower subcutis with few eosinophils, consistent with EF at early stage (hemalun-eosin stain, ×250 magnification). [This figure can be viewed in color online at http://www.md-journal.com].
FIGURE 3.
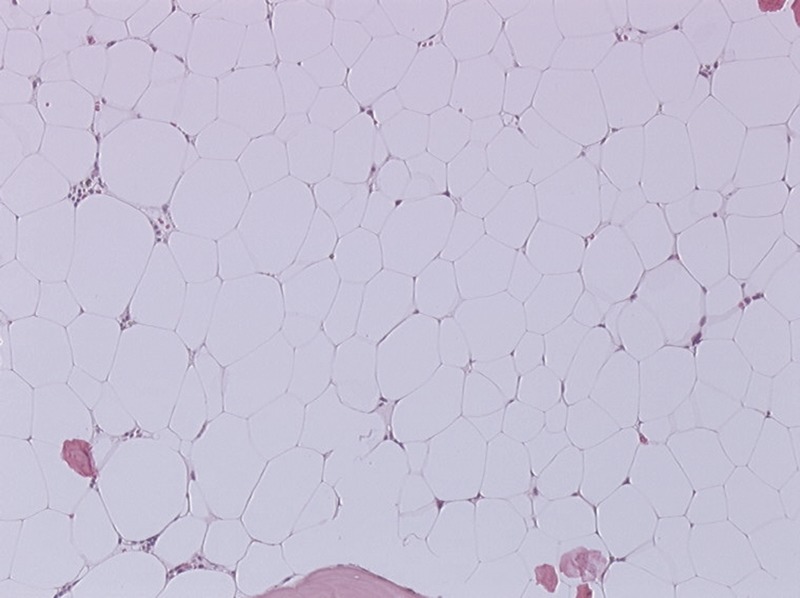
Patient 2. Bone marrow biopsy. Fatty, severely hypoplastic marrow with complete absence of megacaryocytes and a discrete lymphocytic inflammatory infiltrate (hemalun-eosin stain, ×100 magnification). [This figure can be viewed in color online at http://www.md-journal.com].
Patient 3
A 35-year-old bricklayer experienced a rapid onset (a few days) of painful swelling of the limbs, arthralgia, and myalgia. He reported no previous physical stress. A clinical examination revealed thickening of the skin and subcutaneous tissue involving the trunk and all 4 limbs, including the digits. Laboratory studies revealed eosinophilia (1.15 g/L), an elevated erythrocyte sedimentation rate (23 mm/h), and polyclonal hypergammaglobulinemia (20 g/L). The antinuclear antibody titer was <1:80, with no antibodies to extractable nuclear antigens, including anti-Scl70 and anticentromere antibodies. A leg fascial biopsy showed a mononuclear infiltration of lymphoplasmocytes and eosinophils involving the perimysial tissue and the fascia, confirming the diagnosis of diffuse EF. The epidermis was normal, whereas thickened collagen bundles were observed in the dermis as were straightening with parallel orientation of the elastic fibers. Daily treatment with 1 mg/kg prednisone was started. Eight months later, he experienced massive hematemesis and was admitted to the intensive care unit. An esophago-gastroduodenoscopy revealed a normal esophagus, hematin-covered gastric lesions and a gastric ulcer with an adherent clot. His full blood count showed aplastic anemia (hemoglobin 8.3 g/dL, reticulocytes 36 g/L), platelets 3 g/L, and leukocytes 1.8 g/L. A bone marrow biopsy confirmed the diagnosis of SAA. There was no PNH clone detected. After 9 months of prednisone therapy, his skin condition did not improve, and the patient required blood and platelet transfusions every 2 weeks. A clinical examination showed diffuse sclerosis of the skin and subcutaneous tissue, including sclerodactyly, affecting 90% of the total body surface area (Figure 4A). Venous furrowing (groove sign) was visible on the forearms, and the skin seemed dimpled with a peau d’orange appearance. The nipples, palms, and plantar surfaces were not affected. The edema and arthralgia resolved. A careful examination of the neck revealed patchy, ivory, atrophic lesions with a pigmented peripheral halo, indicative of guttate morphea. Raynaud phenomenon was present. Microhemorrhages, edema, giant capillaries, a decreased number of capillary loops and avascular areas were found on nail-fold capillaroscopy consistent with an organic microangiopathy. Neither gastroesophageal reflux nor renal failure was observed. Echocardiography revealed increased systolic pulmonary arterial pressure (41 mm Hg) with normal left ventricular function. The lung diffusion capacity for carbon monoxide was reduced (47% of the predictive value). High-resolution computed tomography of the chest was normal. The skin abnormalities gradually subsided (Figure 4B), but immunosuppressive therapy (2 courses of ATG plus CsA 5 mg/kg) failed to improve the aplasia after 24 months of follow-up. Unfortunately, no compatible donor was found for allogeneic hematopoietic stem cell transplantation (HSCT). The patient had severe Pseudomonas aeruginosa mastoiditis that required surgery and antibiotic treatment with partial sterilization; he then developed invasive pulmonary aspergillosis. He was still alive at the last follow-up.
FIGURE 4.
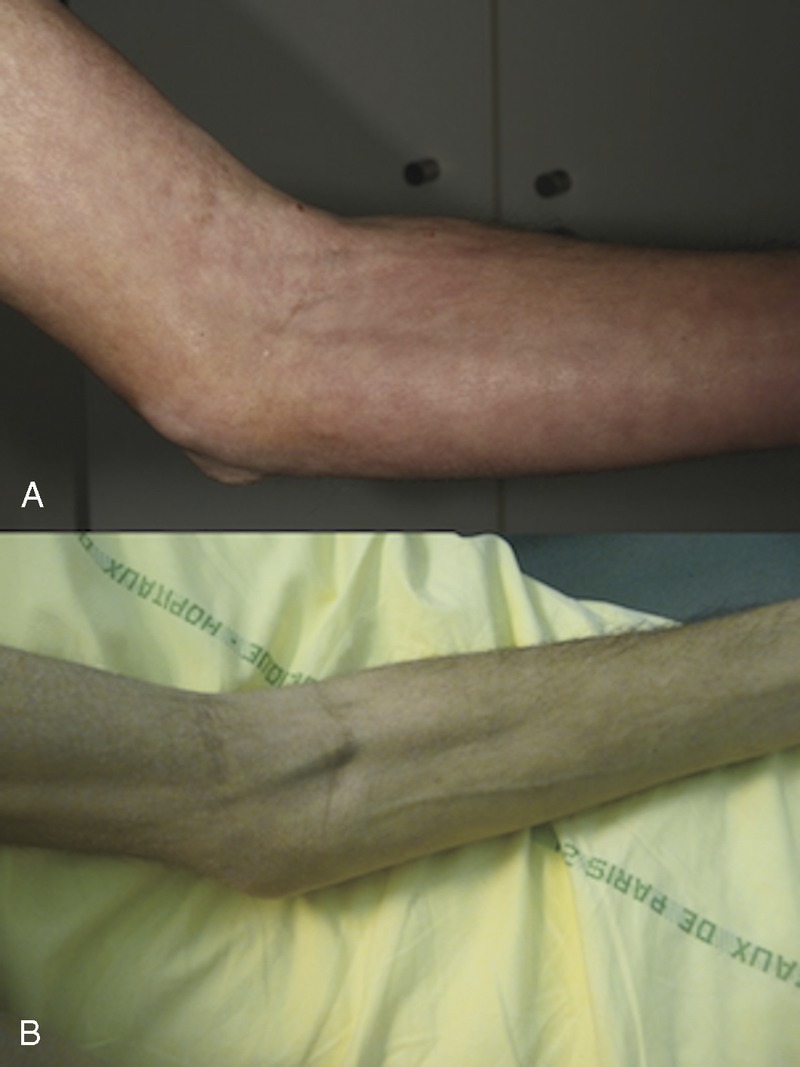
Patient 3. Redness, warmth, and woody induration of the skin of the left forearm (top). After 12 months of immunosuppressive therapy (bottom). Marked softening of the skin; veins have become visible. [This figure can be viewed in color online at http://www.md-journal.com].
Patient 4
A 60-year-old man developed asthenia, weight loss, myalgia, pruritus, and progressive skin stiffness of his abdomen, limbs, and the superior part of the back. He was an insurer and reported no long-term exposure to toxins and no previous physical stress. His past history included a deep vein thrombosis of the left leg 3 months earlier and ulcerative colitis in remission. He was diagnosed with ulcerative colitis 6 years ago and was treated with corticosteroids; the dose had been decreased from 20 to 1 mg daily during the previous year. A physical examination showed thickening of the subcutaneous thigh tissue and deep morphea of the legs and forearms. Hypo- and hyperpigmentation of the thighs were also observed. His face and digits were not affected, and there was no Raynaud phenomenon. The nail-fold capillaroscopy was normal. Fluctuating eosinophilia (maximum: 2.5 g/L) and an elevated erythrocyte sedimentation rate (50 mm/h) had been found during the onset of symptoms. Antinuclear antibodies, antibodies to extractable nuclear antigens, including anti-Scl70 and anticentromere antibodies, were absent. A deep skin biopsy showed edema and thickened collagen bundles in the hypodermis as well as a dense lymphoplasmacytic infiltrate, mainly around the capillaries in the hypodermis, fascia, and muscle walls. The same alterations were also noticed in the deep dermis, whereas the epidermis appeared normal. The corticosteroid dose was increased to 80 mg daily with no efficacy after 3 months. He was readmitted for fever 8 months after the onset of these symptoms, and a complete blood count disclosed aplastic anemia (Hb: 7.6 g/dL), leukopenia (leukocytes: 2.9 g/L), neutropenia (neutrophils: 0.7 g/L), and severe thrombocytopenia (platelets: 30 g/L). Antiplatelet glycoprotein IgG antibodies, detected using the monoclonal antibody-specific immobilization of platelet antigens assay, were present. Bone marrow aspiration revealed severe hypocellularity (<20%) and an absence of megakaryocytes. Fever resolution occurred after the patient was placed on empirical antibiotic therapy. He received ATG (300 mg/d) and CsA (4 mg/kg per d) for 5 days. One year later, his skin and hematologic conditions had normalized. The CsA dosage was gradually decreased and was stopped 8 years later. Four years after CsA withdrawal, he developed autoimmune hemolytic anemia (Hb: 7.6 g/dL; reticulocytes: 101 g/L; LDH: 552 IU/L, Direct Coombs test: positive using anti-IgG antibody reagent), which responded well to oral corticosteroids.
DISCUSSION AND LITERATURE REVIEW
Age, Sex, and Environmental Factors
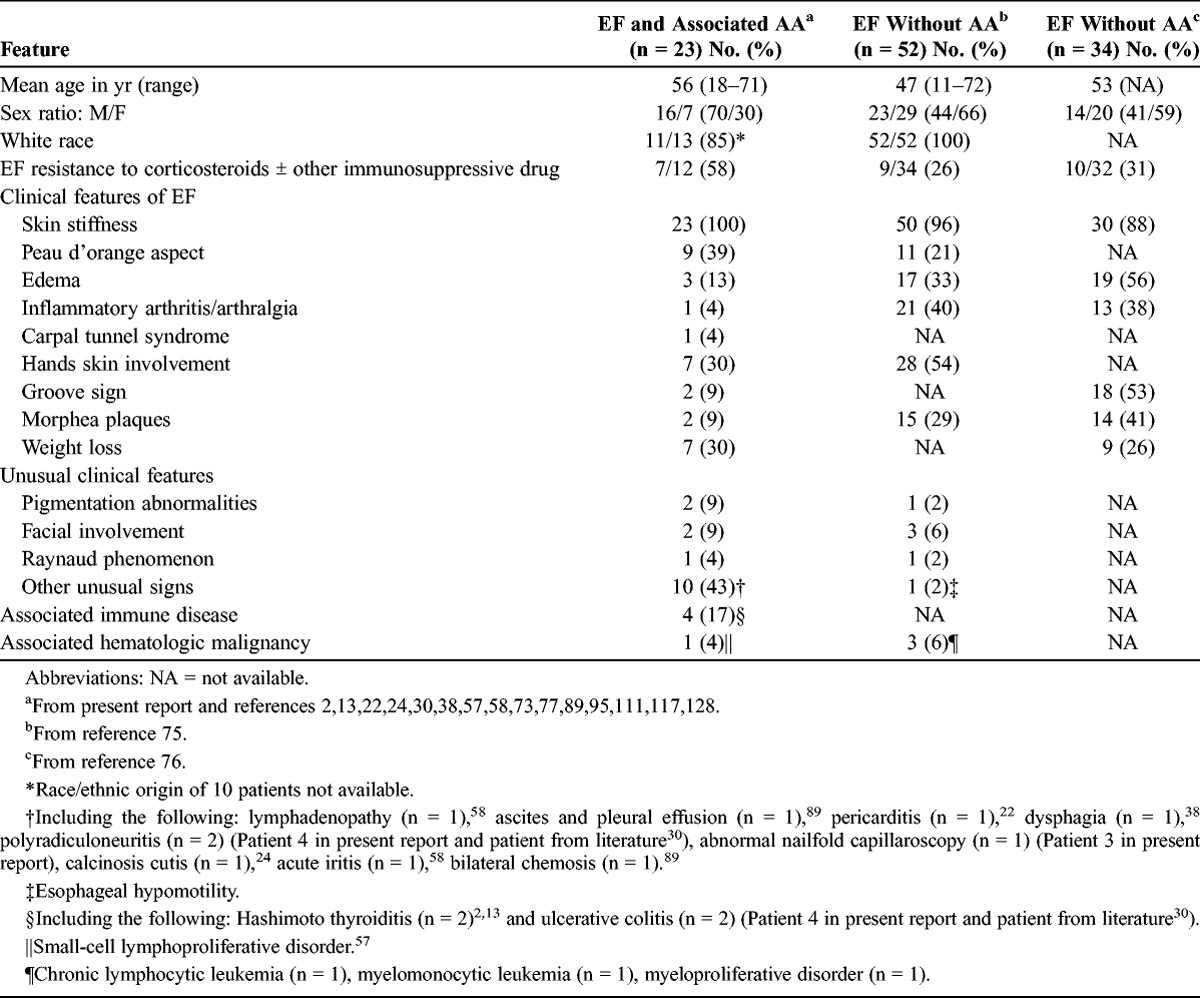
Among the 23 patients with EF and concomitant AA from previous and present reports, most were aged more than 60 years (Table 1). The mean age was 56 years (range, 18–71 yr). Only 3 patients were under 40 years of age when they were diagnosed with EF. Of the 23 patients, 16 (70%) were men. These results contrast with the clinical characteristics of EF patients without associated AA,75,76 who tended to be younger (mean age, 47 yr75 or 53 yr76) and were more often women (66%75 or 59%76). As in the Lakhanpal case series, patients with EF and AA were most often white.
TABLE 1.
Clinical Features of Patients With EF and Associated AA and Patients With EF Without Associated AA
The professions of 11 patients were specified. Eight were manual workers: 3 were farm workers (Patient 2 and patients reported previously58,77), 1 a foundry worker,58 1 a dry cleaner,58 1 a mechanic,38 1 a brick mason (Patient 3), and 1 a beautician (Patient 1). Patient 1 reported exposure to lindane and pentachlorophenol for 20 years, which are 2 organochlorine pesticides used for wood preservation; these agents have been implicated in other reported cases of isolated AA.16,99 We are unaware of other reported cases of EF and AA following exposure to pesticides, but 1 case of AA and diffuse scleroderma following exposure to paradichlorobenzene and naphthalene in an employee of a clothing resale shop has been reported.50 These observations led us to speculate on the potential role of toxic exposure in the onset of the disease, although it must be remembered that no definitive conclusion can be drawn based on relatively few cases.
Benzene exposure80 and exposure to agricultural pesticides60 have been clearly shown to be linked to AA in case-control studies. The role of environmental factors has also been suspected in the context of EF, although no case-control study is available. In case reports, EF has been linked to infection with Borrelia species,2,9,47,51,87,107,116 brucellosis,90 chronic hepatitis C,90 tertiary syphilis,90 chemical exposure,11 trichloroethylene,52 insect bites,79,90 irradiation,109 and various pharmaceuticals, including simvastatin,28,108 phenytoin,18 atorvastatin,31 fosinopril,10 alpha-methyldopa,90 subcutaneous heparin use,20 and antituberculosis therapy.104 Geographical clustering of scleroderma-like syndromes, including EF, was reported in a rural area in Italy.124 More importantly, eosinophilia-myalgia syndrome, which includes myalgia, scleroderma, constant peripheral eosinophilia, fasciitis in 25%–55% of cases, and sometimes severe peripheral neuropathies, is linked to L-tryptophan ingestion.54 Similarly, adulterated cooking oils have induced toxic-oil syndrome, which presents with myalgia, diffuse scleroderma, and peripheral eosinophilia, yet to our knowledge no associated fasciitis has ever been reported.70
Clinical Features at EF Diagnosis
Progressive skin stiffness was present in all 23 patients (see Table 1), with peau d’orange presentation in 9 cases. The digits were involved in 7 cases. Seven patients also reported weight loss, although this symptom was not reported in patients with EF from the Lakhanpal case series.75 It is likely, however, that it was simply not recorded in that particular case series; it was reported in 9 of 34 patients with EF (26%) in the retrospective clinical study from Lebeaux et al.76
Other clinical features of EF, such as pitting edema of the lower legs, inflammatory arthritis, a groove sign, carpal tunnel syndrome, and morphea plaques, were rarely reported in patients with EF and AA, but this finding could be related to the scarcity of in-depth clinical descriptions.
Uncommon clinical features of EF were found in 10 patients. One patient had lymphadenopathy.58 A lymph-node biopsy showed lymphoid and reticular hyperplasia. Lymphadenopathy is rarely associated with EF; in a case series of 10 patients with EF and peripheral lymphadenopathy,91 lymph-node biopsies revealed lymphoma in 6 cases.
Two patients exhibited seritis, including ascites and pleural effusion89 or pericarditis.22 Rare cases of EF with pleural71 or pericardial92 involvement, or both,97 have been described previously. Two patients had peripheral neuropathy (our Patient 2 and a patientfrom the literature30). However, in our patient, the role of cyclosporine toxicity could not be excluded. Three cases of EF with associated peripheral neuropathy have been reported.86,101,106
One patient had acute, unilateral nongranulomatous iritis,58 and 1 patient had bilateral chemosis.89
One patient had dyschromia, microstomia, and dysphagia.38 Similarly, Patient 3 had dyschromia, Raynaud phenomenon, and nail-fold capillaroscopy abnormalities. Finally, 1 patient exhibited calcinosis cutis.24 These features are unusual in EF and can complicate the differentiation of this disease from systemic sclerosis. Moreover, apart from EF, 9 cases of diffuse scleroderma have been reported in association with AA7,14,21,29,39,41,66,118,122 or amegakaryocytic thrombocytopenia.67 None of these patients had been diagnosed with EF. One patient had Raynaud phenomenon.39 One had lung carcinoma and esophageal hypomotility.41 Another patient29 had Crohn disease, Raynaud phenomenon, esophageal dysmotility, and interstitial lung disease. These findings further blur the boundaries between EF, diffuse morphea, and systemic sclerosis, as these 3 diseases seem to be associated with AA; however, AA is more frequently associated with EF than the 2 other conditions.
Associated Immune Diseases
Four patients (17%) had prior immune disease diagnoses. Two patients had Hashimoto thyroiditis,2,13 and 2 others (Patient 4 and a patient reported previously30) had ulcerative colitis. Hashimoto thyroiditis5,13,59,114 and Crohn disease82 have been previously reported as associated with EF but not AA in some case reports. AA is rarely associated with other immune diseases; in a retrospective survey of 1251 patients with AA, only 50 (4%) had prior autoimmune disease diagnoses.23 AA has been reported, however, in association with autoimmune enteropathy.100
Hematologic Involvement
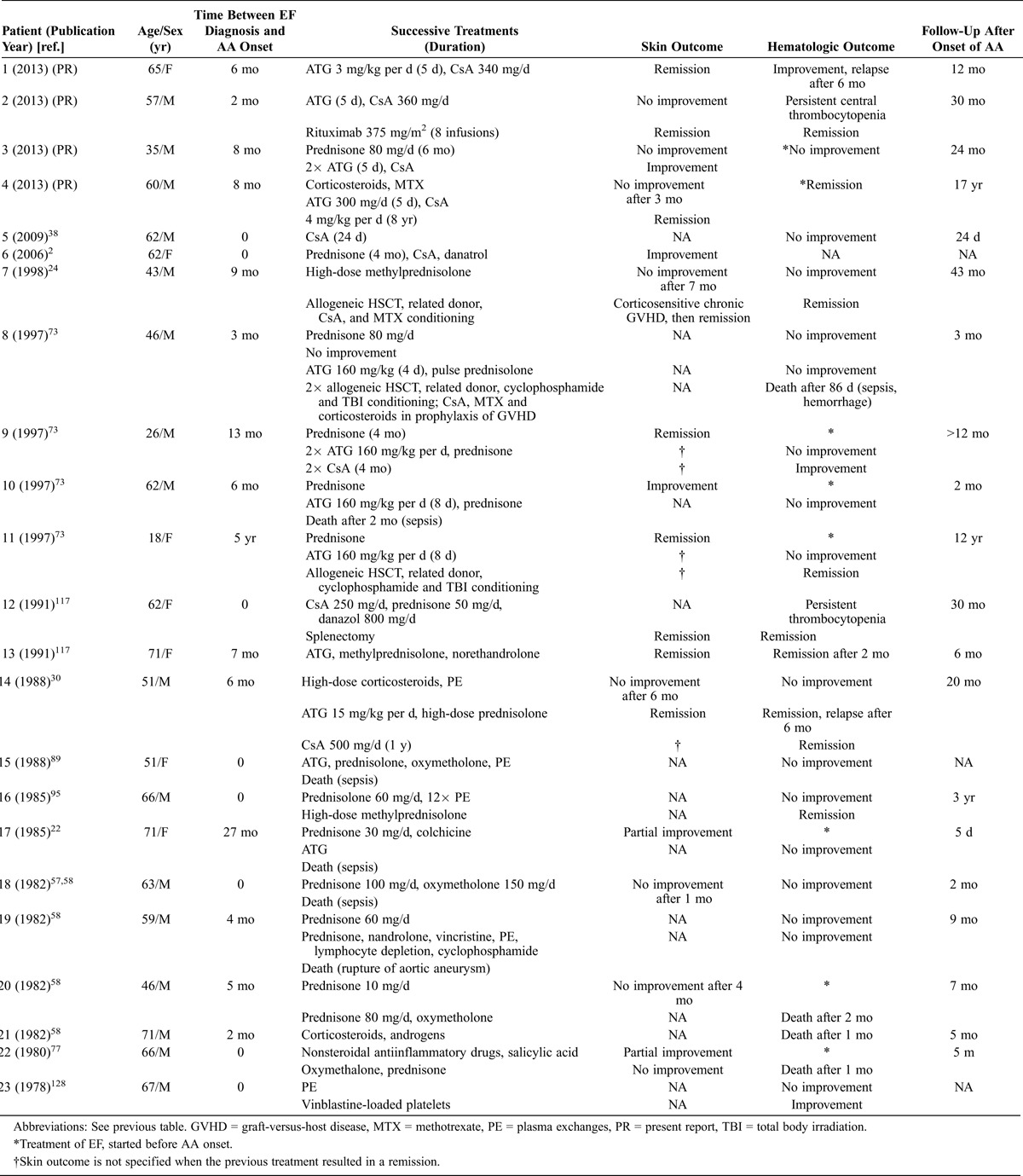
In most cases, the interval between the diagnosis of EF and the onset of AA was less than 6 months (mean delay, 7.2 mo; median, 4 mo; range, 0–60 mo) (Table 2). In 8 cases, EF and AA were diagnosed simultaneously. AA was revealed by cutaneomucosal hemorrhagic syndrome in 14 cases. Among these cases, 2 patients experienced massive gastrointestinal bleeding (hematemesis in Patient 3, hematemesis and melena in a patient from the literature24). In 3 cases, the patients presented with sepsis caused by undocumented pneumonia (Patient 4 and from the literature73) and Streptococcus fecalis periorbital cellulitis.77 In 5 patients, AA was discovered fortuitously by a complete blood count. Marrow hypocellularity was described as “profound,”30 “marked,”58 or “severe”58,73,89 in 7 cases. Marrow cellularity was <20% in 6 additional cases (4 patients in the current study and 2 previously described22,24) and was 30% in another case;128 cellularity was not specified in the remaining cases. Two patients also had positive direct Coombs test results (Patient 1 and a patient previously described58). Patient 4 tested positive for antiplatelet glycoprotein IgG antibodies, detected using the monoclonal antibody-specific immobilization of platelet antigens assay, and this patient developed autoimmune hemolytic anemia several years later. Yet another patient128 had platelet IgG antibodies as determined by complement lysis inhibition.
TABLE 2.
Clinical Characteristics, Management, and Outcome of 23 Patients With EF-Associated AA
Normal marrow cultures from healthy donors in the presence of diseased patient’s serum were performed in 4 cases.57,73,128 There was 90% inhibition of CFU-GM growth in the first case,73 no inhibition of myeloid progenitor cell growth in the second,73 inhibition of BFU-E and CFU-E growth in the third,128 and 92%, 100%, and 83% inhibition of CFU-M, CFU-E, and BFU-E growth, respectively, in the fourth case.57 Inhibition of myeloid progenitor cell growth was found to be restricted to the IgG fraction of the patient’s serum in the latter experimental condition.57 Co-cultures of the patient’s and healthy donors marrow cells were performed in 5 patients,73,128 and stem cell colony growth suppression was absent in all but 1 case, in which growth of CFU-E was 40%–75% of the predicted value.128 Finally, AA can coexist or evolve into clonal disorders such as PNH, myelodysplasia, or acute myeloblastic leukemia.130 Detection of a PNH clone was sought but not found in 3 of our patients (Patients 1, 3, and 4). No clonal myeloid disorders were described in 23 patients with EF and AA. However, apart from this literature review, there have been 3 reports of EF patients who experienced pancytopenia in whom an excess of blast cells,48 an evolving myeloproliferative process,34 or myelomonocytic leukemia34 were found upon bone marrow examination.
Pathophysiology
In general, AA is considered an autoimmune disease involving interferon-γ-secreting Th1113 and IL-17-producing Th1732 CD4 T cells as well as oligoclonal cytotoxic CD8 T cells,96 leading to the destruction of autologous hematopoietic stem and progenitor cells.130 Moreover, regulatory T cells (Tregs), which control and suppress autoreactive T cells, are decreased at disease presentation in almost all AA patients, suggesting their involvement in AA pathophysiology.115 Specific autoantibodies to kinectin,56 diazepam-binding inhibitor-related protein 1,40 postmeiotic segregation-increased protein 1,55 and moesin120 have been found in the serum of patients with AA. Moesin is a cytoskeleton-membrane linker protein that is expressed on the surface of T cells, NK cells, and monocytes. Antimoesin antibodies have been shown to stimulate interferon-γ secretion from peripheral mononuclear cells retrieved from AA patients in vitro;119 therefore, these antibodies may contribute to AA pathophysiology. Abnormalities in telomere repair have also been identified in acquired AA patients.17 Telomeres are nucleotide repeats at the ends of the chromosomes that function as protective caps to prevent erosion of genomic DNA during cell division. Each time a cell divides, the telomeres shorten. When they become too short, the cell can no longer divide and becomes inactive. Critically short telomeres produce apoptosis, cell senescence, and chromosomal instability. Mutations in telomerase complex genes resulting in extremely short telomeres have been described in some patients with AA.129 In acquired AA, independent of known genetic alterations, the presence of short telomeres in leukocytes at the time of presentation affects the clinical course: patients with short telomeres respond to immunosuppressive interventions, but their relapse rate is almost double that of cases with normal telomere length. Additionally, virtually all clonal evolution occurs in patients in the lowest quartile of telomere length.103
EF is also believed to be an immune-mediated disease. This conclusion is supported by the following evidence: 1) histologic features (cytotoxic CD8 T-lymphocytic, monocytic, and eosinophilic infiltrates123 and IgG and C3 deposits8); 2) the disease’s association with other autoimmune diseases (autoimmune cytopenias, thyroiditis, or systemic lupus erythematosus); 3) the disease’s association with biologic abnormalities (polyclonal hypergammaglobulinemia75 and circulating immune complexes105); and 4) reported cases of fasciitis in the context of alloimmunity during chronic graft-versus-host disease after allogeneic stem cell transplantation.62 To date, few specific studies have been performed on the immune regulation of EF. However, this disease belongs to the group of localized scleroderma, also referred to as morphea or skin scleroderma,93 as illustrated by the association of plaque morphea with EF.75,76 Localized scleroderma is characterized by increased collagen deposition (fibrosis), which differs from systemic sclerosis in which collagen deposition and vasculopathy are predominant features. Nonetheless, systemic sclerosis shares clinical and pathophysiologic features with EF, such as increased serum levels of TGF-β, a potent profibrotic cytokine,35,42 and elevated levels of inhibitors of metalloproteinases in the affected tissues.64,69 Increased production of Th1 (that is, IL-2 and interferon-γ) and Th2 (that is, IL-5 and IL-10) cytokines have been found in EF patients after stimulation.126 Fewer circulating Tregs have been described in patients with systemic sclerosis and localized scleroderma in comparison with healthy individuals.3 Defects in telomerase biology have been described in systemic sclerosis with contradictory results.4,78 Dysregulation of B-cell homeostasis has been described in chronic graft-versus-host disease,110 and fasciitis similar in symptomology to EF is frequent in chronic graft-versus-host disease.62
To summarize, pathophysiologic links between EF and AA may involve the following: 1) increased CD8 and Th1/Th17 T-cell responses, 2) decreased Tregs, 3) abnormal telomere repair homeostasis, and 4) dysregulated B-cell responses with autoantibody production, which may explain the potential benefit of rituximab in 1 of our patients.
Treatment and Outcome
At least 8 patients died of complications from AA22,58,73,77,89 (Table 3). Death occurred after a mean interval of 4 months after the onset of AA (median, 2.5 mo; range, 1–9 mo). Six patients died of sepsis secondary to the following: gram-negative sepsis,22,73 Zygomycetes pneumonia,89 unspecified pneumonia,77 disseminated sepsis with cellulitis,58 and Escherichia coli and Streptococcus pneumoniae.58 One patient died of an unspecified infection and bleeding,73 1 died of an aortic aneurysm rupture,58 and 1 died of an intracranial hemorrhage.58 Five of these patients were treated with corticosteroids and various other drugs, including androgens but not ATG or CsA for AA.58,77 Additionally, 4 patients were treated with corticosteroids, ATG, and other drugs, excluding CsA.22,73,89 Among these 4 patients, 1 underwent allogeneic HSCT from an unrelated donor73 but eventually died 86 days after the transplantation.
TABLE 3.
Treatment Regimens and Outcome in 19 Patients With EF-Associated AA, Present and Previous Reports
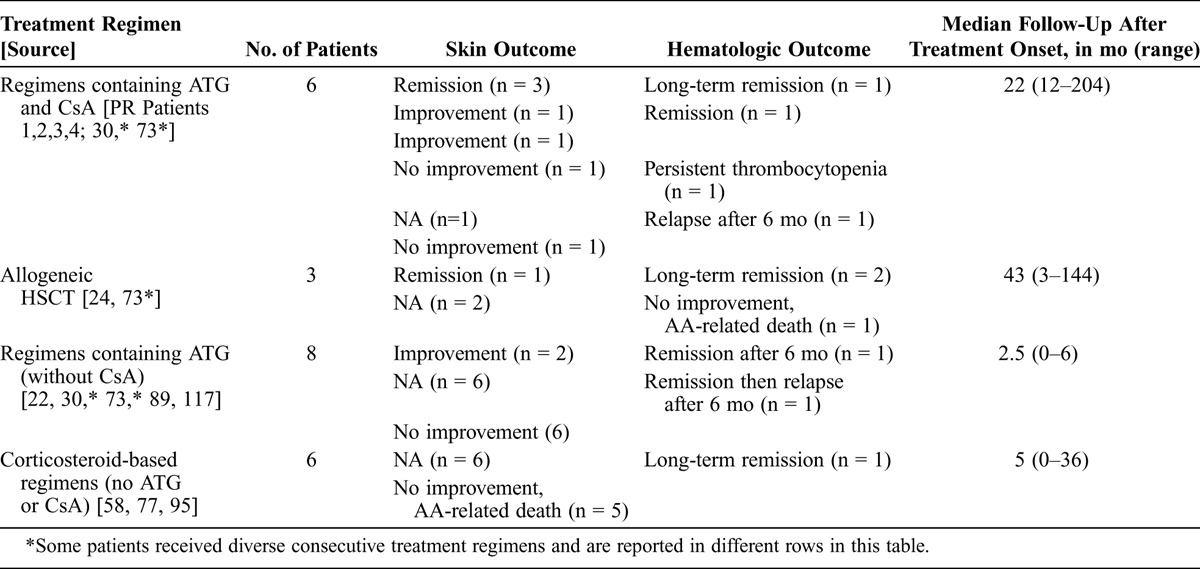
Long-term remission (>2 yr) was reported in only 5 patients. One was treated with high-dose corticosteroids and plasma exchanges (3 yr of follow-up),95 whereas corticosteroids alone or together with androgens failed to improve the aplasia in all other cases. Two patients were treated with HSCT from a sibling donor24,73 and had remission over 43 months of follow-up in the first case and 12 years in the second case. Patient 4 received a combination of CsA and ATG (17 yr of follow-up). The last patient with long-term remission was treated with CsA, corticosteroids and splenectomy (30 mo of follow-up after starting treatment).117
The combination of ATG and CsA is the current standard first-line therapy for patients with SAA and no available HLA-matched sibling donor.130 It has proven effective in randomized controlled trials, and an overall response is achieved in two-thirds of the patients, with a cumulative incidence of relapse of 20%–30% among responders. Allogeneic HSCT is considered a first-line therapy in patients with available HLA-matched related donors.130 Of the 6 patients with EF and AA who received ATG and CsA, 5 (83%) were partial (n = 3) or complete (n = 2) responders. Therefore, in this small number of patients with EF and SAA, the prognosis after standard immunosuppressive therapy seemed to be similar to that of isolated SAA in clinical trials.
Finally, Patient 2 underwent 2 courses of rituximab therapy (375 mg/m2 per infusion, 4 monthly infusions during each course) with remission of both the skin and hematologic disorders 15 months after treatment, whereas ATG and CsA alone did not show efficacy after 15 months. To our knowledge, rituximab has shown efficacy in EF treatment in only 1 other single reported case102 and in AA in 3 cases with 5 months,49 4 years,121 and 6 months1 of follow-up. The last case had AA related to systemic lupus erythematosus.
The evolutions of EF and AA were not always correlated. Unlike systemic sclerosis, EF is usually corticosteroid-sensitive. Thus, prednisone monotherapy proved effective in 42 of 55 patients (76%) in 4 recent studies2,12,76,88 and in 25 of 34 patients (74%) in the Lakhanpal case series.75 Among the 12 patients demonstrating AA associated with EF who were treated with corticosteroids as either monotherapy73 or in addition to ATG,117 CsA,2 or colchicine,22 and for whom skin outcome data were available, only 5 (42%) showed EF improvement2,22,73 or remission.117 In the 7 remaining cases (Patients 3 and 4 and patients from the literature24,30,58,77), no skin improvement was observed after corticosteroid therapy. It could be assumed that patients with EF and associated AA had more corticosteroid-resistant EF than patients with isolated EF; however, the mean delay between treatment initiation and the assessment of efficacy was short (mean, 3.5 mo; range, 1–6 mo), and the small number of patients does not allow us to draw firm conclusions.
Among the 4 patients who died of AA and for whom skin outcomes were available, 3 achieved partial improvement of EF,22,73,77 and 1 did not show improvement.58 In the same way, 2 of our patients achieved complete (Patient 1) or almost complete (Patient 3) skin remission; however, they were still reliant on transfusions after ATG and CsA therapy. The 8 remaining patients for whom AA and EF outcomes were available (Patients 2 and 4 and patients reported previously13,24,30,73,117) achieved both hematologic and skin improvement or remission. In the patients who had relapses of AA, no concomitant relapse of EF was described.
A small-cell lymphoproliferative disorder was revealed after the autopsy of 1 patient,57 but no other cases of evolving malignancy were described in the 18 remaining cases, nor were any found in our patients even after long-term follow-up.
Summary and Conclusions
We studied 4 patients with EF and associated SAA and reviewed 19 cases retrospectively. According to the data in the literature, AA seems to be the most frequently recorded hematologic disease associated with EF. It was the direct cause of death in at least 8 of 23 cases (35%). Compared to patients with isolated EF, patients with EF and associated AA were more likely to be men (70%) and older (mean age, 56 yr; range, 18–71 yr). It should be noted that unusual clinical features in the context of EF, such as systemic involvement (n = 7), Raynaud phenomenon (n = 1), calcinosis cutis (n = 1), and facial skin sclerosis (n = 2), were not rare in patients with EF and associated AA, even though they are usually considered to be the hallmark of systemic sclerosis. Four patients had other associated immune diseases: ulcerative colitis (n = 2), autoimmune thyroiditis (n = 2), and autoimmune cytopenias (n = 4). No clonal myeloid disorders were detected in these 23 patients.
The evolutions of EF and AA were not always correlated: remission of EF was not predictive of AA improvement, and no relapse of EF was observed in patients with AA relapses. Corticosteroid-containing regimens improved the skin condition in 5 of 12 cases (42%) but were ineffective in treating aplasia in 5 of 6 cases (83%). Among our 3 patients who were refractory to ATG and CsA, 1 had 2 courses of rituximab therapy with both skin and hematologic improvement. Finally, long-term remission (>2 yr) was reported in 5 cases with the following treatments: corticosteroid-containing regimen (n = 1), allogeneic HSCT from sibling donor (n = 2), and CsA-containing regimen (n = 2). In conclusion, patients with EF should be carefully monitored for associated AA, which occurs mostly in the first year after EF diagnosis. AA in this setting is usually severe and rapidly life-threatening in the absence of early immunosuppressive therapy. The response of AA to immunosuppressive therapy can be slow, even after EF remission, and AA relapse does occur. Therefore, long-term suppressive treatment with CsA is recommended. Allogeneic HSCT should be considered in patients with an available HLA-matched related donor.
Abbreviations
- AA
aplastic anemia
- ATG
antithymocyte globulin
- CsA
cyclosporine A
- EF
eosinophilic fasciitis
- HSCT
hematopoietic stem cell transplantation
- PNH
paroxysmal nocturnal hemoglobinuria
- SAA
severe aplastic anemia
- Tregs
regulatory T cells
Footnotes
*These authors contributed equally and are co-first authors.
†These authors contributed equally and are co-last authors.
All figures can be viewed in color online at http://www.md-journal.com.
The authors have no funding or conflicts of interest to disclose.
REFERENCES
- 1. Alishiri GH, Saburi A, Bayat N, Saadat AR, Saburi E. The initial presentation of systemic lupus erythematosis with aplastic anemia successfully treated with rituximab. Clin Rheumatol. 2012; 31: 381– 384. [DOI] [PubMed] [Google Scholar]
- 2. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Report of 11 patients and review of the literature. Dermatology. 2006; 213: 93– 101. [DOI] [PubMed] [Google Scholar]
- 3. Antiga E, Quaglino P, Bellandi S, Volpi W, Del Bianco E, Comessatti A, Osella-Abate S, De Simone C, Marzano A, Bernengo MG, Fabbri P, Caproni M. Regulatory T cells in the skin lesions and blood of patients with systemic sclerosis and morphoea. Br J Dermatol. 2010; 162: 1056– 1063. [DOI] [PubMed] [Google Scholar]
- 4. Artlett CM, Black CM, Briggs DC, Stevens CO, Welsh KI. Telomere reduction in scleroderma patients: a possible cause for chromosomal instability. Br J Rheumatol. 1996; 35: 732– 737. [DOI] [PubMed] [Google Scholar]
- 5. Bachmeyer C, Monge M, Dhote R, Sanguina M, Aractingi S, Mougeot-Martin M. Eosinophilic fasciitis following idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia and Hashimoto’s disease. Dermatology. 1999; 199: 282. [DOI] [PubMed] [Google Scholar]
- 6. Baffoni L, Frisoni M, Maccaferri M, Ferri S. Systemic lupus erythematosus and eosinophilic fasciitis: an unusual association. Clin Rheumatol. 1995; 14: 591– 592. [DOI] [PubMed] [Google Scholar]
- 7. Balaban EP, Sheehan RG, Lipsky PE, Frenkel EP. Treatment of cutaneous sclerosis and aplastic anemia with antithymocyte globulin. Ann Intern Med. 1987; 106: 56– 58. [DOI] [PubMed] [Google Scholar]
- 8. Barnes L, Rodnan GP, Medsger TA, Short D. Eosinophilic fasciitis. A pathologic study of twenty cases. Am J Pathol. 1979; 96: 493– 518. [PMC free article] [PubMed] [Google Scholar]
- 9. Belot V, Mulleman D, Perrinaud A, Abdallah-Lotf M, Machet MC, Machet L. [Eosinophilic fasciitis associated with Borrelia burgdorferi infection.] Ann Dermatol Venereol. 2007; 134: 673– 677. [DOI] [PubMed] [Google Scholar]
- 10. Biasi D, Caramaschi P, Carletto A, Bambara LM. Scleroderma and eosinophilic fasciitis in patients taking fosinopril. J Rheumatol. 1997; 24: 1242. [PubMed] [Google Scholar]
- 11. Biasi D, Carletto A, Caramaschi P, Pacor ML, Spinaci E, Bambara LM. [Scleroderma induced by chemical agents. Description of a case and review of the literature.] Recenti Prog Med. 1995; 86: 155– 158. [PubMed] [Google Scholar]
- 12. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics, disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008; 47: 29– 35. [DOI] [PubMed] [Google Scholar]
- 13. Blaser KU, Steiger U, Wursch A, Speck B. [Eosinophilic fasciitis with aplastic anemia and Hashimoto’s thyroiditis. Review of the literature and report of a typical example.] Schweiz Med Wochenschr. 1989; 119: 1899– 1906. [PubMed] [Google Scholar]
- 14. Bonati A, Aielli F, Carnevali C. [A case of scleroderma associated with total medullary aplasia.] Ateneo Parmense Acta Biomed. 1977; 48: 499– 504. [PubMed] [Google Scholar]
- 15. Bonnotte B, Chauffert B, Caillot D, Martin F, Lorcerie B. Successful treatment with antithymocyte globulin and cyclosporin A of a severe aplastic anaemia associated with an eosinophilic fasciitis. Br J Rheumatol. 1998; 37: 1358– 1359. [DOI] [PubMed] [Google Scholar]
- 16. Brahams D. Lindane exposure and aplastic anaemia. Lancet. 1994; 343: 1092. [DOI] [PubMed] [Google Scholar]
- 17. Brummendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001; 97: 895– 900. [DOI] [PubMed] [Google Scholar]
- 18. Buchanan RR, Gordon DA, Muckle TJ, McKenna F, Kraag G. The eosinophilic fasciitis syndrome after phenytoin (dilantin) therapy. J Rheumatol. 1980; 7: 733– 736. [PubMed] [Google Scholar]
- 19. Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975; 45: 355– 363. [PubMed] [Google Scholar]
- 20. Cantini F, Salvarani C, Olivieri I, Padula A, Senesi C, Bellandi F, Truglia MC, Niccoli L, Palchetti R. Possible association between eosinophilic fasciitis and subcutaneous heparin use. J Rheumatol. 1998; 25: 383– 385. [PubMed] [Google Scholar]
- 21. Carcassonne Y, Gastaut JA. Pancytopenia and scleroderma. Br Med J. 1976; 1: 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cayla J, Rondier J, Toubert A, Leblond Missenard V, Chomette G. [A new case of eosinophilic fasciitis complicated by bone marrow aplasia.] Rev Rhum Mal Osteoartic. 1985; 52: 263– 265. [PubMed] [Google Scholar]
- 23. Cesaro S, Marsh J, Tridello G, Rovo A, Maury S, Montante B, Masszi T, Van Lint MT, Afanasyev B, Iriondo Atienza A, Bierings M, Carbone C, Doubek M, Lanino E, Sarhan M, Risitano A, Steinerova K, Wahlin A, Pegoraro A, Passweg J. Retrospective survey on the prevalence and outcome of prior autoimmune diseases in patients with aplastic anemia reported to the registry of the European group for blood and marrow transplantation. Acta Haematol. 2010; 124: 19– 22. [DOI] [PubMed] [Google Scholar]
- 24. Cetkovsky P, Koza V, Cetkovska P, Svojgrova M. Successful treatment of severe Shulman’s syndrome by allogeneic bone marrow transplantation. Bone Marrow Transplant. 1998; 21: 637– 639. [DOI] [PubMed] [Google Scholar]
- 25. Chan LS, Hanson CA, Cooper KD. Concurrent eosinophilic fasciitis and cutaneous T-cell lymphoma. Eosinophilic fasciitis as a paraneoplastic syndrome of T-cell malignant neoplasms? Arch Dermatol. 1991; 127: 862– 865. [PubMed] [Google Scholar]
- 26. Chaudhary UB, Eberwine SF, Hege KM. Acquired amegakaryocytic thrombocytopenia purpura and eosinophilic fasciitis: a long relapsing and remitting course. Am J Hematol. 2004; 75: 146– 150. [DOI] [PubMed] [Google Scholar]
- 27. Chevalier X, Hermine O, Authier FJ, Gaulard P, Gherardi RK. Carpal tunnel syndrome due to T cell lymphoma. Arthritis Rheum. 1995; 38: 1707– 1709. [DOI] [PubMed] [Google Scholar]
- 28. Choquet-Kastylevsky G, Kanitakis J, Dumas V, Descotes J, Faure M, Claudy A. Eosinophilic fasciitis and simvastatin. Arch Intern Med. 2001; 161: 1456– 1457. [DOI] [PubMed] [Google Scholar]
- 29. Davies JM, Dunn HG. Interstitial lung disease developing during treatment with cyclosporine in a patient with diffuse scleroderma, aplastic anemia and Crohn’s disease: implications for pathogenesis and treatment. J Clin Rheumatol. 1995; 1: 287– 291. [PubMed] [Google Scholar]
- 30. Debusscher L, Bitar N, De Maubeuge J, De Conninck G, Stryckmans P. Eosinophilic fasciitis and severe aplastic anemia: favorable response to either antithymocyte globulin or cyclosporine A in blood and skin disorders. Transplant Proc. 1988; 20: 310– 313. [PubMed] [Google Scholar]
- 31. DeGiovanni C, Chard M, Woollons A. Eosinophilic fasciitis secondary to treatment with atorvastatin. Clin Exp Dermatol. 2006; 31: 131– 132. [DOI] [PubMed] [Google Scholar]
- 32. de Latour RP, Visconte V, Takaku T, Wu C, Erie AJ, Sarcon AK, Desierto MJ, Scheinberg P, Keyvanfar K, Nunez O, Chen J, Young NS. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood. 2010; 116: 4175– 4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Masson A, Bouaziz JD, Rybojad M, Peffault de Latour R, Robin M, Rodriguez-Otero P, Durant C, Socie G, Bagot M. EF/SSc overlap syndrome and aplastic anaemia resistant to immunosuppressive therapy. Rheumatology (Oxford). 2012; 51: 762– 764. [DOI] [PubMed] [Google Scholar]
- 34. Doyle JA, Connolly SM, Hoagland HC. Hematologic disease in scleroderma syndromes. Acta Derm Venereol. 1985; 65: 521– 525. [PubMed] [Google Scholar]
- 35. Dziadzio L, Kelly EA, Panzer SE, Jarjour N, Huttenlocher A. Cytokine abnormalities in a patient with eosinophilic fasciitis. Ann Allergy Asthma Immunol. 2003; 90: 452– 455. [DOI] [PubMed] [Google Scholar]
- 36. Eklund KK, Anttila P, Leirisalo-Repo M. Eosinophilic fasciitis, myositis and arthritis as early manifestations of peripheral T-cell lymphoma. Scand J Rheumatol. 2003; 32: 376– 377. [DOI] [PubMed] [Google Scholar]
- 37. Endo Y, Tamura A, Matsushima Y, Iwasaki T, Hasegawa M, Nagai Y, Ishikawa O. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol. 2007; 26: 1445– 1451. [DOI] [PubMed] [Google Scholar]
- 38. Falcao S, Mouro AF, Ribeiro C, Pinto TL, Mateus M, Araujo P, Nero P, Pimentao JB, Branco JC. [Eosinophilic fasciitis and aplastic anemia.] Acta Reumatol Port. 2009; 34: 120– 126. [PubMed] [Google Scholar]
- 39. Fenaux P, Merignargues S, Pagniez D, Janin A, Lucidarme D, Bauters F. Cutaneous sclerosis, aplastic anemia, and antithymocyte globulin. Ann Intern Med. 1989; 110: 575– 576. [DOI] [PubMed] [Google Scholar]
- 40. Feng X, Chuhjo T, Sugimori C, Kotani T, Lu X, Takami A, Takamatsu H, Yamazaki H, Nakao S. Diazepam-binding inhibitor-related protein 1: a candidate autoantigen in acquired aplastic anemia patients harboring a minor population of paroxysmal nocturnal hemoglobinuria-type cells. Blood. 2004; 104: 2425– 2431. [DOI] [PubMed] [Google Scholar]
- 41. Focan C, Swale JL, Borlee-Hermans G, Claessens JJ. Systemic sclerosis, aplastic anemia and amyloidosis associated with lung carcinoma. Acta Clin Belg. 1985; 40: 204– 205. [DOI] [PubMed] [Google Scholar]
- 42. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009; 360: 1989– 2003. [DOI] [PubMed] [Google Scholar]
- 43. Gallardo F, Vadillo M, Mitjavila F, Servitje O. Systemic lupus erythematosus after eosinophilic fasciitis: a case report. J Am Acad Dermatol. 1998; 39: 283– 285. [DOI] [PubMed] [Google Scholar]
- 44. Garcia VP, de Quiros JF, Caminal L. Autoimmune hemolytic anemia associated with eosinophilic fasciitis. J Rheumatol. 1998; 25: 1864– 1865. [PubMed] [Google Scholar]
- 45. Garcia-Morteo O, Nitsche A, Maldonado-Cocco JA, Barcelo HA. Eosinophilic fasciitis and retroperitoneal fibrosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1987; 30: 1314– 1315. [DOI] [PubMed] [Google Scholar]
- 46. Gourvil J, Wechsler J, Revuz J, Touraine R. [Eosinophilic fasciitis (Shulman syndrome) associated with insulin-dependent diabetes. 1 case (author’s transl).] Nouv Presse Med. 1979; 8: 4087– 4090. [PubMed] [Google Scholar]
- 47. Granter SR, Barnhill RL, Duray PH. Borrelial fasciitis: diffuse fasciitis and peripheral eosinophilia associated with Borrelia infection. Am J Dermatopathol. 1996; 18: 465– 473. [DOI] [PubMed] [Google Scholar]
- 48. Griffin AJ. Eosinophilic fasciitis with megakaryocyte aplasia. J R Soc Med. 1979; 72: 779– 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hansen PB, Lauritzen AM. Aplastic anemia successfully treated with rituximab. Am J Hematol. 2005; 80: 292– 294. [DOI] [PubMed] [Google Scholar]
- 50. Harden RA, Baetjer AM. Aplastic anemia following exposure to paradichlorobenzene and naphthalene. J Occup Med. 1978; 20: 820– 822. [PubMed] [Google Scholar]
- 51. Hashimoto Y, Takahashi H, Matsuo S, Hirai K, Takemori N, Nakao M, Miyamoto K, Iizuka H. Polymerase chain reaction of Borrelia burgdorferi flagellin gene in Shulman syndrome. Dermatology. 1996; 192: 136– 139. [DOI] [PubMed] [Google Scholar]
- 52. Hayashi N, Igarashi A, Matsuyama T, Harada S. Eosinophilic fasciitis following exposure to trichloroethylene: successful treatment with cyclosporin. Br J Dermatol. 2000; 142: 830– 832. [DOI] [PubMed] [Google Scholar]
- 53. Herson S, Brechignac S, Piette JC, Mouthon JM, Coutellier A, Bletry O, Godeau P. Capillary microscopy during eosinophilic fasciitis in 15 patients: distinction from systemic scleroderma. Am J Med. 1990; 88: 598– 600. [DOI] [PubMed] [Google Scholar]
- 54. Hertzman PA, Blevins WL, Mayer J, Greenfield B, Ting M, Gleich GJ. Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med. 1990; 322: 869– 873. [DOI] [PubMed] [Google Scholar]
- 55. Hirano N, Butler MO, Guinan EC, Nadler LM, Kojima S. Presence of anti-kinectin and anti-PMS1 antibodies in Japanese aplastic anaemia patients. Br J Haematol. 2005; 128: 221– 223. [DOI] [PubMed] [Google Scholar]
- 56. Hirano N, Butler MO, Von Bergwelt-Baildon MS, Maecker B, Schultze JL, O’Connor KC, Schur PH, Kojima S, Guinan EC, Nadler LM. Autoantibodies frequently detected in patients with aplastic anemia. Blood. 2003; 102: 4567– 4575. [DOI] [PubMed] [Google Scholar]
- 57. Hoffman R, Dainiak N, Sibrack L, Pober JS, Waldron JA., Jr Antibody-mediated aplastic anemia and diffuse fasciitis. N Engl J Med. 1979; 300: 718– 721. [DOI] [PubMed] [Google Scholar]
- 58. Hoffman R, Young N, Ershler WB, Mazur E, Gewirtz A. Diffuse fasciitis and aplastic anemia: a report of four cases revealing an unusual association between rheumatologic and hematologic disorders. Medicine (Baltimore). 1982; 61: 373– 381. [PubMed] [Google Scholar]
- 59. Hur JW, Lee HS, Uhm WS, Jun JB, Bae SC, Park CK, Yoo DH. Eosinophilic fasciitis associated with autoimmune thyroiditis. Korean J Intern Med. 2005; 20: 180– 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Issaragrisil S, Kaufman DW, Anderson T, Chansung K, Leaverton PE, Shapiro S, Young NS. The epidemiology of aplastic anemia in Thailand. Blood. 2006; 107: 1299– 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jacob SE, Lodha R, Cohen JJ, Romanelli P, Kirsner RS. Paraneoplastic eosinophilic fasciitis: a case report. Rheumatol Int. 2003; 23: 262– 264. [DOI] [PubMed] [Google Scholar]
- 62. Janin A, Socie G, Devergie A, Aractingi S, Esperou H, Verola O, Gluckman E. Fasciitis in chronic graft-versus-host disease. A clinicopathologic study of 14 cases. Ann Intern Med. 1994; 120: 993– 998. [DOI] [PubMed] [Google Scholar]
- 63. Janzen L, Jeffery JR, Gough J, Chalmers IM. Response to methotrexate in a patient with idiopathic eosinophilic fasciitis, morphea, IgM hypergammaglobulinemia, and renal involvement. J Rheumatol. 1995; 22: 1967– 1970. [PubMed] [Google Scholar]
- 64. Jinnin M, Ihn H, Yamane K, Asano Y, Yazawa N, Tamaki K. Serum levels of tissue inhibitor of metalloproteinase-1 and 2 in patients with eosinophilic fasciitis. Br J Dermatol. 2004; 151: 407– 412. [DOI] [PubMed] [Google Scholar]
- 65. Junca J, Cuxart A, Tural C, Ojanguren I, Flores A. Eosinophilic fasciitis and non-Hodgkin lymphoma. Eur J Haematol. 1994; 52: 304– 306. [DOI] [PubMed] [Google Scholar]
- 66. Kamada K, Kobayashi Y, Katada K, Takahashi Y, Chikayama S, Ikeda M, Kondo M. Scleroderma associated with anemia and thrombocytopenia that responded well to cyclosporin. Acta Haematol. 2000; 104: 106– 109. [DOI] [PubMed] [Google Scholar]
- 67. Katsumata Y, Suzuki T, Kuwana M, Hattori Y, Akizuki S, Sugiura H, Matsuoka Y. Anti-c-Mpl (thrombopoietin receptor) autoantibody-induced amegakaryocytic thrombocytopenia in a patient with systemic sclerosis. Arthritis Rheum. 2003; 48: 1647– 1651. [DOI] [PubMed] [Google Scholar]
- 68. Khanna D, Verity A, Grossman JM. Eosinophilic fasciitis with multiple myeloma: a new haematological association. Ann Rheum Dis. 2002; 61: 1111– 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kikuchi K, Kadono T, Furue M, Tamaki K. Tissue inhibitor of metalloproteinase 1 (TIMP-1) may be an autocrine growth factor in scleroderma fibroblasts. J Invest Dermatol. 1997; 108: 281– 284. [DOI] [PubMed] [Google Scholar]
- 70. Kilbourne EM, Rigau-Perez JG, Heath CW, Jr, Zack MM, Falk H, Martin-Marcos M, de Carlos A. Clinical epidemiology of toxic-oil syndrome. Manifestations of a New Illness. N Engl J Med. 1983; 309: 1408– 1414. [DOI] [PubMed] [Google Scholar]
- 71. Killen JW, Swift GL, White RJ. Eosinophilic fasciitis with pulmonary and pleural involvement. Postgrad Med J. 2000; 76: 36– 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim H, Kim MO, Ahn MJ, Lee YY, Jung TJ, Choi IY, Kim IS, Park CK. Eosinophilic fasciitis preceding relapse of peripheral T-cell lymphoma. J Korean Med Sci. 2000; 1: 346– 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim SW, Rice L, Champlin R, Udden MM. Aplastic anemia in eosinophilic fasciitis: responses to immunosuppression and marrow transplantation. Haematologia (Budap). 1997; 28: 131– 137. [PubMed] [Google Scholar]
- 74. Kitamura Y, Hatamochi A, Hamasaki Y, Ikeda H, Yamazaki S. Association between eosinophilic fasciitis and systemic lupus erythematosus. J Dermatol. 2007; 34: 150– 152. [DOI] [PubMed] [Google Scholar]
- 75. Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988; 17: 221– 231. [DOI] [PubMed] [Google Scholar]
- 76. Lebeaux D, Frances C, Barete S, Wechsler B, Dubourg O, Renoux J, Maisonobe T, Benveniste O, Gatfosse M, Bourgeois P, Amoura Z, Cacoub P, Piette JC, Sene D. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford). 2012; 51: 557– 561. [DOI] [PubMed] [Google Scholar]
- 77. Littlejohn GO, Keystone EC. Eosinophilic fasciitis and aplastic anaemia. J Rheumatol. 1980; 7: 730– 732. [PubMed] [Google Scholar]
- 78. MacIntyre A, Brouilette SW, Lamb K, Radhakrishnan K, McGlynn L, Chee MM, Parkinson EK, Freeman D, Madhok R, Shiels PG. Association of increased telomere lengths in limited scleroderma, with a lack of age-related telomere erosion. Ann Rheum Dis. 2008; 67: 1780– 1782. [DOI] [PubMed] [Google Scholar]
- 79. Mallepalli JR, Quinet RJ, Sus R. Eosinophilic fasciitis induced by fire ant bites. Ochsner J. 2008; 8: 114– 118. [PMC free article] [PubMed] [Google Scholar]
- 80. Maluf E, Hamerschlak N, Cavalcanti AB, Junior AA, Eluf-Neto J, Falcao RP, Lorand-Metze IG, Goldenberg D, Santana CL, Rodrigues Dde O, Passos LN, Rosenfeld LG, Pitta M, Loggetto S, Ribeiro AA, Velloso ED, Kondo AT, Coelho EO, Pintao MC, de Souza HM, Borbolla JR, Pasquini R. Incidence and risk factors of aplastic anemia in Latin American countries: the LATIN case-control study. Haematologica. 2009; 94: 1220– 1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Markusse HM, Breedveld FC. Rheumatoid arthritis with eosinophilic fasciitis and pure red cell aplasia. J Rheumatol. 1989; 16: 1383– 1384. [PubMed] [Google Scholar]
- 82. Martin JR, Williams JP, Barrowman JA. Diffuse (? eosinophilic) fasciitis, atypical rash, and chronic inflammatory disease of the colon (? Crohn’s disease).[sic] J Rheumatol. 1980; 7: 928– 929. [PubMed] [Google Scholar]
- 83. Masuoka H, Kikuchi K, Takahashi S, Kakinuma T, Hayashi N, Furue M. Eosinophilic fasciitis associated with low-grade T-cell lymphoma. Br J Dermatol. 1998; 139: 928– 930. [DOI] [PubMed] [Google Scholar]
- 84. Michaels RM. Eosinophilic fasciitis complicated by Hodgkin’s disease. J Rheumatol. 1982; 9: 473– 476. [PubMed] [Google Scholar]
- 85. Michet CJ, Jr, Doyle JA, Ginsburg WW. Eosinophilic fasciitis: report of 15 cases. Mayo Clin Proc. 1981; 56: 27– 34. [PubMed] [Google Scholar]
- 86. Moriguchi M, Terai C, Kuroki S, Tanaka E, Someya N, Tsunoda Y, Kashiwazaki S. Eosinophilic fasciitis complicated with peripheral polyneuropathy. Intern Med. 1998; 37: 417– 420. [DOI] [PubMed] [Google Scholar]
- 87. Mosconi S, Streit M, Bronimann M, Braathen LR. Eosinophilic fasciitis (Shulman syndrome). Dermatology. 2002; 205: 204– 206. [DOI] [PubMed] [Google Scholar]
- 88. Naoui A, Bouslama K, Abdallah M, Hamzaoui S, Arbi T, Bahri F, M’zabi S, Harmel A, Ennafaa M, Ben Dridi M, M’rad S. [Eosinophilic fasciitis (Shulman’s disease): a case series of 11 patients.] Rev Med Interne. 2010; 31: 535– 539. [DOI] [PubMed] [Google Scholar]
- 89. Narayanan MN, Liu Yin JA, Love EM, Geary CG, Holt PJ, Freemont AJ, Feinmann R. Eosinophilic fasciitis and aplastic anaemia. Clin Lab Haematol. 1988; 10: 471– 474. [DOI] [PubMed] [Google Scholar]
- 90. Naschitz JE, Boss JH, Misselevich I, Yeshurun D, Rosner I. The fasciitis-panniculitis syndromes. Clinical and pathologic features. Medicine (Baltimore). 1996; 75: 6– 16. [DOI] [PubMed] [Google Scholar]
- 91. Naschitz JE, Misselevich I, Rosner I, Yeshurun D, Weiner P, Amar M, Amato L, Ciompi ML, Boss JH. Lymph-node-based malignant lymphoma and reactive lymphadenopathy in eosinophilic fasciitis. Am J Med Sci. 1999; 318: 343– 349. [DOI] [PubMed] [Google Scholar]
- 92. Naschitz JE, Yeshurun D, Misselevich I, Boss JH. Colitis and pericarditis in a patient with eosinophilic fasciitis. A contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989; 16: 688– 692. [PubMed] [Google Scholar]
- 93. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995; 70: 1068– 1076. [DOI] [PubMed] [Google Scholar]
- 94. Philpott H, Hissaria P, Warrren L, Singhal N, Brown M, Proudman S, Cleland L, Gillis D. Eosinophilic fasciitis as a paraneoplastic phenomenon associated with metastatic colorectal carcinoma. Australas J Dermatol. 2008; 49: 27– 29. [DOI] [PubMed] [Google Scholar]
- 95. Quilichini R, Chaffanjon P, Aubert L, Mugnier C, Pelissier JF, Gastaut JA, Carcassonne Y. [A new case of eosinophilic fasciitis with bone marrow aplasia. Cure by high doses of corticoids.] Presse Med. 1985; 14: 427. [PubMed] [Google Scholar]
- 96. Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004; 364: 355– 364. [DOI] [PubMed] [Google Scholar]
- 97. Rizzo S. Eosinophilic pleuropericarditis and fasciitis. A new case. Monaldi Arch Chest Dis. 2002; 57: 311– 313. [PubMed] [Google Scholar]
- 98. Rodat O, Harousseau JL, Reynaud C, Milpied N, Stalder JF, Chupin M. [Eosinophilic fasciitis and Hodgkin’s disease.] Sem Hop. 1982; 58: 2207– 2209. [PubMed] [Google Scholar]
- 99. Rugman FP, Cosstick R. Aplastic anaemia associated with organochlorine pesticide: case reports and review of evidence. J Clin Pathol. 1990; 4: 98– 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Salmeron G, Patey N, de Latour RP, Raffoux E, Gluckman E, Brousse N, Socie G, Robin M. Coeliac disease and aplastic anaemia: a specific entity? Br J Haematol. 2009; 146: 122– 124. [DOI] [PubMed] [Google Scholar]
- 101. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992; 239: 91– 92. [DOI] [PubMed] [Google Scholar]
- 102. Scheinberg M, Hamerschlak N, Kutner JM, Ribeiro AA, Ferreira E, Goldenberg J, Kiss MH, Chahade WH. Rituximab in refractory autoimmune diseases: Brazilian experience with 29 patients (2002–2004). Clin Exp Rheumatol. 2006; 24: 65– 69. [PubMed] [Google Scholar]
- 103. Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA. 2010; 304: 1358– 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seaman JM, Goble M, Madsen L, Steigerwald JC. Fasciitis and polyarthritis during antituberculous therapy. Arthritis Rheum. 1985; 28: 1079– 1084. [DOI] [PubMed] [Google Scholar]
- 105. Seibold JR, Rodnan GP, Medsger TA, Jr, Winkelstein A. Circulating immune complexes in eosinophilic fasciitis. Arthritis Rheum. 1982; 25: 1180– 1185. [DOI] [PubMed] [Google Scholar]
- 106. Seko Y, Tomiya T, Kuro-o M, Takano K, Nojima Y, Terai C, Yamada A, Shimizu T, Inoue K, Takaku F. [A case of eosinophilic fasciitis with peripheral nerve disorder.] Nihon Naika Gakkai Zasshi. 1988; 77: 370– 376. [DOI] [PubMed] [Google Scholar]
- 107. Sepp N, Schmutzhard E, Fritsch P. Shulman syndrome associated with Borrelia burgdorferi and complicated by carpal tunnel syndrome. J Am Acad Dermatol. 1988; 18: 1361– 1362. [DOI] [PubMed] [Google Scholar]
- 108. Serrano-Grau P, Mascaro-Galy JM, Iranzo P. [Eosinophilic fasciitis after taking simvastatin.] Actas Dermosifiliogr. 2008; 99: 420– 421. [PubMed] [Google Scholar]
- 109. Sherber NS, Wigley FM, Paget SA. Diffuse fasciitis with eosinophilia developing after local irradiation for breast cancer. Clin Rheumatol. 2009; 28: 729– 732. [DOI] [PubMed] [Google Scholar]
- 110. Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, von Bergwelt-Baildon MS. The role of B cells in the pathogenesis of graft-versus-host disease. Blood. 2009; 114: 4919– 4927. [DOI] [PubMed] [Google Scholar]
- 111. Shulman LE, Hoffman R, Dainiak N, Nesbitt J, Adelman HM, Florida T, Lawless OJ, Lindsay SM. Antibody-mediated aplastic anemia and thrombocytopenic purpura in diffuse eosinophilic fasciitis. Arthritis Rheum. 1979; 22: 659. [Google Scholar]
- 112. Shulman LE. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia: a new syndrome? J Rheumatol. 1984; 11: 569– 570. [PubMed] [Google Scholar]
- 113. Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. 2002; 100: 1185– 1191. [DOI] [PubMed] [Google Scholar]
- 114. Smiley AM, Husain M, Indenbaum S. Eosinophilic fasciitis in association with thyroid disease: a report of three cases. J Rheumatol. 1980; 7: 871– 876. [PubMed] [Google Scholar]
- 115. Solomou EE, Rezvani K, Mielke S, Malide D, Keyvanfar K, Visconte V, Kajigaya S, Barrett AJ, Young NS. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood. 2007; 110: 1603– 1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Stanek G, Konrad K, Jung M, Ehringer H. Shulman syndrome, a scleroderma subtype caused by Borrelia burgdorferi? Lancet. 1987; 1: 1490. [DOI] [PubMed] [Google Scholar]
- 117. Stebler C, Tichelli A, Gratwohl A, Dazzi H, Nissen C, Steiger U, Speck B. [Aplastic anemia combined with an autoimmune disease (eosinophilic fasciitis or glomerulonephritis).] Schweiz Med Wochenschr. 1991; 121: 873– 876. [PubMed] [Google Scholar]
- 118. Suematsu E, Miyamura T, Idutsu K, Minami R, Yamamoto M. [Efficacy of anti-thymocyte globulin and cyclosporin A combined therapy in aplastic anemia complicated with limited cutaneous systemic sclerosis.] Nihon Rinsho Meneki Gakkai Kaishi. 2005; 28: 99– 103. [DOI] [PubMed] [Google Scholar]
- 119. Takamatsu H, Espinoza JL, Lu X, Qi Z, Okawa K, Nakao S. Anti-moesin antibodies in the serum of patients with aplastic anemia stimulate peripheral blood mononuclear cells to secrete TNF-alpha and IFN-gamma. J Immunol. 2009; 182: 703– 710. [DOI] [PubMed] [Google Scholar]
- 120. Takamatsu H, Feng X, Chuhjo T, Lu X, Sugimori C, Okawa K, Yamamoto M, Iseki S, Nakao S. Specific antibodies to moesin, a membrane-cytoskeleton linker protein, are frequently detected in patients with acquired aplastic anemia. Blood. 2007; 109: 2514– 2520. [DOI] [PubMed] [Google Scholar]
- 121. Takamatsu H, Yagasaki H, Takahashi Y, Hama A, Saikawa Y, Yachie A, Koizumi S, Kojima S, Nakao S. Aplastic anemia successfully treated with rituximab: the possible role of aplastic anemia-associated autoantibodies as a marker for response. Eur J Haematol. 2011; 86: 541– 545. [DOI] [PubMed] [Google Scholar]
- 122. Tooze JA, Marsh JC, Wickham N, Duke OL, Behrens J, Gordon-Smith EC. Response of aplastic anaemia and scleroderma to cyclosporin. Br J Haematol. 1993; 85: 829– 831. [DOI] [PubMed] [Google Scholar]
- 123. Toquet C, Hamidou MA, Renaudin K, Jarry A, Foulc P, Barbarot S, Laboisse C, Mussini JM. In situ immunophenotype of the inflammatory infiltrate in eosinophilic fasciitis. J Rheumatol. 2003; 30: 1811– 1815. [PubMed] [Google Scholar]
- 124. Valesini G, Litta A, Bonavita MS, Luan FL, Purpura M, Mariani M, Balsano F. Geographical clustering of scleroderma in a rural area in the province of Rome. Clin Exp Rheumatol. 1993; 11: 41– 47. [PubMed] [Google Scholar]
- 125. Veyssier-Belot C, Zuech P, Lumbroso-Le Rouic L, Recanati G, Dendale R. [Eosinophilic fasciitis and metastatic choroidal melanoma: a paraneoplastic syndrome?] Rev Med Interne. 2008; 29: 1013– 1016. [DOI] [PubMed] [Google Scholar]
- 126. Viallard JF, Taupin JL, Ranchin V, Leng B, Pellegrin JL, Moreau JF. Analysis of leukemia inhibitory factor, type 1 and type 2 cytokine production in patients with eosinophilic fasciitis. J Rheumatol. 2001; 28: 75– 80. [PubMed] [Google Scholar]
- 127. Watts RA, Merry P. Familial eosinophilic fasciitis and breast cancer. Br J Rheumatol. 1994; 33: 93– 94. [DOI] [PubMed] [Google Scholar]
- 128. Weltz M, Salvado A, Rosse W, Berenberg J. Humoral suppression of hematopoiesis in eosinophilic fasciitis. Blood. 1978; 52: 218. [Google Scholar]
- 129. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005; 352: 1413– 1424. [DOI] [PubMed] [Google Scholar]
- 130. Young NS, Scheinberg P, Calado RT. Aplastic anemia. Curr Opin Hematol. 2008; 15: 162– 168. [DOI] [PMC free article] [PubMed] [Google Scholar]