

Abstract
Idiopathic retroperitoneal fibrosis (RPF) is a periaortic sclerotic disease that encases adjacent retroperitoneal structures, particularly the ureters. A subset of idiopathic RPF cases can be associated with IgG4-related disease, but the frequency of this association is not clear. We selected 23 cases of idiopathic RPF and identified IgG4-related RPF cases based on the presence of IgG4+ plasma cells in the tissue, using an IgG4/IgG ratio cutoff of >40%. We then compared the IgG4-related RPF patients and the non-IgG4-related RPF patients in terms of both the presence of histopathologic features typical of IgG4-related disease and the simultaneous occurrence (or history) of other organ manifestations typical of IgG4-related disease. The IgG4-related RPF and non-IgG4-related RPF groups were also analyzed in terms of clinical, laboratory, and radiologic features and treatment review.
We identified 13 cases of IgG4-related RPF (57% of the total cohort). The distinguishing features of IgG4-related RPF were histopathologic and extra-organ manifestations of IgG4-related disease. The IgG4-related RPF patients were statistically more likely than non-IgG4-related RPF patients to have retroperitoneal biopsies showing lymphoplasmacytic infiltrate (p = 0.006), storiform fibrosis (p = 0.006), or tissue eosinophilia (p = 0.0002). Demographics of the 2 groups, including a middle-aged, male predominance (mean age, 58 yr; 73% male), were similar.
IgG4-related disease accounts for a substantial percentage of patients with “idiopathic” RPF. Histopathologic features such as storiform fibrosis, obliterative phlebitis, and tissue eosinophilia are critical to identifying this disease association. Extraretroperitoneal manifestations of IgG4-related disease are also often present among patients with IgG4-related RPF. Elevated IgG4/total IgG ratios in tissue biopsies are more useful than the number of IgG4+ plasma cells per high-power field in cases of RPF that are highly fibrotic.
INTRODUCTION
Retroperitoneal fibrosis (RPF), sometimes termed “Ormond’s disease,” is an enigmatic disorder characterized by sclerotic tissue in the periaortic or periiliac retroperitoneum that encases adjacent structures.50 A urologist, Dr. John Ormond, described RPF in 1948 upon observing intraoperatively the fibrous tissue encasement of both ureters in a patient with renal failure.32 The most common symptoms of RPF include abdominal or flank pain, weight loss, fatigue, and urinary frequency.39,51 Specific serologic markers for RPF do not exist, but acute-phase reactants such as the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are frequently elevated. Imaging studies show a soft tissue density that envelops the abdominal aorta or iliac vessels, often leading to hydronephrosis of 1 or both kidneys.3
RPF can be divided into idiopathic and secondary subsets. Idiopathic RPF is essentially a diagnosis of exclusion after secondary causes of RPF, for example, drug exposure, infection, and malignancy, have been eliminated.49,50 Definitive diagnosis generally requires histopathologic confirmation by biopsy.
IgG4-related disease (IgG4-RD) is an immune-mediated disease characterized by unique histopathologic features in affected organs. These features are a lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis.5 Mild to moderate tissue eosinophilia is also present in many patients, consistent with the strong history of allergic disease or atopy that frequently accompanies (or is an integral part of) IgG4-RD.15 Immunostaining of tissue lesions in IgG4-RD demonstrates an enrichment with IgG4+ plasma cells, indicated by either an increase in their overall concentration in tissue (number per high-power field [HPF]), an elevated IgG4/total IgG ratio, or both. Characteristic organs affected in IgG4-RD include the pancreas, salivary glands, orbits, lung, kidney, and aorta, but the disease has also been described in the thyroid gland (Riedel thyroiditis),4 the prostate gland,46 the pachymeninges,23 skin,18 and nearly every other organ system.
An association between RPF and “multifocal fibrosclerosis” has been acknowledged for decades.2 Multifocal fibrosclerosis is now known to be synonymous with IgG4-RD. However, there have been few studies of the retroperitoneum during the era in which IgG4-RD has been recognized. These studies are contradictory with regard to any potential relationship of IgG4-RD to “idiopathic” RPF. Zen et al54 observed the typical histopathologic features and immunostaining characteristics of IgG4-RD in 10 of 17 RPF patients from Japan, suggesting that a proportion of idiopathic RPF cases are part of the IgG4-RD spectrum. In contrast, other investigators writing on idiopathic RPF did not comment on the potential contribution of IgG4-RD to their cases.39,51
We conducted the current study to address the possible role of IgG4-RD in the clinical entity known as idiopathic RPF. We identified 23 cases of idiopathic RPF and evaluated them for the possibility of IgG4-related RPF on the basis of their IgG4/total IgG ratios within tissue. We then compared the presence of histopathologic features typical of IgG4-RD, the simultaneous occurrence or previous history of other organ manifestations typical of IgG4-RD, and other clinical and radiologic findings in the 2 categories of idiopathic RPF.
METHODS
Case Selection
This study was approved by the institutional review board of our hospital. We searched the Massachusetts General Hospital Department of Pathology database for the keyword phrase “retroperitoneal fibrosis.” One hundred nine patients had been diagnosed with RPF between the years 1990 and 2011 and had both clinical and pathology records available for review. Secondary RPF cases were excluded by review of the patients’ medical records, including clinical, pathologic, and radiologic data. The mean clinical follow-up on this group of patients was 6 years (range, 1–15 yr). Twenty-three of the idiopathic RPF cases had pathology blocks were available for further immunohistochemical analysis. These cases included 2 patients referred to our group specifically because of suspected IgG4-RD. The 23 cases included 8 surgical biopsy specimens and 15 needle biopsy specimens. In all cases, both imaging studies and tissue biopsies were obtained before the institution of immunosuppressive therapy.
IgG4-Related RPF: Histopathologic Classification
The original hematoxylin and eosin-stained slides were reviewed in a blinded manner by one of the authors (VD) to document the histopathologic features. The potential features assessed specifically included lymphoplasmacytic infiltration, storiform fibrosis, obliterative phlebitis, and tissue eosinophilia (defined as presence of any eosinophils). Immunohistochemistry was performed using antibodies to IgG4 (Zymed; 1:200 dilution) and IgG (Dako; 1:3000 dilution). We used an IgG4/IgG ratio cutoff of >40% to separate patients into 2 groups: IgG4-related RPF and non-IgG4-related RPF. This cutoff is consistent with a consensus statement on the pathology of IgG4-RD.5
The degree of IgG4+ plasma cells within biopsy samples was determined for all 23 patients. For each case, the number of plasma cells staining for IgG4 was assessed in 3 nonoverlapping HPFs (magnification 400×). The 3 fields with the highest degree of IgG4 reactivity were selected for counting. The number of IgG4+ plasma cells was then divided by the total number of IgG-bearing plasma cells in these fields to determine the IgG4/IgG ratio.
Clinical Features of the Overall Cohort
We retrospectively reviewed the patients’ electronic medical records. Patients with secondary causes of RPF such as metastatic carcinoma, lymphoma, and sarcoma were excluded. The date of biopsy was considered to be the patient’s date of RPF diagnosis. The clinical features determined for each patient included age, sex, and comorbidities present at the time of diagnosis, including cardiovascular disease, hypertension, diabetes, pelvic malignancy, and any known immune-mediated inflammatory processes. Laboratory data from the time of RPF diagnosis included the ESR, CRP, white blood cell count, hematocrit, serum albumin and creatinine concentrations, the results of antinuclear antibody (ANA) assays, and, when available, IgG subclass concentrations.
The treatments administered to each patient were determined by chart review. We defined first-line therapy as the medication prescribed initially following confirmation of the RPF diagnosis, and maintenance therapy as a medication used continuously for more than 6 months. Chronic kidney disease was defined as a glomerular filtration rate of <60 mL/min for more than 3 months.29
Radiologic Features
Imaging evaluations of the abdomen and pelvis on 21 patients were reviewed in a blinded fashion by one of the authors (NS). Images were reviewed on a picture archiving and communication system workstation (PACS, Agfa, v. 4.0; Agfa, Richmond, VA). Contrast-enhanced computed tomography (CT) examinations of the abdomen and pelvis were available on 18 patients. 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) studies with contrast-enhanced CT (PET/CT) were available on 5 patients, and contrast-enhanced magnetic resonance imaging (MRI) studies of the abdomen and pelvis were performed on 2 patients. All imaging studies reported herein were obtained within 3 months before the biopsy procedure was performed.
The extent and location of soft tissue in the periaortic, pericaval, presacral, retrovesical, perirectal, pelvic side-wall, and periureteral regions were recorded. The maximum thickness of the soft tissue in axial dimension was recorded. In addition, the presence and absence of abdominal aortic aneurysms (defined as a maximal infrarenal axial diameter of 3 cm), unilateral or bilateral hydronephrosis, renal atrophy, and localized or diffuse lymphadenopathy were recorded.
Statistical Methods
The clinical features of the IgG4-related RPF and non-IgG4-related idiopathic RPF were compared. Continuous variables, including age and laboratory values, were compared using unpaired t-tests. The Fisher exact test was used to compare sex distribution, comorbidities, ANA positivity, and radiologic features.
RESULTS
Pathologic Features
Immunohistochemistry
An IgG4/IgG ratio cutoff of >40% was used to divide the patients into IgG4-related RPF and non-IgG4-related RPF disease subsets. There were 13 patients in the IgG4-related RPF subset (57%) and 10 in the non-IgG4-related RPF subset (43%). Patients in the IgG4-related RPF group had a mean biopsy IgG4/total IgG ratio of 81%, compared to 8% in the non-IgG4-related RPF group (p = 0.0001) (Figure 1). The mean number of IgG4+ plasma cells per HPF in the IgG4-related group was 13 IgG4+ cells/HPF (range, 2–55 IgG4+ cells/HPF), and the mean number in the non-IgG4-related group was 1 IgG4+ cell/HPF (range, 0–8 cells/HPF; p = 0.02).
FIGURE 1.
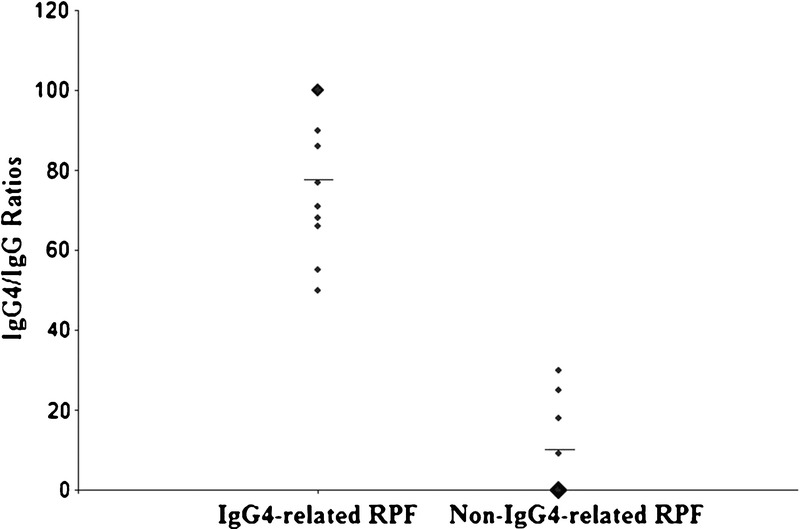
IgG4/IgG ratios of IgG4-related RPF and non-IgG4-related RPF groups. The mean IgG4/IgG ratios, designated by the horizontal lines, are 78% for the IgG4-related RPF group and 8.2% for the non-IgG4-related RPF group.
Histopathology
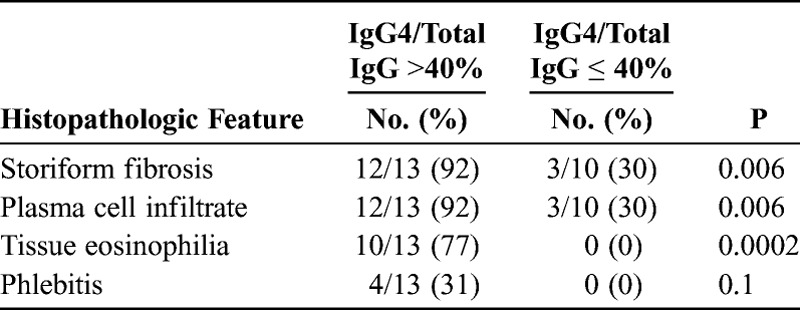
We compared the histologic features of cases in the 2 groups of RPF selected by their IgG4/IgG ratio. The characteristic IgG4-related RPF cases demonstrated a disproportionately IgG4+ enriched lymphoplasmactyic infiltrate (Figure 2). Patients with IgG4/total IgG ratios >40% were more likely to have the histopathologic features of lymphoplasmacytic infiltrates, storiform fibrosis, and eosinophilia than were those with IgG4/IgG ratios <40% (see below)(Table 1). The IgG4-related RPF cases demonstrated the morphologic appearance of IgG4-RD but the retroperitoneal tissue typically had more fibrosis than is observed in most extraretroperitoneal organs involved in this condition. A storiform pattern of fibrosis was noted significantly more often among cases in the IgG4-related RPF group (92% vs. 30%; p = 0.006). The non-IgG4-related RPF cases showed a nonspecific histologic appearance, generally an acellular, hyalinizing type of fibrosis. The lymphoplasmacytic infiltrate was more dense in the IgG4-related RPF group than in the non-IgG4-related RPF group, and plasma cells were identified easily within biopsies from the former. In addition, tissue eosinophilia was an important distinguishing feature between the 2 groups (10/13 IgG4-related RPF and 0/10 non-IgG4-related RPF; p = 0.0002) (see Figure 2). Obliterative phlebitis was observed in 4 cases, all in the IgG4-related RPF group (see Table 1).
FIGURE 2.
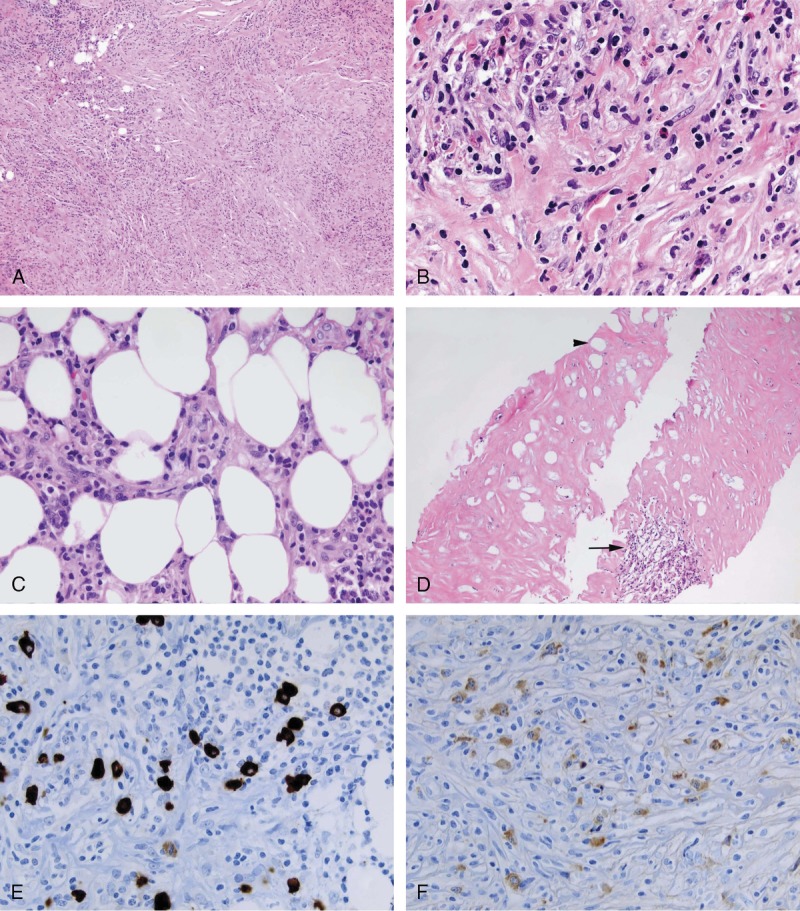
IgG4-related RPF showing storiform type of fibrosis (A & B). The inflammatory infiltrate is composed of lymphocytes, plasma cell, and eosinophils (B). The inflammatory infiltrate frequently extends into the adipose tissue (C). Non-IgG4-related RPF cases typically showed acellular fibrosis with entrapped necrotic fat cell (arrow head). A lymphoid aggregate is also seen (arrow) (D). Immunohistochemical stain for IgG4. IgG4-related RPF showing elevated numbers of IgG4+ plasma cells (E). Immunohistochemical stain for IgG (F). The IgG4/IgG ratio was close to 100%. [This figure can be viewed in color online at http://www.md-journal.com].
TABLE 1.
Histopathologic Features in IgG4-RD Specimens With IgG4/IgG Cutoffs of Greater Than or Less Than 40%
Clinical Features
The mean age of the overall cohort was 58 years old (range, 33–71 yr), and 73% of the patients were male. There were no statistical differences between the IgG4-related and non-IgG4-related groups in terms of age, sex, and comorbidities including cardiovascular disease, hypertension, diabetes, pelvic malignancy, and the presence of other immune-mediated conditions (Table 2).
TABLE 2.
Clinical Characteristics of Patients With IgG4-Related RPF and Non-IgG4-Related RPF
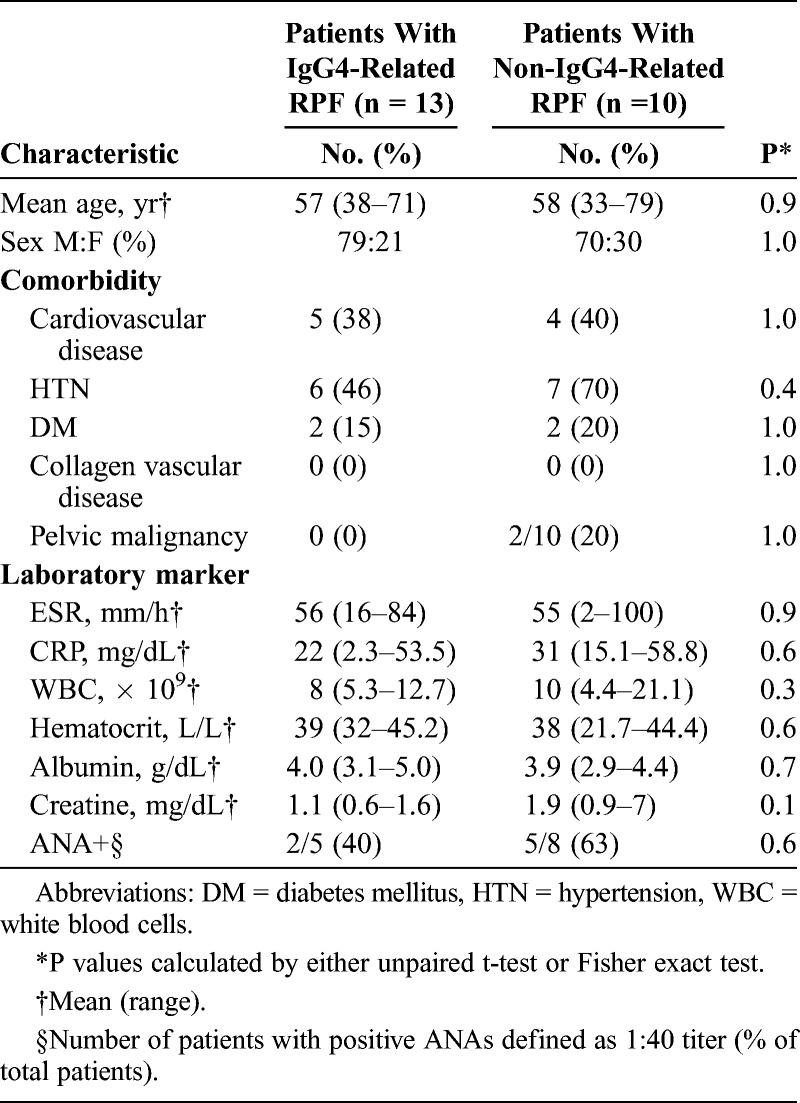
The clinical symptoms were not different between IgG4-related and non-IgG4-related groups. The most common presenting symptoms in both groups were back and flank pain, followed by lower extremity swelling. The laboratory values revealed no statistical differences between the 2 groups. The ESR and CRP were elevated in both groups: 56 mm/h (normal range, 0–11 mm/h) in the IgG4-related group and 55 mm/h in the non-IgG4-related group. The complete blood counts and serum albumin concentrations did not differ between the groups. The serum creatinine concentration was higher in the non-IgG4-related group (1.9 mg/dL vs. 1.1 mg/dL; normal <1.4 mg/dL), but this comparison did not achieve statistical significance.
The patients in the IgG4-related group had a mean serum IgG4 concentration of 107 mg/dL (range, 3–386 mg/dL; normal <135 mg/dL), but serum concentrations were measured in only 8 patients. There were only 2 serum IgG4 concentrations (16 and 18 mg/dL, respectively) measured in the non-IgG4-related group.
Radiologic Features
The radiologic features of the 2 groups are summarized in Table 3. No radiologic features were specific for IgG4-related RPF. The location of the retroperitoneal masses in both groups was periaortic (Figure 3A), and, in the majority of cases, also involved the periiliac arterial region. A minority of patients in both groups had additional involvement in pericaval, presacral, retrovesicular, or perirectal locations. The maximal thickness, defined as the broadest dimension measured in 3 different directions, was nearly identical in the IgG4-related and non-IgG4-related groups: 0.9 cm and 1.0 cm, respectively. Of the 19 patients who underwent thoracic imaging, none was found to have concurrent thoracic aortitis or periaortitis.
TABLE 3.
Radiologic Features of the IgG4-Related RPF and Non-IgG4-Related RPF Groups
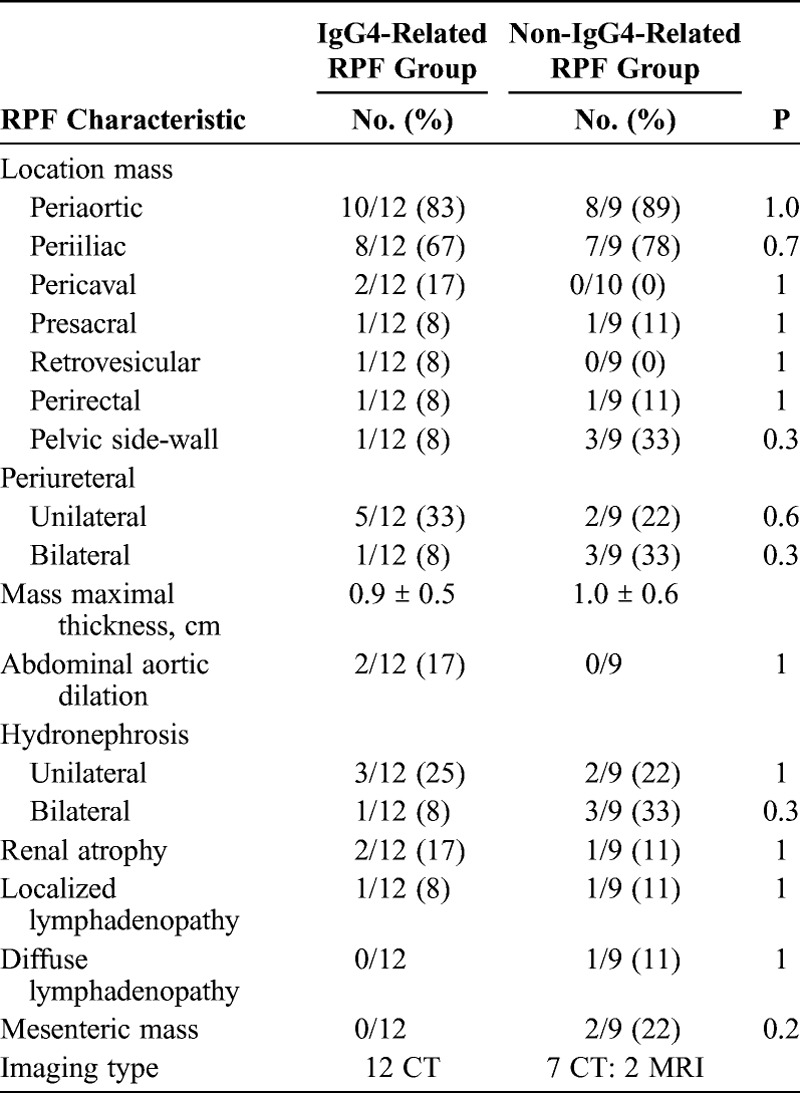
FIGURE 3.
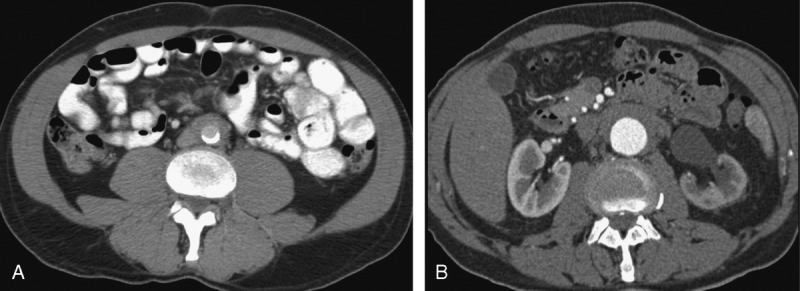
A, Abdominal and pelvic CT scan of a patient with known IgG4-related RPF displaying a soft tissue periaortic mantle. B, Abdominal and pelvic CT scan of a patient with IgG4-related RPF showing a periaortic soft tissue and left hydronephrosis with asymmetric nephrogram on the left side.
Two patients with IgG4-related RPF had aortic dilatation. This finding can be considered as periaortitis, which has been reported in association with IgG4-RD.3,37 Unilateral or bilateral hydronephrosis was found in 33% of IgG4-related RPF patients and 55% of non-IgG4-related RPF cases (Figure 3B). Renal atrophy was present in only 3 cases, and lymphadenopathy was detected in 2.
Other Organ Involvement
Six patients (46%) with IgG4-related RPF had biopsy-proven disease manifestations of IgG4-RD beyond the retroperitoneum. The extraretroperitoneal organ involvement included the orbits, lungs, salivary glands, kidneys, and lymph nodes. The non-IgG4-RD groups did not have IgG4-RD involvement recognized beyond the retroperitoneum.
Treatment
The most common first-line therapy, glucocorticoid treatment, was employed in 68% of the cases. Four patients, 2 in each group, did not receive any treatment at all for RPF. Patients with idiopathic RPF received a number of maintenance therapies (Table 4). The types of maintenance medications used in the IgG4-related RPF and non-IgG4-related RPF groups were similar and included prednisone (15 patients), rituximab (5 patients), tamoxifen (1 patient), azathioprine (1 patient), methotrexate (1 patient), and mycophenolate (1 patient).
TABLE 4.
Treatment Summary of IgG4-Related RPF and Non-IgG4-Related RPF
Six total patients (4 with IgG4-related RPF) were treated with rituximab. Of these, 4 (2 with IgG4-related RPF; 2 with non-IgG4-related RPF) tapered their prednisone successfully and had remained off of glucocorticoids for periods of 6, 12, 14, and 25 months at the end of study. The other 2 patients, both of whom had IgG4-related RPF, tapered their prednisone significantly, and their ureteral stents were removed.
Nearly half of the patients, 10 of 22 (45%), required the placement of ureteral stents. Seven patients (32%) underwent ureterolysis procedures. Despite these interventions, chronic kidney disease developed in 7 patients (32%): 2 in the IgG4-related RPF group and 5 in the non-IgG4-related RPF group. Of these 7 patients with chronic kidney disease, 6 developed renal failure due to recurrent obstruction, and 1 patient developed tubulointerstitial nephritis. One patient progressed to end-stage renal disease and required dialysis.
Cases Reports From the IgG4-Related RPF Group
Two cases from the IgG4-related RPF group illustrate important points pertaining to the multiorgan nature of IgG4-RD and the response of IgG4-related RPF to treatment even after the emergence of extensive fibrosis.
Case 1
A 59-year-old-man with a 15-year history of RPF presented for consultation on treatment options. He had presented initially with bilateral flank pain, and, after 6 months of severe pain, a CT scan of the abdomen revealed a rind of soft tissue extending from the renal arteries to the iliac bifurcation, without hydronephrosis. A needle biopsy performed at that time showed chronic inflammation without signs of malignancy.
Idiopathic RPF was diagnosed. The patient was started and maintained on prednisone 10 mg daily. Several years after his initial presentation, he developed worsening flank pain and a repeat abdominal CT revealed right-sided hydronephrosis. A ureteral stent was placed, which required changing every 6 months for a decade. His stent requirements were associated with recurrent urinary tract infections.
Several months before referral to Rheumatology Clinic, the patient experienced a disease flare, characterized by worsening flank pain and an abdominal CT scan that revealed extension of the previously stable soft tissue density to the renal arteries bilaterally and the right iliac artery. His prednisone dose was increased to 60 mg and could not be tapered below 30 mg daily without recurrent flank pain.
Upon referral to Rheumatology Clinic, review of the patient’s retroperitoneal biopsy from 15 years earlier showed a lymphoplasmacytic infiltrate, storiform fibrosis, and mild tissue eosinophilia (Figure 4). Immunostaining for IgG4 and IgG were also performed on the extant tissue blocks. There were 4 IgG4+ plasma cells/HPF, and the IgG4/total IgG ratio was 50%. He received rituximab 1 g for 2 doses spaced 14 days apart. This was accompanied by resolution of flank pain and the successful taper of his prednisone dose to discontinuation. He was also able to stop using daily narcotics for the first time in a decade. His ureteral stents were removed and have not been replaced. He has not had a urinary tract infection since his rituximab infusion 2 years ago.
FIGURE 4.
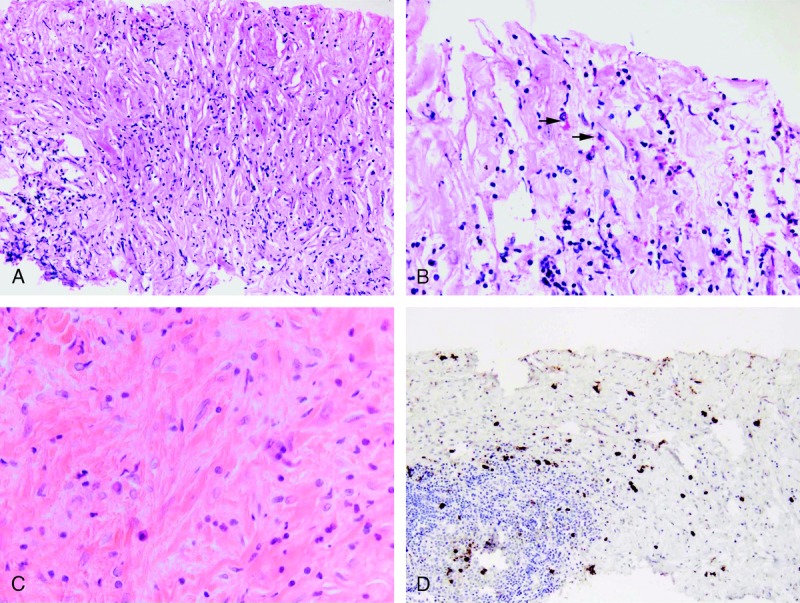
Histopathology of Cases 1 and 2. Case 1. Cellular fibroinflammatory infiltrate arranged in a storiform pattern (A). The infiltrate was also rich in eosinophils (arrows) (B). Case 2. The biopsy is dominated by fibrosis with only a few lymphocytes and plasma cells (C). Immunohistochemical stain for IgG4 (D): 13 IgG4+ plasma cells were identified per hpf. [This figure can be viewed in color online at http://www.md-journal.com].
Case 2
A 65-year-old woman was referred for suspicion of IgG4-RD. Fifteen years before her presentation, she had developed bilateral submandibular gland swelling. She underwent removal of 1 gland, and the histopathology was interpreted as chronic inflammation with lymphocytes, plasma cells, and fibrosis. This tissue had been discarded several years before and could not be examined further.
The patient also had a 10-year history of idiopathic RPF. She had presented with acute renal failure caused by bilateral renal entrapment. Her renal function stabilized following bilateral stent placements, and the patient underwent a series of medical treatments over the next 4 years, including courses of prednisone, tamoxifen, azathioprine, and cyclophosphamide, none of which appeared to help her RPF. She continued on 8 mg/d of prednisone for the control of constitutional symptoms. A ureterolysis procedure led to injury of the left ureter. The patient required several stent changes and even a nephrostomy tube in the left kidney following an exacerbation of postobstructive renal failure.
Review of her retroperitoneal biopsy specimen from a decade earlier showed a moderate lymphoplasmacytic infiltrate, storiform fibrosis, and moderate tissue eosinophilia (>5 cells/hpf). There was a mean of 13 IgG4+ plasma cells/hpf and an IgG4/IgG ratio of 68% (see Figure 4). Despite years of continuous glucocorticoid use, her serum IgG4 level was 154 mg/dL (reference range, >135 mg/dL).
The patient received 2 doses of rituximab (1 g each), 2 weeks apart. Her IgG4 serum declined to 47 mg/dL 1 month after her infusion. Her constitutional symptoms improved such that she was able to taper her prednisone to 2 mg/d, a dose she maintained because of adrenal insufficiency. Her ureteral stents remained in place at the time of this writing.
DISCUSSION
To our knowledge, the current study is the first in a North American population to demonstrate that a significant proportion of “idiopathic” RPF cases are in fact part of the spectrum of IgG4-RD.11,13,30,52,54,55 More than half of the cases in our study (13/23; 57%) were found to be IgG4-related RPF. The small number of other studies have reported disparate findings on the relationship between RPF and IgG4-RD. Our data provide important indications about why the association between these overlapping conditions was difficult to identify for so long and why the link is often overlooked even now.
The current study demonstrates that the IgG4/total IgG ratio within the affected retroperitoneal tissue is particularly helpful in making the diagnosis in the retroperitoneum, where abundant fibrosis often militates against an extensive cellular infiltrate. We used a minimum IgG4/total IgG ratio of 40% in this study to distinguish initially between cases of idiopathic RPF that might be associated with IgG4-RD, an approach consistent with that used in another major study of this condition.54 Although immunostaining and the IgG4/total IgG ratio were essential to the design of this study, it must be emphasized that immunostaining features of retroperitoneal tissue (and all tissues potentially affected by IgG4-RD) must be interpreted in the context of the histopathologic findings. The findings of lymphoplasmacytic infiltrates, storiform fibrosis, obliterative phlebitis, and eosinophilia generally play central roles. Striking correlations existed in our series between the histopathologic findings of storiform fibrosis, lymphoplasmacytic infiltrates, eosinophilia, and elevated IgG4/total IgG ratios.
In a study of 29 patients, Yamashita et al52 demonstrated that patients with RPF occurring in the setting of multifocal fibrosclerosis had greater numbers of IgG4+ plasma cells within tissue compared to those whose RPF was associated with other conditions. Zen et al54 reported 10 IgG4-related RPF cases out of 17 cases with a high prevalence of IgG4-RD manifestations beyond the retroperitoneum in patients with IgG4-related RPF (5 of 10 patients; 50%). In a 2011 randomized clinical trial of prednisone and tamoxifen by Vaglio et al48—to our knowledge the only prospective study of this question performed to date—IgG4-related RPF counted for only 29% of the RPF cases (4 of 14 patients).
Our data are compatible with those of studies from Japan by Yamashita et al52 and Zen et al,54 and are also consistent with the findings of studies in many other organ systems published in recent years: IgG4-RD accounts for substantial proportions—sometimes the majority—of cases of idiopathic inflammation occurring within the orbit,24,38 salivary glands,10,21 biliary tree,14,16,31,47,56 aorta,42 and pachymeninges,23 among others.1,41 The potential for IgG4-RD to cause “idiopathic” inflammation in virtually any organ system, particularly that associated with lymphoplasmacytic inflammation at an early stage of the disease process, makes it critical for pathologists, clinicians, and radiologists to be familiar with the protean manifestations of this condition.
Careful pathologic evaluation of an adequate tissue biopsy is essential for pinpointing more precise etiologies in idiopathic RPF. Causes of secondary RPF such as lymphoma, carcinoma, or sarcoma require tissue biopsies to differentiate these causes from each other and from inflammatory causes. Our series demonstrates that the clinical, serologic, and radiologic characteristics are seldom useful in distinguishing among cases of idiopathic RPF. Patients in both of our disease subsets—that is, those with and without IgG4-related RPF—tended to be middle-aged males with similar comorbid medical conditions. Serum IgG4 concentrations were not dramatically elevated in most of our IgG4-RPF patients, and other serologic markers did not distinguish reliably between the subsets. Finally, radiologic features such as the location of the retroperitoneal mass, the presence of hydronephrosis, and the occurrence of regional lymphadenopathy were not sufficiently distinctive to distinguish the 2 groups clearly.
Although it is possible that idiopathic RPF and IgG4-related RPF are phases of the same disease, their respective histopathologic features are distinctive even at stages of advanced fibrosis: storiform fibrosis is prominent in IgG4-RD, compared with the nonspecific hyalinized sheets of fibrosis typical of non-IgG4-related RPF. This finding, as well as other findings typical of IgG4-RD—such as obliterative phlebitis and eosinophilia—was enriched among the cases with high IgG4/total IgG ratios and frequent evidence of IgG4-RD in other organs. The mean times between initial symptoms and biopsy were similar between the IgG4-related and non-IgG4-related RPF groups: 6 and 4 months, respectively.
Inspection of the histopathologic and immunostaining features of the involved tissues offers the most direct and often the only way of delineating IgG4-related RPF cases from those that are truly idiopathic. Needle biopsies are an appropriate first approach to obtaining a tissue diagnosis and are particularly effective at excluding cancer. However, low thresholds should be maintained for laparoscopic biopsies if needle biopsies do not provide a tissue sample adequate for investigating inflammatory causes of RPF. One likely explanation for the higher tissue concentrations of IgG4+ plasma cells in the study of Zen et al54 stems from the fact that most of the retroperitoneal samples investigated in that study were obtained at surgery, thereby offering substantially larger sections of tissue than those provided from needle biopsies. Counts of IgG4-positive cells are typically performed by pathologists in areas with the highest concentrations of IgG4 staining, and larger samples therefore diminish the risk of low counts caused by sampling error.5 We note, however, that the mean IgG4/IgG ratios among patients with IgG4-related RPF were similar between our study and that of Zen et al, 57% and 59%, respectively.
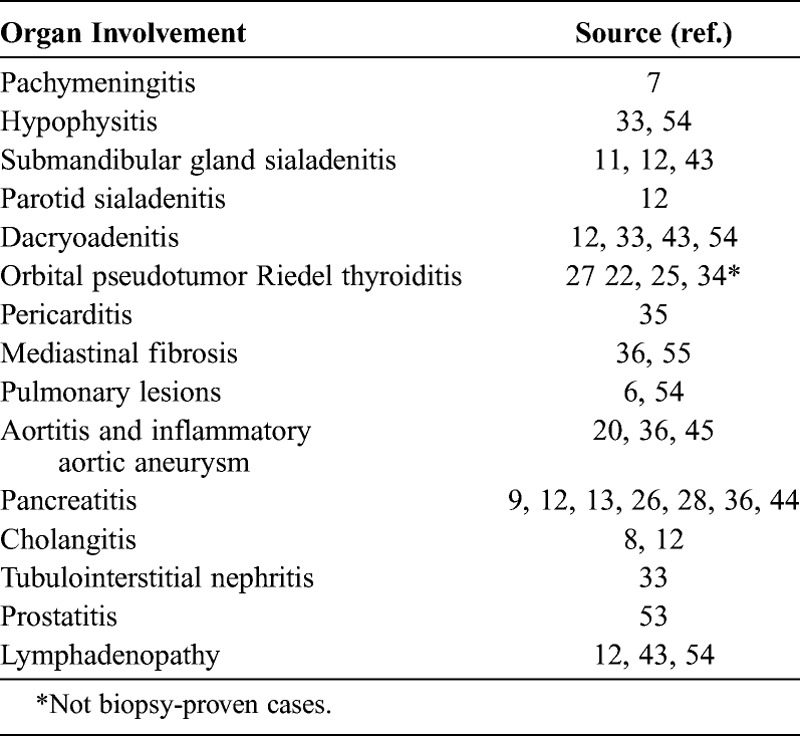
The possibility of securing a diagnosis through biopsy of an organ outside the retroperitoneum should not be overlooked. As examples, the lacrimal, parotid, or submandibular glands offer easily accessible sites for biopsy, as do the skin and lymph nodes when these organs are involved. The extraretroperitoneal organ manifestations associated with IgG4-related RPF that have been reported in the literature are summarized in Table 5. Half of the patients with IgG4-related RPF in this study had extraretroperitoneal disease. Retroperitoneal disease was recognized as the sole site of involvement in 7 of the 13 patients (54%).
TABLE 5.
Extraretroperitoneal Organ Manifestations of IgG4-Related RPF, Previous Reports
The absolute numbers of IgG4+ plasma cells were low in our IgG4-related RPF group compared to other organs that are classic sites for IgG4-RD involvement, such as the lacrimal or submandibular glands. Our patients with IgG4-related RPF had a mean of only 13 IgG4+ plasma cells/HPF in their retroperitoneal biopsies. The use of needle biopsies to sample the retroperitoneum is a partial explanation for this, but it is also possible that the nonspecific clinical presentation of RPF ultimately makes parsing the histopathologic diagnosis more challenging. In contrast to the lacrimal or submandibular glands, in which the organ dysfunction is visually obvious to the patient and to others, retroperitoneal disease likely has a significantly longer interval between the onset of inflammation in the retroperitoneum and the performance of biopsies to establish the diagnosis. One result of the diagnostic delay in the retroperitoneum is that biopsies from RPF patients are likely to be less cellular and more fibrotic than are biopsies from other organs. The lower numbers of IgG4+ plasma cells in cases of IgG4-related RPF correspond with the overall lower degree of cellular inflammation as opposed to fibrosis. Although the numbers of IgG4+ cells in the retroperitoneum are low compared to numbers in other organs involved in IgG4-RD, the histopathologic features appear to be preserved in IgG4-related RPF, particularly the storiform type of fibrosis. Thus, the morphologic appearance of the tissues is the key to the diagnosis of IgG4-RPF, and represents a more sensitive and specific indicator than do the absolute numbers of IgG4-positive cells.
The characteristic IgG4-related histopathologic features of lymphoplasmacytic infiltrates, storiform fibrosis, and eosinophilia correlated strongly with higher IgG4/IgG ratios. These histopathologic features are essential in distinguishing IgG4-related RPF from non-IgG4-related RPF. A recent pathology consensus statement highlights these features as the key histopathologic features of IgG4-RD.5 Tissue eosinophilia and phlebitis without obliteration are also sometimes striking features of this condition. Our experience with RPF suggests that eosinophilia is a useful discriminator between IgG4-related and non-IgG4-related RPF, and that obliterative phlebitis, although less common (at least in the needle biopsies studied here), is likely to be a marker of IgG4-RD when present.
The treatment approaches used in our patients were varied, and no treatments—even glucocorticoids—were employed often enough to permit firm conclusions. However, our limited experience to date with the use of a B-cell depletion strategy in IgG4-related RPF was consistent with that observed in other parts of the IgG4-RD disease spectrum.17,19
In conclusion, the current series provides support for the concept that a substantial proportion of patients with idiopathic RPF have a diagnosis that falls within the spectrum of IgG4-RD. We propose the term IgG4-related RPF for this subset of patients.40 Detailed, prospective study of RPF is necessary to distinguish the exact clinical and treatment characteristics of the IgG4-related subset from the non-IgG4-related RPF subset. Our study was limited due to its retrospective nature with variable access to serologic data, clinical follow-up, and treatment experience.
Abbreviations
- ANA
antinuclear antibody
- CRP
C-reactive protein
- CT
computed tomography
- ESR
erythrocyte sedimentation rate
- HPF
high-power field
- IgG4-RD
IgG4-related disease
- RPF
retroperitoneal fibrosis
Footnotes
Drs. Khosroshahi and Carruthers contributed equally to this manuscript.
Dr. Khosroshahi’s current address/affiliaton is Rheumatology Division, Department of Medicine, Emory University, Atlanta, Georgia.
Figures 2 and 4 can be viewed in color online at http://www.md-journal.com.
Financial support and conflicts of interest: Dr. Stone reports financial support for work outside this study from Genentech (visiting lectureship and grant support to his institution for open label trial of rituximab in IgG4-related disease) and Roche (consultancy and grant support to his institution for multicenter clinical trial). Dr. Hasserjian reports financial support for work outside this study from Genzyme (consultancy) and Amgen (consultancy), as well as support from the Colorado Society of Pathologists, Elsevier, American Society for Clinical Pathology, United States and Canadian Academy of Pathology, and Harvard Medical School (for lectures, royalties, and/or travel expenses).
The other authors have no funding or conflicts of interest to disclose.
REFERENCES
- 1. Carruthers MN, Stone JH, Khosroshahi A. The latest on IgG4-RD: a rapidly emerging disease. Curr Opin Rheumatol. 2012; 24: 60– 69. [DOI] [PubMed] [Google Scholar]
- 2. Comings DE, Skubi KB, Van Eyes J, Motulsky AG. Familial multifocal fibrosclerosis. Findings suggesting that retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel’s thyroiditis, and pseudotumor of the orbit may be different manifestations of a single disease. Ann Intern Med. 1967; 66: 884– 892. [DOI] [PubMed] [Google Scholar]
- 3. Corradi D, Maestri R, Palmisano A, Bosio S, Greco P, Manenti L, Ferretti S, Cobelli R, Moroni G, Dei Tos AP, Buzio C, Vaglio A. Idiopathic retroperitoneal fibrosis: clinicopathologic features and differential diagnosis. Kidney Int. 2007; 72: 742– 753. [DOI] [PubMed] [Google Scholar]
- 4. Dahlgren M, Khosroshahi A, Nielsen GP, Deshpande V, Stone JH. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken). 2010; 62: 1312– 1318. [DOI] [PubMed] [Google Scholar]
- 5. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Kloppel G, Heathcote JG, Khosroshahi A, Ferry JA, Aalberse RC, Bloch DB, Brugge WR, Bateman AC, Carruthers MN, Chari ST, Cheuk W, Cornell LD, Fernandez-Del Castillo C, Forcione DG, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Lauwers GY, Masaki Y, Nakanuma Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani DV, Smyrk TC, Stone JR, Takahira M, Webster GJ, Yamamoto M, Zamboni G, Umehara H, Stone JH. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012; 25: 1181– 1192. [DOI] [PubMed] [Google Scholar]
- 6. Duvic C, Desrame J, Leveque C, Nedelec G. Retroperitoneal fibrosis, sclerosing pancreatitis and bronchiolitis obliterans with organizing pneumonia. Nephrol Dial Transplant. 2004; 19: 2397– 2399. [DOI] [PubMed] [Google Scholar]
- 7. Fukuda W, Kimura M, Akaogi T, Sako M, Ohiwa K, Yamamoto Y, Kato G, Hayashi H, Yoshikawa T. Multifocal fibrosclerosis: retroperitoneal fibrosis associated with a suprasellar tumor and pachymeningitis. Intern Med. 2003; 42: 1006– 1010. [DOI] [PubMed] [Google Scholar]
- 8. Fukui T, Okazaki K, Yoshizawa H, Ohashi S, Tamaki H, Kawasaki K, Matsuura M, Asada M, Nakase H, Nakashima Y, Nishio A, Chiba T. A case of autoimmune pancreatitis associated with sclerosing cholangitis, retroperitoneal fibrosis and Sjogren’s syndrome. Pancreatology. 2005; 5: 86– 91. [DOI] [PubMed] [Google Scholar]
- 9. Fukukura Y, Fujiyoshi F, Nakamura F, Hamada H, Nakajo M. Autoimmune pancreatitis associated with idiopathic retroperitoneal fibrosis. AJR Am J Roentgenol. 2003; 181: 993– 995. [DOI] [PubMed] [Google Scholar]
- 10. Geyer JT, Ferry JA, Harris NL, Stone JH, Zukerberg LR, Lauwers GY, Pilch BZ, Deshpande V. Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol. 2010; 34: 202– 210. [DOI] [PubMed] [Google Scholar]
- 11. Gill J, Taylor G, Carpenter L, Lewis C, Chiu W. A case of hyperIgG4 disease or IgG4-related sclerosing disease presenting as RPF, chronic sclerosing sialadenitis and mediastinal lymphadenopathy. Pathology. 2009; 41: 297– 300. [DOI] [PubMed] [Google Scholar]
- 12. Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol. 2006; 41: 1197– 1205. [DOI] [PubMed] [Google Scholar]
- 13. Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, Shimojo H, Kiyosawa K. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002; 359: 1403– 1404. [DOI] [PubMed] [Google Scholar]
- 14. Hirano K, Shiratori Y, Komatsu Y, Yamamoto N, Sasahira N, Toda N, Isayama H, Tada M, Tsujino T, Nakata R, Kawase T, Katamoto T, Kawabe T, Omata M. Involvement of the biliary system in autoimmune pancreatitis: a follow-up study. Clin Gastroenterol Hepatol. 2003; 1: 453– 464. [DOI] [PubMed] [Google Scholar]
- 15. Kamisawa T, Anjiki H, Egawa N, Kubota N. Allergic manifestations in autoimmune pancreatitis. Eur J Gastroenterol Hepatol. 2009; 21: 1136– 1139. [DOI] [PubMed] [Google Scholar]
- 16. Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology. 2006; 6: 132– 137. [DOI] [PubMed] [Google Scholar]
- 17. Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012; 91: 57– 66. [DOI] [PubMed] [Google Scholar]
- 18. Khosroshahi A, Carruthers MD, Deshpande V, Leb L, Reed JI, Stone JH. Cutaneous immunoglobulin G4-related systemic disease. Am J Med. 2011; 124: e7– e8. [DOI] [PubMed] [Google Scholar]
- 19. Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010; 62: 1755– 1762. [DOI] [PubMed] [Google Scholar]
- 20. Khosroshahi A, Stone JR, Pratt DS, Deshpande V, Stone JH. Painless jaundice with serial multi-organ dysfunction. Lancet. 2009; 373: 1494. [DOI] [PubMed] [Google Scholar]
- 21. Kitagawa S, Zen Y, Harada K, Sasaki M, Sato Y, Minato H, Watanabe K, Kurumaya H, Katayanagi K, Masuda S, Niwa H, Tsuneyama K, Saito K, Haratake J, Takagawa K, Nakanuma Y. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner’s tumor). Am J Surg Pathol. 2005; 29: 783– 791. [DOI] [PubMed] [Google Scholar]
- 22. Kruit WH, den Ottolander GJ. Riedel’s thyroiditis in a patient with retroperitoneal fibrosis. Neth J Med. 1991; 39: 17– 19. [PubMed] [Google Scholar]
- 23. Lindstrom KM, Cousar JB, Lopes MB. IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol. 2010; 120: 765– 776. [DOI] [PubMed] [Google Scholar]
- 24. Mehta M, Jakobiec F, Fay A. Idiopathic fibroinflammatory disease of the face, eyelids, and periorbital membrane with immunoglobulin G4-positive plasma cells. Arch Pathol Lab Med. 2009; 133: 1251– 1255. [DOI] [PubMed] [Google Scholar]
- 25. Miro I, Bakir R, Chanu B, Brocheriou C, Brenot F, Rouffy J. [Riedel’s thyroiditis and retroperitoneal fibrosis. Apropos of a case of multiple fibrosing disease.] Ann Med Interne (Paris). 1984; 135: 212– 216. [PubMed] [Google Scholar]
- 26. Miyajima N, Koike H, Kawaguchi M, Zen Y, Takahashi K, Hara N. Idiopathic retroperitoneal fibrosis associated with IgG4-positive plasmacyte infiltrations and idiopathic chronic pancreatitis. Int J Urol. 2006; 13: 1442– 1444. [DOI] [PubMed] [Google Scholar]
- 27. Murawska J, Lipowski P, Raczynska K. [Orbital pseudotumor due to Ormond’s disease. Klin Oczna. 2008; 110: 75– 77. [PubMed] [Google Scholar]
- 28. Naitoh I, Nakazawa T, Ohara H, Ando T, Hayashi K, Tanaka H, Okumura F, Miyabe K, Yoshida M, Sano H, Takada H, Joh T. Clinical significance of extrapancreatic lesions in autoimmune pancreatitis. Pancreas. 2010; 39: e1– e5. [DOI] [PubMed] [Google Scholar]
- 29.National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002; 39 (2 Suppl 1): S1– S266. [PubMed] [Google Scholar]
- 30. Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med. 2006; 4: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishino T, Toki F, Oyama H, Oi I, Kobayashi M, Takasaki K, Shiratori K. Biliary tract involvement in autoimmune pancreatitis. Pancreas. 2005; 30: 76– 82. [PubMed] [Google Scholar]
- 32. Ormond JK. Bilateral ureteral obstruction due to envelopment and compression by an inflammatory process. J Urol. 1948; 59: 1072– 1079. [DOI] [PubMed] [Google Scholar]
- 33. Patel SM, Szostek JH. IgG4-related systemic disease in a Native American man. Intern Med. 2011; 50: 931– 934. [DOI] [PubMed] [Google Scholar]
- 34. Rao CR, Ferguson GC, Kyle VN. Retroperitoneal fibrosis associated with Riedel’s struma. Can Med Assoc J. 1973; 108: 1019– 1021. [PMC free article] [PubMed] [Google Scholar]
- 35. Rebelo M, Lima J, Ramos L, Vieira JD, Carvalho L, Xavier C, Costa JN. The intriguing co-existence of a chronic periaortitis, a pericarditis and a pancreatitis: case report. Acta Reumatol Port. 2011; 36: 160– 166. [PubMed] [Google Scholar]
- 36. Sakamoto A, Nagai R, Saito K, Imai Y, Takahashi M, Hosoya Y, Takeda N, Hirano K, Koike K, Enomoto Y, Kume H, Homma Y, Maeda D, Yamada H, Fukayama M, Hirata Y, Ishizaka N. Idiopathic retroperitoneal fibrosis, inflammatory aortic aneurysm, and inflammatory pericarditis—retrospective analysis of 11 case histories. J Cardiol. 2012; 59: 139– 146. [DOI] [PubMed] [Google Scholar]
- 37. Sakata N, Tashiro T, Uesugi N, Kawara T, Furuya K, Hirata Y, Iwasaki H, Kojima M. IgG4-positive plasma cells in inflammatory abdominal aortic aneurysm: the possibility of an aortic manifestation of IgG4-related sclerosing disease. Am J Surg Pathol. 2008; 32: 553– 559. [DOI] [PubMed] [Google Scholar]
- 38. Sato Y, Ohshima K, Ichimura K, Sato M, Yamadori I, Tanaka T, Takata K, Morito T, Kondo E, Yoshino T. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008; 58: 465– 470. [DOI] [PubMed] [Google Scholar]
- 39. Scheel PJ, Jr, Feeley N. Retroperitoneal fibrosis: the clinical, laboratory, and radiographic presentation. Medicine (Baltimore). 2009; 88: 202– 207. [DOI] [PubMed] [Google Scholar]
- 40. Stone JH, Khosroshahi A, Deshpande V, Chan J, Heathcote J, Aalberse R, Azumi A, Bloch DB, Brugge WR, Carruthers MN, Cheuk W, Cornell L, Castillo CF, Ferry JA, Forcione D, Kloppel G, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Masaki Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani D, Sato Y, Smyrk T, Stone JR, Takahira M, Umehara H, Webster G, Yamamoto M, Yi E, Yoshino T, Zamboni G, Zen Y, Chari S. IgG4-related disease: recommendations for the nomenclature of this condition and its individual organ system manifestations. Arthritis Rheum. 2012; 64: 3061– 3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012; 366: 539– 551. [DOI] [PubMed] [Google Scholar]
- 42. Stone JH, Khosroshahi A, Hilgenberg A, Spooner A, Isselbacher EM, Stone JR. IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum. 2009; 60: 3139– 3145. [DOI] [PubMed] [Google Scholar]
- 43. Takenaka K, Takada K, Kobayashi D, Moriguchi M, Harigai M, Miyasaka N. A case of IgG4-related disease with features of Mikulicz’s disease, and retroperitoneal fibrosis and lymphadenopathy mimicking Castleman’s disease. Mod Rheumatol. 2011; 21: 410– 414. [DOI] [PubMed] [Google Scholar]
- 44. Takuma K, Kamisawa T, Anjiki H, Egawa N, Igarashi Y. Metachronous extrapancreatic lesions in autoimmune pancreatitis. Intern Med. 2010; 49: 529– 533. [DOI] [PubMed] [Google Scholar]
- 45. Trinidad-Hernandez M, Duncan AA. Contained ruptured paravisceral aortic aneurysm related to immunoglobulin G4 aortitis. Ann Vasc Surg. 2012; 26: 108.e1-e4. [DOI] [PubMed] [Google Scholar]
- 46. Uehara T, Hamano H, Kawakami M, Koyama M, Kawa S, Sano K, Honda T, Oki K, Ota H. Autoimmune pancreatitis-associated prostatitis: distinct clinicopathological entity. Pathol Int. 2008; 58: 118– 125. [DOI] [PubMed] [Google Scholar]
- 47. Uehara T, Hamano H, Kawa S, Sano K, Honda T, Ota H. Distinct clinicopathological entity “autoimmune pancreatitis-associated sclerosing cholangitis”. Pathol Int. 2005; 55: 405- 411. [DOI] [PubMed] [Google Scholar]
- 48. Vaglio A, Palmisano A, Alberici F, Maggiore U, Ferretti S, Cobelli R, Ferrozzi F, Corradi D, Salvarani C, Buzio C. Prednisone versus tamoxifen in patients with idiopathic retroperitoneal fibrosis: an open-label randomised controlled trial. Lancet. 2011; 378: 338– 346. [DOI] [PubMed] [Google Scholar]
- 49. Vaglio A, Palmisano A, Corradi D, Salvarani C, Buzio C. Retroperitoneal fibrosis: evolving concepts. Rheum Dis Clin North Am. 2007; 33: 803– 817, vi-vii. [DOI] [PubMed] [Google Scholar]
- 50. Vaglio A, Salvarani C, Buzio C. Retroperitoneal fibrosis. Lancet. 2006; 367: 241– 251. [DOI] [PubMed] [Google Scholar]
- 51. Van Bommel EF, Jansen I, Hendriksz TR, Aarnoudse AL. Idiopathic retroperitoneal fibrosis: prospective evaluation of incidence and clinicoradiologic presentation. Medicine (Baltimore). 2009; 88: 193– 201. [DOI] [PubMed] [Google Scholar]
- 52. Yamashita K, Haga H, Mikami Y, Kanematsu A, Nakashima Y, Kotani H, Ogawa O, Manabe T. Degree of IgG4+ plasma cell infiltration in retroperitoneal fibrosis with or without multifocal fibrosclerosis. Histopathology. 2008; 52: 387– 411. [DOI] [PubMed] [Google Scholar]
- 53. Zaidan M, Adam J, Cervera-Pierot P, Joly D. The case a 69-year-old man with a 10-year history of idiopathic retroperitoneal fibrosis. Kidney Int. 2011; 80: 1379– 1380. [DOI] [PubMed] [Google Scholar]
- 54. Zen Y, Onodera M, Inoue D, Kitao A, Matsui O, Nohara T, Namiki M, Kasashima S, Kawashima A, Matsumoto Y, Katayanagi K, Murata T, Ishizawa S, Hosaka N, Kuriki K, Nakanuma Y. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol. 2009; 33: 1833– 1839. [DOI] [PubMed] [Google Scholar]
- 55. Zen Y, Sawazaki A, Miyayama S, Notsumata K, Tanaka N, Nakanuma Y. A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis). Hum Pathol. 2006; 37: 239– 243. [DOI] [PubMed] [Google Scholar]
- 56. Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, Kurumaya H, Katayanagi K, Masuda S, Niwa H, Morimoto H, Miwa A, Uchiyama A, Portmann BC, Nakanuma Y. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004; 28: 1193– 1203. [DOI] [PubMed] [Google Scholar]