

Abstract
Epigenetics refers to changes in cell characteristics that occur independently of modifications to the deoxyribonucleic acid (DNA) sequence. Alterations mediated by epigenetic mechanisms are important factors in cancer progression. Although an exciting prospect, the identification of early epigenetic markers associated with clinical outcome in premalignant and malignant disorders remains elusive. We examined alterations in chromatin acetylation in oral lichen planus (OLP) with distinct clinical behavior and compared the alterations to the levels of DNA double-strand breaks (DSBs). We analyzed 42 OLP patients, who had different responses to therapy, for acetyl-histone H3 at lys9 (H3K9ac), which is associated with enhanced transcription and nuclear decondensation, and the presence of DSBs, as determined by accumulation of phosphorylated γH2AX foci. Patients with high levels of H3K9ac acetylation failed to respond to therapy or experienced disease recurrence shortly after therapy. Similar to H3K9ac, patients who responded poorly to therapy had increased accumulation of DNA DSB, indicating genomic instability. These findings suggest that histone modifications occur in OLP, and H3K9ac and γH2AX histones may serve as epigenetic markers for OLP recurrence.
INTRODUCTION
From the discovery of oncogenes in the late 1980s to cancer genome sequencing during the Cancer Genome Project to the latest findings using next-generation sequencing technology, it is evident that the cancer genomic landscape is far more complex than anticipated. Known as a genetic disease, cancers are also susceptible to epigenetic events that regulate gene expression independently of deoxyribonucleic acid (DNA) mutations. New evidence suggests that epigenetic alterations including histone modifications are associated with the initial steps of carcinogenesis.
Histone modifications occur by acetylation, methylation, ubiquitination, phosphorylation, and sumoylation to directly influence DNA packaging and increase transcription.1,2 Histone modifications are detected during normal cellular plasticity in neurons and lymphocytes and play a major role in tumor behavior.3–5 In cancer cells, histone modifications dynamically promote transcription of prosurvival genes and silence tumor suppressor genes to support the deregulated cancer physiology. Therefore, by identifying early epigenetic modifications in lesions at risk for malignancy will help us to understand epigenetic events that dictate tumor formation and progression.
It has been challenging to identify early epigenetic markers associated with clinical outcome of potentially malignant disorder. Oral lichen planus (OLP) is a relatively common disease that affects the oral mucosa and is classified as potentially malignant disorder by the World Health Organization (WHO).6,7 The OLP clinical presentation can range from reticular, atrophic, to erosive lesions. The reticular lesions are asymptomatic and appear as bilateral white striae located specially in the buccal mucosa, tongue, and lips. Erosive and ulcerative lesions are associated with symptoms that range from a burning sensation to severe pain.8,9 The histopathologic aspects of OLP consist of atrophy of the surface epithelium with hyperkeratosis (except for erosive lesions), absent or saw-toothed rete ridges, and a band-like infiltrate of lymphocytes immediately subjacent to the basement membrane with associated destruction of the basal layer. Diagnosis of OLP should be made by evaluating both clinical and histological features.10–12 Several treatments have been proposed to OLP including topical or systemic corticosteroids, immunosuppressors, immunomodulators, and laser phototherapy (LPT).11,13
Although controversial, 1% to 2% of OLP becomes malignant. However, at present, there are no reliable predicting factors of malignant transformation that can be used. Interestingly, the etiology of OLP is unknown, but consensus agrees that this disorder involves the immune system; thus, OLP is characterized as an autoimmune disease. The presence of a substantial chronic inflammatory infiltrate primarily composed by T lymphocytes localized juxta epithelial have elicited comparing OLP to other inflammatory diseases that have greater potential for cellular transformation,14 including colon polyps, stomach gastritis, bronchial preneoplastic lesions, and Barret esophagus.15 Interestingly, epigenetic events have also been linked to the development of chronic inflammation by upregulating proinflammatory cytokines,16–20 reviewed by Coussens and Werb,15 and Lonkar and Dedon.21
Histones are the most abundant proteins associated with DNA and are related with the regulation of nuclear gene expression in several tissue types. The pattern of histone modifications determines chromatin status (euchromatin or heterochromatin), the accessibility of DNA to nuclear factors, and ultimately transcription.2,22 Alterations in chromatin structure due to histone modifications have been correlated with gene expression, the cell cycle, DNA replication and damage, DNA repair, and chromosome stability.2,23 Among all histone modifications, the process of global chromatin remodeling driven by acetylation of histones is still largely unknown. Histone acethylation results in a switch from repressive heterochromatin to permissive euchromatin.
Nonetheless, histone hypoacetylation (H4K12ac) is an effective epigenetic marker for colorectal cancer staging and for tumor recurrence of prostate and non-small cell lung cancers.20,24,25 In contrast, histone hyperacetylation occurs in hepatocellular carcinoma and head and neck squamous cell carcinomas.26,27 We have recently shown that histone modifications play a central role in the aggressiveness and resistance to chemotherapy observed in head and neck squamous cell carcinoma via upregulation of nuclear factor kappa-light-chain-enhancer of activated B cells, a molecule involved in cancer development, autoimmune diseases, and inflammation.5,28
In a previous randomized controlled trial performed by our group, we compared the efficacy of LPT to topical clobetasol propionate 0.05% for the treatment of 42 patients with atrophic and erosive OLP. We observed that, independent of the treatment used, the OLP lesions exhibited different clinical behavior. Based on these findings we decided to investigate the clinical relevance of hyperacetylation of histone H3 lys9 (H3K9ac) as an epigenetic marker for OLP disease behavior and response to therapy. Acetylation of H3K9 controls chromatin decondensation, chromatin assembly, and gene activation thereby being a great marker of transcriptionally active chromatin.29 Furthermore, we decided to associate the expression of H3K9ac with the identification of γ-H2AX phosphorylated on serine 139 (γH2AX) as an indirect monitor of DNA damage and DNA double-strand breaks (DSBs). Also, we focus on γH2AX histone foci because it is accepted that it reflects the number of DSBs and is a powerful tool to analyze DSB repair.30–32 Surprisingly, patients with high levels of H3K9ac either failed to respond to therapy or experienced disease recurrence shortly after therapy. Furthermore, similar to H3K9ac, patients who responded poorly to therapy had increased phosphorylation of γH2AX. These findings suggest that histone modifications are early events in OLP and that H3K9ac and γH2AX histones are promising epigenetic markers for OLP recurrence. Our findings also suggest that increased acetylation of H3K9ac and accumulation of phosphorylated γH2AX may help identify OLP patients with genomic instability above the average. Although the potential for OLP to become malignant is controversial, it is disturbing that increased genomic instability is associated with a poor response to therapy. We discuss these findings in light of recent developments in the molecular circuitries that regulate OLP.
METHODS
Ethics Statement
All human samples were derived from our previously published single-center, randomized, controlled, single-blind study and approved by the Human Research Ethics Committee (HCPA protocol 11-0365), as previously reported by our group.13
Patients and OLP Clinical Follow-Up
Medical records and histopathological slides from 42 patients with atrophic/erosive OLP were analyzed. All participants were clinically examined and submitted to oral biopsy during our previous study13 to establish the diagnosis of OLP. Inclusion criteria were age ≥21 years, symptomatic atrophic/erosive OLP, and histopathological diagnosis of OLP based on the criteria proposed by the WHO. The exclusion criteria were pregnant or nursing women, histological signs of dysplasia, OLP therapy in the previous 3 months, amalgam restoration near the lesions, and the use of medications associated with oral lichenoid reaction. The patients received topical clobetasol propionate 0.05% or applications of InGaAlP diode laser during the initial 30 days of the trial.13 The patients were evaluated at baseline (day 0), once a week during treatment (days 7, 14, 21, and 30) as well as at 8 weeks (day 90) after the discontinuation of treatment (follow-up period).13 At the end of outcome period (day 90), we observed different clinical behavior of OLP lesions independent of the treatment. Then, we decided in the present study to examine the histopathologic and cellular alterations according to the clinical behavior of this cohort of OLP. The patients were divided into 3 groups according to their clinical outcome after therapy. The groups included the following: Type I patients presenting complete resolution without recurrence, Type II presenting complete resolution associated with recurrence, and Type III presenting no response to treatment.
Histology and Immunofluorescence of OLP Tissues
We reassess the histopathological slides of the 42 patients that received diagnosis of OLP based on the criteria proposed by the WHO.22 Histological signs of dysplasia were exclusion criteria. Immunofluorescence was performed as previously reported5 using a double staining with anti acetyl-histone H3 (cell signaling) and vimentin (DAKO, Carpinteria, CA) and anti phospho-histone H2A.X (Millipore, Billerica, CA) as primary antibodies followed by FITC or TRITC-conjugated secondary antibody (Covance, Berkeley, CA). DNA was stained using Hoechst 33342. Images of 4 to 10 fields of each case were captured at 400× magnification using a QImaging-ExiAqua monochrome digital camera attached to a Nikon Microscope (Nikon, Melville, NY) and visualized with QCapturePro 7 software (Surrey, BC, Canada). We performed morphometric image analysis for γH2AX using the software ImageJ (Version 1.38s; NIH, Bethesda, MD). All positive and negative cells were counted in each field and the percentage of total number of cells in each case was calculated. For ac.H3K9ac staining intensity analysis, all cases were classified as negative (0), weak (+), moderate (++), or strong (+++). The intensity of H3K9ac was graduated independently by 3 oral pathologists.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA). Statistical analyses of the total number of ac.H3 (Lys9)-positive nuclei, OLP type, and comparison between γH2AX and ac.H3 (Lys9) staining was performed using 1-way analysis of variance followed by Tukey multiple comparison tests. We determined the mean of OLP clinical types and the total number of nuclei expressing γH2AX. We also calculated standard error of the mean. Asterisks denote statistical significance (∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; and nsP > 0.05).
RESULTS
OLP Patients Show Distinct Response to Therapy
Current therapeutic strategies for OLP patients include corticosteroids, immunosuppressors, retinoids, antifungal agents, and low-level laser therapy, among others.13,33,34 All of these treatments improve symptomatic OLP, but none are curative. The clinical response to therapy is heterogeneous, and disease relapse and resistance to treatment is common in OLP patients.13 OLP lesions have a range of clinical appearances that vary from reticular to atrophic and erosive lesions. With the goal of identifying markers associated with disease progression, we classified the patients treated into 3 groups based on response to therapy. We found that half of patients had a complete clinical resolution of symptoms (Type I), which was determined by total absence of symptoms and remission of all atrophic/erosive lesions regardless of persisting hyperkeratotic lesions. This group of patients was classified as responsive to therapy. Thirty-two percent of patients achieved partial clinical resolution (Type II), which was determined by a decrease in, but not the complete remission of, atrophic/erosive areas and symptoms; these patients were classified as partially responsive. Finally, all remaining patients were nonresponsive (Type III) and did not benefit from therapy and/or experienced worsening of symptoms (Figure 1A). Histologically, all patients enrolled in this trial had clinical manifestations and histological findings compatible with OLP (Figure 1A).
FIGURE 1.
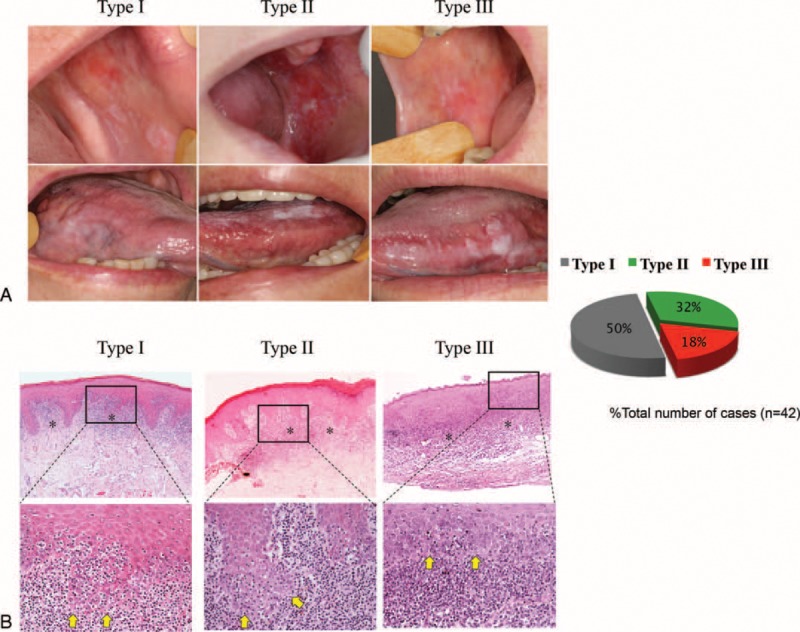
OLP cases exhibit differential responses to therapy. (A) Clinical aspects of response of OLP patients after outcome period (8 wk after the discontinuation of treatment—day 90). Type I patients (50%) have complete remission of erosive lesions. Type II patients (32%) have partial clinical resolution. Type III patients (18%) are nonresponsive to therapy. (B) H&E-stained sections of OLP lesions from patients with differential responses to therapy depict similar histological aspects. Upper panel illustrates the presence of a well-defined band-like zone of inflammatory cell infiltration (∗) (40× magnification). Lower panel shows signs of degeneration of the basal cell layer (arrows) associated with lymphocytes infiltrate and exocytosis and absence of epithelial dysplasia (200× magnification). H&E = hematoxylin and eosin, OLP = oral lichen planus.
Poor Response to Therapy Correlates With Increased Histone H3 Acetylation
We next reevaluated the histological samples of OLP patients in light of their responsiveness to therapy and histone modifications. Emerging evidences indicate the involvement of histone modifications in the inflammatory conditions such as rheumatoid arthritis, chronic obstructive pulmonary disease, and severe asthma.35,36 Functional acetylation of histone H3 at Lys 9 results in chromatin decondensation, chromatin assembly, and gene activation thereby being considered a marker of transcriptionally active chromatin.37–39 Here we decided to explore the transcriptional activity of OLP in patients who had different responses to therapy (Type I, Type II, and Type III), using a H3K9ac antibody. Interestingly, we observed high levels of histone H3 acetylation (Lys9) in the basal layer of tissue samples from Types II and III patients compared to Type I patients (Figure 2B). The correlation between increased acetylation levels of histone H3 (Lys9) and poor response to therapy is observed in Types II and III patients (Figure 2C) (∗∗∗P < 0.001).
FIGURE 2.
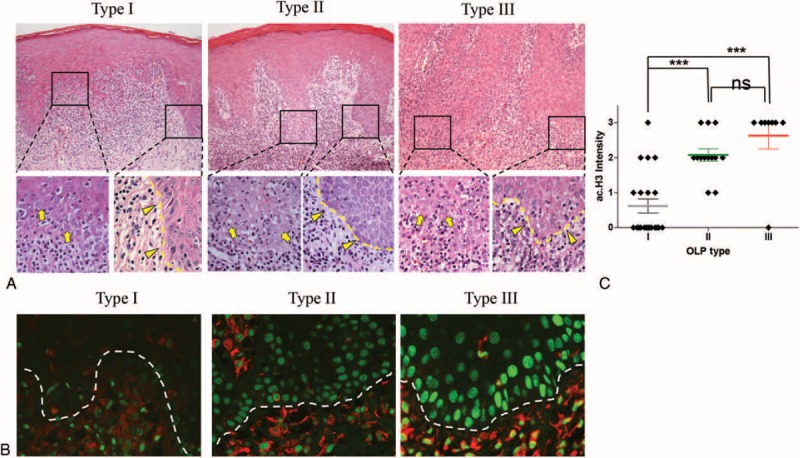
Acetylation of histone 3 correlates to poor response to therapy. (A) H&E staining of OLP patients (upper panel—100× magnification; lower panel—400× magnification). Modification in basal cell morphology and nuclear shape is evident in all OLP types in areas with different degree of exocytosis (arrows). Note that all OLP lesions present focal areas of well-preserved basal architecture (arrowhead). (B) Immunofluorescence staining of well-preserved basal layers demonstrates increased acetylation of histone 3 in the nucleus of Types II and III OLP patients (green labeling). Vimentin staining (red staining) was used to differentiate the epithelial from mesenchymal tissues. (C) Diamond shape represents each patient distributed by OLP response to therapy (OLP type) and histone 3 acetylation. Type I patients have an average ac.H3 intensity of 0.61 compared to 2.07 and 2.62 in Types II and III patients, respectively (error bar, SEM; P = 0.15, ∗∗∗P<0.001). H&E = hematoxylin and eosin, OLP = oral lichen planus, SEM = standard error of the mean.
Enhanced DNA Damage in OLP Samples From Patients Partially Responsive or Nonresponsive to Therapy
The transformation potential of OLP is largely unknown. However, genetic aberrations have been reported in OLP40–43 and provide the rational for the presence of a subset of genetically instable OLP lesions. Our previous results suggest that the regulation of OLP response to therapy is directly associated with histone modifications, which enhance gene transcription. However, is unknown whether histone acetylation correlates with increased genomic instability. We next examined for chronic genomic injuries to OLP chromatin by identifying DSBs. Histone γH2AX detect DSBs in chromatin and initiates the DNA damage response complex. In response to phosphorylation at serine 139 by ATM, γH2AX recruits repair proteins, including p53, BRCA1 and 2, FANCD2, and others, to DSBs.44,45 However, sustained phosphorylation of γH2AX denotes chronic injury to the genome. Early-stage cancer development is associated with DNA replication stress, which enhances DNA DSBs and genomic instability, following increased pressure for p53 mutations.46 We found that the majority of OLP lesions were positive for γH2AX in the basal and parabasal layer of epithelial cells from the oral mucosa (Figure 3A). Interestingly, Type I patients had the lowest rate of γH2AX-postive cells (8.71%) (Figure 3B) compared to 49.10% and 45.30% in Types II (Figure 3C) and III patients (Figure 3D), respectively. Compared to Type I patients, the amount of DNA damage in Type II and Type III patients was 4.8 and 5.2-fold higher, respectively.
FIGURE 3.
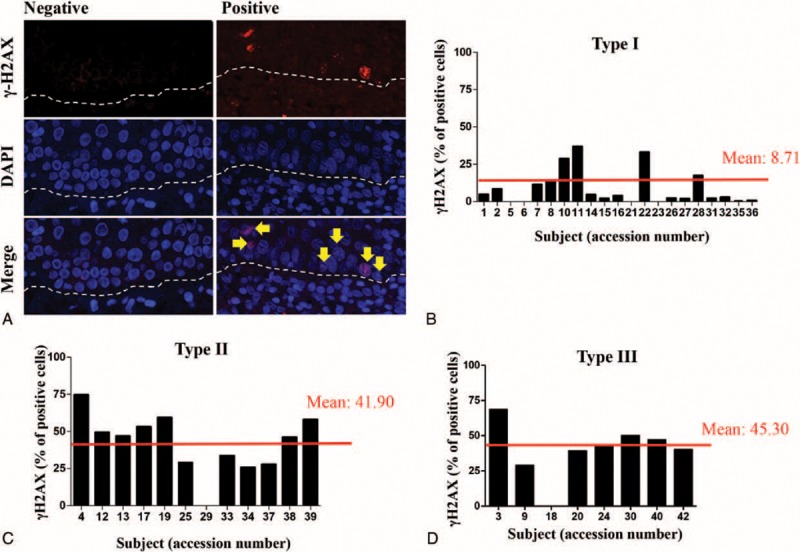
Increased DNA damage correlates to poor response to therapy. (A) Representative examples of positive and negative OLP lesions for the γH2AX DNA double-strand break marker. Note the presence of γH2AX-positive cells next to the basal layer (arrow) of the oral mucosa (dashed line defines limit between connective and epithelial tissue). Quantification of γH2AX positive cells in (B) Type I OLP patients (mean value of 8.71% of positive cells), (C) Type II OLP patients (mean value of 41.90% positive cells), and (D) Type III OLP patients (mean value of 45.30% positive cells). OLP = oral lichen planus.
Enhanced DNA Damage Correlates to Histone Modifications Observed in OLP Cases
We next examined the potential correlation between the accumulation of DNA DSBs and the progressive acetylation of OLP chromatin. By crossing the ac.H3 (Lys9) staining data with the γH2AX data, we observed that progressive chromatin decondensation correlates to accumulation of DNA DSBs (Figure 4). In general, the majority of OLP Type I (n = 13/15) did not have chromatin decondensation (negative for ac.H3K9ac) or a substantial number of γH2AX-positive cells (mean 7.46). Low expression of ac.H3K9ac was associated with a modest and nonsignificant accumulation on DNA DSBs (mean 18.78, nsP > 0.05). However, OLP Types II and III were characterized by a robust accumulation of DNA DSBs and increased chromatin acetylation (ac.H3 intensity 2 and 3, ∗∗∗P < 0.001). In both the groups, DNA DSBs had a mean value of 40.72 and 38.98, respectively (Figure 4).
FIGURE 4.
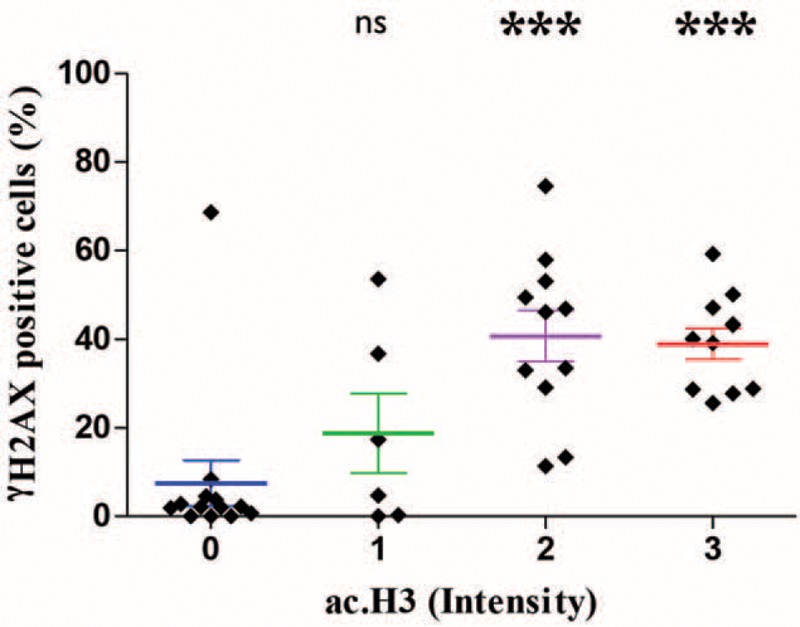
Correlation between histone acetylation and accumulation of DNA double-strand breaks in OLP. Each diamond shape represents 1 OLP patient. Increased acetylation of histone 3 correlates with accumulation of γH2AX. Note that OLP cases lacking histone acetylation (ac.H3 intensity “0”) have low levels of γH2AX (mean of 7.46). Patients with low levels of histone 3 acetylation (ac.H3 intensity “1”) have γH2AX expression in 18.78% of cells (P = 0.25). Patients with moderate and high expression of histone 3 acetylation are characterized by the accumulation of high levels of γH2AX (mean 40.72% and 38.98%, respectively) compared to OLP patients lacking histone 3 acetylation (∗∗∗P < 0.001). OLP = oral lichen planus.
DISCUSSION
OLP is an inflammatory mucocutaneous disease that affects the oral mucosa. Many studies report the potential for OLP to become malignant (summarized by Gonzalez-Moles et al47), which is reflected in the latest WHO classification of OLP as a potentially malignant disorder. The controversial transformation potential of OLP is likely based on inconsistent criteria used to diagnose OLP.47 Indeed, OLP has been misdiagnosed as dysplastic lesions with lichenoid features.48 Follow-up studies of OLP that was diagnosed using more stringent criteria suggests that 0.5% to 2% of OLP becomes malignant.49–54 Given that premalignant lesions can become malignant, patients with OLP require careful follow-up. This strategy has been effective in managing premalignant polyps associated with genetic trails, such as familial adenomatous polyposis in which the APC gene is mutated and MYH-associated polyposis in which the MUTYH gene is mutated. The question remains as to how to identify a potentially malignant disorder in which the clinical and microscopic features are not sufficient to predict disease evolution. Emerging evidence suggests epigenetic mechanisms silence tumor suppressors and activate pro-survival genes prior to cellular transformation and independent of changes in the DNA sequence (mutations).55–62 In fact, the term epigenetic disease refers to the development of cancer driven by epigenetic deregulation of genes.63 Epigenetic alterations are mediated by DNA methylation, posttranslational silencing of RNA, or histone modifications. Oral dysplasia and proliferative verrucous leukoplakia are associated with increased methylation of the p16 tumor suppressor,60–62 whereas global hypomethylation and compromised 5-hydroxymethylcytosine occur in cervical dysplasia and dysplastic nevi, respectively.64,65 The effects of histone modifications partially explain the common alterations to nuclear hyperchromatin observed in several pathologies. Histones H2A, H2B, H3, and H4 assemble the nucleosome, which is responsible for packaging DNA inside the nucleus. Acetylation and deacetylation of histones control gene transcription by exposing specific areas of the DNA to the transcriptional machinery.66 The acetylation of histone H3 observed at Lys9, 14, 18, 23, 27, and 56 is associated with gene activity.67,68 Of interest, functional acetylation of histone H3 at Lys 9 has been extensively studied and is associated with chromatin assembly and gene activation.37–39 We recently reported changes in the behavior of head and neck squamous cell carcinomas (HNSCCs) upon chromatin acetylation (reviewed by Martins and Castilho2 and Le et al22). We showed that modulation of tumor histones is sufficient to control tumor invasion5 and resistance to chemotherapy.28 Similar to these findings, global levels of histone modifications can predict the prognosis of prostate and kidney cancer.25,69 However, the role of histone acetylation in the behavior of a potentially malignant disorder and its potential value as a predictor of disease progression and resistance to conventional therapy is unknown. To address this gap in knowledge, we analyzed OLP patients who were responsive, partially responsive or nonresponsive to therapy. Rather than attempt to identify OLP lesions with the potential to transform, we focused on identifying patients with epigenetic alterations characterized by the acetylation of histone H3 (Lys9). Interestingly, this approach revealed that chromatin decondensation and enhanced histone acetylation correlates to lesions that respond poorly to therapy and are clinically “aggressive.” These results align with our previous findings in HNSCC, which become more aggressive upon global chromatin acetylation in response to treatment with histone deacetylase inhibitors.5 Further analysis also revealed that enhanced acetylation of histone H3 (Lys9) was accompanied by substantial accumulation of DNA DSBs in refractory OLP lesions. Accumulation of DNA DSBs is a well-known molecular event observed in premalignant lesions.46,70–72 One explanation for this event is the oncogene-induced stalling and collapsing of DNA replication forks, resulting in the formation of DNA DSBs.73–76 The reason that DNA DSBs accumulate in OLP lesions can be interpreted in several ways. In support of an oncogene-induced senescence hypothesis, the accumulation of DNA DSBs may represent a protective mechanism that activates cellular senescence and protects against the development of tumors. Also known as replicative stress, activation of cellular senescence requires fast cellular proliferation. Indeed, the presence of a fast cellular turnover has been reported in OLP lesions, which corroborates with the hypothesis that accumulation of DNA DSBs is indeed associated to the development of cellular senescence in OLP.77 The hypothesis of activation of cellular senescence in OLP is also supported by the presence of normal levels of loss of heterozygosity and microsatellite instability comparable to benign fibromas.78,79 However, the OLP senescence hypothesis also implies that OLP have a constitutively active oncogene, or a deregulated tumor suppressor gene, capable of triggering the senescence protective mechanisms, suggesting the presence of genetic abnormalities rather than a simple inflammatory condition. Such consideration is sustained by the identification of increased aneuploidy cells in patients with OLP.40 A second interpretation would favor the concept of early cancer as an epigenetic disease. In this case, epigenetic cues lead to progressive stress on the DNA repair system, resulting in the accumulation of unrepaired DNA DSBs. This scenario suggests that a fraction of OLP lesions have genomically instable chromatin.
Altogether, these findings suggest that histone modifications occur in OLP, and H3K9ac and γH2AX histones may serve as epigenetic markers for OLP recurrence.
Footnotes
Abbreviations: APC = adenomatous polyposis coli, ATM = ataxia telangiectasia mutated, BRCA1 = breast cancer 1, early onset, BRCA2 = breast cancer 2, early onset, DNA = deoxyribonucleic acid, DSB = double-strand break, FANCD2 = Fanconi anemia group D2 protein, HNSCC = head and neck squamous cell carcinoma, OLP = oral lichen planus, WHO = World Health Organization.
CSD and MATM contributed equally to this work.
The work was partially funded by CNPQ, and the University of Michigan faculty grant.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Tan M, Luo H, Lee S, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011; 146:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martins MD, Castilho RM. Histones: controlling tumor signaling circuitry. J Carcinog Mutagen 2013; 1 suppl 5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guan J-S, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009; 459:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter J, Sauer S, Peters A, et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J 2004; 23:4462–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giudice FS, Pinto DS, Jr, Nör JE, et al. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PloS One 2013; 8:e58672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol 2009; 45:317–323. [DOI] [PubMed] [Google Scholar]
- 7.Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. Part 2. Clinical management and malignant transformation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005; 100:164–178. [DOI] [PubMed] [Google Scholar]
- 8.Eisen D. The clinical manifestations and treatment of oral lichen planus. Dermatol Clin 2003; 21:79–89. [DOI] [PubMed] [Google Scholar]
- 9.Scully C, Carrozzo M. Oral mucosal disease: lichen planus. Br J Oral Maxillofac Surg 2008; 46:15–21. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Gonzalez F, Vazquez-Alvarez R, Reboiras-Lopez D, et al. Histopathological findings in oral lichen planus and their correlation with the clinical manifestations. Med Oral Patol Oral Cirugia Bucal 2011; 16:e641–e646. [PubMed] [Google Scholar]
- 11.Crincoli V, Di Bisceglie MB, Scivetti M, et al. Oral lichen planus: update on etiopathogenesis, diagnosis and treatment. Immunopharmacol Immunotoxicol 2011; 33:11–20. [DOI] [PubMed] [Google Scholar]
- 12.van der Meij EH, van der Waal I. Lack of clinicopathologic correlation in the diagnosis of oral lichen planus based on the presently available diagnostic criteria and suggestions for modifications. J Oral Pathol Med 2003; 32:507–512. [DOI] [PubMed] [Google Scholar]
- 13.Dillenburg CS, Martins MA, Munerato MC, et al. Efficacy of laser phototherapy in comparison to topical clobetasol for the treatment of oral lichen planus: a randomized controlled trial. J Biomed Optics 2014; 19:068002. [DOI] [PubMed] [Google Scholar]
- 14.Georgakopoulou EA, Achtari MD, Achtaris M, et al. Oral lichen planus as a preneoplastic inflammatory model. J Biomed Biotechnol 2012; 2012:759626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shanmugam MK, Sethi G. Role of epigenetics in inflammation-associated diseases. Subcellular Biochem 2013; 61:627–657. [DOI] [PubMed] [Google Scholar]
- 17.Niwa T, Ushijima T. Induction of epigenetic alterations by chronic inflammation and its significance on carcinogenesis. Adv Genet 2010; 71:41–56. [DOI] [PubMed] [Google Scholar]
- 18.Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Investig 2008; 118:2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palucka K, Coussens LM, O'Shaughnessy J. Dendritic cells, inflammation, and breast cancer. Cancer J (Sudbury, Mass) 2013; 19:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Den Broeck A, Brambilla E, Moro-Sibilot D, et al. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin Cancer Res 2008; 14:7237–7245. [DOI] [PubMed] [Google Scholar]
- 21.Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer 2011; 128:1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le JM, Squarize CH, Castilho RM. Histone modifications: targeting head and neck cancer stem cells. World J Stem Cells 2014; 6:511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolffe AP, Guschin D. Review: chromatin structural features and targets that regulate transcription. J Struct Biol 2000; 129:102–122. [DOI] [PubMed] [Google Scholar]
- 24.Barlési F, Giaccone G, Gallegos-Ruiz MI, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol 2007; 25:4358–4364. [DOI] [PubMed] [Google Scholar]
- 25.Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005; 435:1262–1266. [DOI] [PubMed] [Google Scholar]
- 26.Bai X, Wu L, Liang T, et al. Overexpression of myocyte enhancer factor 2 and histone hyperacetylation in hepatocellular carcinoma. J Cancer Res Clin Oncol 2008; 134:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arif M, Vedamurthy BM, Choudhari R, et al. Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chem Biol 2010; 17:903–913. [DOI] [PubMed] [Google Scholar]
- 28.Almeida LO, Abrahao AC, Rosselli-Murai LK, et al. NFκB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio 2014; 4:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrari P, Strubin M. Uncoupling histone turnover from transcription-associated histone H3 modifications. Nucleic Acids Res 2015; 43:3972–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin M, Terradas M, Hernandez L, et al. gammaH2AX foci on apparently intact mitotic chromosomes: not signatures of misrejoining events but signals of unresolved DNA damage. Cell Cycle 2014; 13:3026–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogakou EP, Boon C, Redon C, et al. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 2003; 100:5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radwan-Oczko M. Topical application of drugs used in treatment of oral lichen planus lesions. Adv Clin Exp Med 2013; 22:893–898. [PubMed] [Google Scholar]
- 34.Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies. Clin Dermatol 2010; 28:100–108. [DOI] [PubMed] [Google Scholar]
- 35.Strietholt S, Maurer B, Peters MA, et al. Epigenetic modifications in rheumatoid arthritis. Arthritis Res Ther 2008; 10:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adcock IM, Tsaprouni L, Bhavsar P, et al. Epigenetic regulation of airway inflammation. Curr Opin Immunol 2007; 19:694–700. [DOI] [PubMed] [Google Scholar]
- 37.Hansen JC, Tse C, Wolffe AP. Structure and function of the core histone N-termini: more than meets the eye. Biochemistry 1998; 37:17637–17641. [DOI] [PubMed] [Google Scholar]
- 38.Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Wang X, He K, et al. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol 2010; 72:585–595. [DOI] [PubMed] [Google Scholar]
- 40.Yarom N, Shani T, Amariglio N, et al. Chromosomal numerical aberrations in oral lichen planus. J Dent Res 2009; 88:427–432. [DOI] [PubMed] [Google Scholar]
- 41.Segura S, Rozas-Munoz E, Toll A, et al. Evaluation of MYC status in oral lichen planus in patients with progression to oral squamous cell carcinoma. Br J Dermatol 2013; 169:106–114. [DOI] [PubMed] [Google Scholar]
- 42.Dang J, Bian YQ, Sun JY, et al. MicroRNA-137 promoter methylation in oral lichen planus and oral squamous cell carcinoma. J Oral Pathol Med 2013; 42:315–321. [DOI] [PubMed] [Google Scholar]
- 43.Fonseca-Silva T, Oliveira MV, Fraga CA, et al. DNMT3B (C46359T) polymorphisms and immunoexpression of DNMT3b and DNMT1 proteins in oral lichen planus. Pathobiol: J Immunopathol Mol Cell Biol 2012; 79:18–23. [DOI] [PubMed] [Google Scholar]
- 44.Deng CX. Tumorigenesis as a consequence of genetic instability in Brca1 mutant mice. Mutat Res 2001; 477:183–189. [DOI] [PubMed] [Google Scholar]
- 45.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997; 386:761–763. [DOI] [PubMed] [Google Scholar]
- 46.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907–913. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Moles MA, Scully C, Gil-Montoya JA. Oral lichen planus: controversies surrounding malignant transformation. Oral Dis 2008; 14:229–243. [DOI] [PubMed] [Google Scholar]
- 48.Krutchkoff DJ, Eisenberg E. Lichenoid dysplasia: a distinct histopathologic entity. Oral Surg Oral Med Oral Pathol 1985; 60:308–315. [DOI] [PubMed] [Google Scholar]
- 49.Mattsson U, Jontell M, Holmstrup P. Oral lichen planus and malignant transformation: is a recall of patients justified? Crit Rev Oral Biol Med 2002; 13:390–396. [DOI] [PubMed] [Google Scholar]
- 50.Silverman S, Jr, Gorsky M, Lozada-Nur F. A prospective follow-up study of 570 patients with oral lichen planus: persistence, remission, and malignant association. Oral Surg Oral Med Oral Pathol 1985; 60:30–34. [DOI] [PubMed] [Google Scholar]
- 51.Rajentheran R, McLean NR, Kelly CG, et al. Malignant transformation of oral lichen planus. Eur J Surg Oncol 1999; 25:520–523. [DOI] [PubMed] [Google Scholar]
- 52.Silverman S., Jr Oral lichen planus: a potentially premalignant lesion. J Oral Maxillofacial Surg 2000; 58:1286–1288. [DOI] [PubMed] [Google Scholar]
- 53.Mignogna MD, Lo Muzio L, Lo Russo L, et al. Clinical guidelines in early detection of oral squamous cell carcinoma arising in oral lichen planus: a 5-year experience. Oral Oncol 2001; 37:262–267. [DOI] [PubMed] [Google Scholar]
- 54.Gandolfo S, Richiardi L, Carrozzo M, et al. Risk of oral squamous cell carcinoma in 402 patients with oral lichen planus: a follow-up study in an Italian population. Oral Oncol 2004; 40:77–83. [DOI] [PubMed] [Google Scholar]
- 55.Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lingen MW, Pinto A, Mendes RA, et al. Genetics/epigenetics of oral premalignancy: current status and future research. Oral Dis 2011; 17 suppl 1:7–22. [DOI] [PubMed] [Google Scholar]
- 57.Diez-Perez R, Campo-Trapero J, Cano-Sanchez J, et al. Methylation in oral cancer and pre-cancerous lesions (review). Oncol Rep 2011; 25:1203–1209. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Li Q, Chen H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis 2013; 34:1756–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakazawa T, Kondo T, Ma D, et al. Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum Pathol 2012; 43:834–842. [DOI] [PubMed] [Google Scholar]
- 60.Takeshima M, Saitoh M, Kusano K, et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J Oral Pathol Med 2008; 37:475–479. [DOI] [PubMed] [Google Scholar]
- 61.Kresty LA, Mallery SR, Knobloch TJ, et al. Alterations of p16(INK4a) and p14(ARF) in patients with severe oral epithelial dysplasia. Cancer Res 2002; 62:5295–5300. [PubMed] [Google Scholar]
- 62.Kresty LA, Mallery SR, Knobloch TJ, et al. Frequent alterations of p16INK4a and p14ARF in oral proliferative verrucous leukoplakia. Cancer Epidemiol Biomarkers Prev 2008; 17:3179–3187. [DOI] [PubMed] [Google Scholar]
- 63.Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res (Phila) 2008; 1:219–222. [DOI] [PubMed] [Google Scholar]
- 64.Kim YI, Giuliano A, Hatch KD, et al. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994; 74:893–899. [DOI] [PubMed] [Google Scholar]
- 65.Larson AR, Dresser KA, Zhan Q, et al. Loss of 5-hydroxymethylcytosine correlates with increasing morphologic dysplasia in melanocytic tumors. Modern Pathol 2014; 27:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693–705. [DOI] [PubMed] [Google Scholar]
- 67.Thiagalingam S, Cheng KH, Lee HJ, et al. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann NY Acad Sci 2003; 983:84–100. [DOI] [PubMed] [Google Scholar]
- 68.Jayani RS, Ramanujam PL, Galande S. Studying histone modifications and their genomic functions by employing chromatin immunoprecipitation and immunoblotting. Methods Cell Biol 2010; 98:35–56. [DOI] [PubMed] [Google Scholar]
- 69.Seligson DB, Horvath S, McBrian MA, et al. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 2009; 174:1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raynaud CM, Hernandez J, Llorca FP, et al. DNA damage repair and telomere length in normal breast, preneoplastic lesions, and invasive cancer. Am J Clin Oncol 2010; 33:341–345. [DOI] [PubMed] [Google Scholar]
- 71.Chang YJ, Byun SW, Kim HK, et al. DNA double strand breaks in gastric epithelium with Helicobacter pylori infection. Korean J Gastroenterol 2012; 60:79–85. [DOI] [PubMed] [Google Scholar]
- 72.Chene G, Cayre A, Raoelfils I, et al. Morphological and immunohistochemical pattern of tubo-ovarian dysplasia and serous tubal intraepithelial carcinoma. Eur J Obstet Gynecol Reprod Biol 2014; 183:89–95. [DOI] [PubMed] [Google Scholar]
- 73.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352–1355. [DOI] [PubMed] [Google Scholar]
- 74.Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638–642. [DOI] [PubMed] [Google Scholar]
- 75.Castilho RM, Squarize CH, Chodosh LA, et al. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 2009; 5:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sedelnikova OA, Bonner WM. GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle 2006; 5:2909–2913. [DOI] [PubMed] [Google Scholar]
- 77.Montebugnoli L, Farnedi A, Marchetti C, et al. High proliferative activity and chromosomal instability in oral lichen planus. Int J Oral Maxillofacial Surg 2006; 35:1140–1144. [DOI] [PubMed] [Google Scholar]
- 78.Zhang L, Michelsen C, Cheng X, et al. Molecular analysis of oral lichen planus. A premalignant lesion? Am J Pathol 1997; 151:323–327. [PMC free article] [PubMed] [Google Scholar]
- 79.Accurso BT, Warner BM, Knobloch TJ, et al. Allelic imbalance in oral lichen planus and assessment of its classification as a premalignant condition. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011; 112:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]