

Abstract
Background:
Focal adhesion kinase (FAK) mediates survival of normal pancreatic islets through activation of AKT. Upon malignant transformation of islet cells into pancreatic neuroendocrine tumors (PanNETs), AKT is frequently overexpressed and mutations in the AKT/mTOR pathway are detected. Because mTOR inhibitors rarely induce PanNET tumor regression, partly because of feedback activation of AKT, novel combination strategies are needed to target FAK/AKT/mTOR signaling.
Methods:
We characterized the activation of FAK in PanNETs using immunohistochemistry and Western blot analysis and tested the FAK inhibitor PF-04554878 in human PanNET cells in vitro and in vivo (at least three mice per group). In addition, we evaluated the effect of combined FAK and mTOR inhibition on PanNET viability and apoptosis. All statistical tests were two-sided.
Results:
We found that FAK is overexpressed and hyperphosphorylated in human PanNETs and that PF-04554878 strongly inhibited FAK (Tyr397) autophosphorylation in a dose-dependent manner. We found that PF-04554878 inhibited cell proliferation and clonogenicity and induced apoptosis in PanNET cells. Moreover, oral administration of PF-04554878 statistically significantly reduced tumor growth in a patient-derived xenograft model of PanNET (P = .02) and in a human PanNET xenograft model of peritoneal carcinomatosis (P = .03). Importantly, PF-04554878 synergized with the mTOR inhibitor everolimus by preventing feedback AKT activation.
Conclusions:
We demonstrate for the first time that FAK is overexpressed in PanNETs and that inhibition of FAK activity induces apoptosis and inhibits PanNET proliferation. We found that the novel FAK inhibitor PF-04554878 synergizes with everolimus, a US Food and Drug Administration–approved agent for PanNETs. Our findings warrant the clinical investigation of combined FAK and mTOR inhibition in PanNETs.
Pancreatic neuroendocrine tumors (PanNETs) are increasing in incidence, and therapeutic options are limited (1). The role of the PI3K/mTOR pathway in these tumors has recently been elucidated (2), and in 2011 the mTOR inhibitor everolimus became the first agent approved for this disease in nearly three decades as a result of the RADIANT-3 study (3). Surprisingly, while everolimus doubled the progression-free survival of PanNET patients, the overall response rates were extremely low (4.8% partial response (PR), 0% complete response (CR)) (4). The lack of tumor regressions observed is proposed to be attributed to the observation that everolimus and other rapalogs are potently cytostatic, but not cytotoxic, in cancer cells (5–7). Therefore, novel therapeutic approaches to enhance the activity of everolimus in PanNET patients are needed.
PanNETs have been historically understudied because of their perceived rarity, and as a result molecular mechanisms underlying their progression and clinical aggressiveness remain to be fully elucidated (1). For example, while overexpression of AKT is observed in the majority of PanNET specimens, with studies reporting that 61% to 76% of PanNETs display increased AKT activity (8–10), only 15% of these tumors have genetic mutations in PI3K/AKT/mTOR pathway genes (2), Therefore, the increased AKT activity observed in most PanNET cases may be explained by aberrations in other oncogenic signaling proteins upstream of AKT, such as focal adhesion kinase (FAK).
Studies of AKT signaling in normal pancreatic islets (the precursor cells of PanNETs) have shown that AKT survival signaling protects normal islet cells from apoptosis (11,12). This prosurvival effect of AKT in normal islets was demonstrated to occur following activation of FAK. For example, in vitro exposure of harvested islets to basement membrane extracellular matrix proteins results in increased phosphorylation of FAK and AKT, inhibition of apoptosis, and increased islet survival (12,13). These findings implicate FAK and AKT in the evasion of apoptosis by normal islet cells and suggest that FAK/AKT survival signaling continues to be functional in PanNET cells. In agreement with this hypothesis, FAK has been shown to activate AKT signaling, resulting in evasion of apoptosis in breast, colon, liver, soft tissue, and brain cancers (14–18) and chemoresistance in prostate and ovarian cancers (19, 20), highlighting the documented role of FAK as a proximal oncogenic signaling protein. In addition, FAK is overexpressed in a wide variety of tumors including gastrointestinal (GI) cancers, such as pancreatic ductal adenocarcinoma (21–26), as well as neuroendocrine tumors (NETs) of the thyroid (27), in some cases because of increased copy number of the FAK gene locus that is observed in GI cancers, thymic NETs, and gastroenteropancreatic neuroendocrine tumors (GEP-NETs) (28–31).
Given the role of FAK signaling in the evasion of apoptosis by normal islet cells, as well as the overexpression of FAK in neuroendocrine and GI cancers, we proposed that FAK provides critical anti-apoptotic and prosurvival signals in PanNETs. In the present study we demonstrate that FAK is overexpressed and hyperphosphorylated in PanNETs, and we show that a novel ATP-competitive kinase inhibitor of FAK inhibits PanNET growth and induces apoptosis. In addition, we identified a novel combination strategy that uses FAK kinase inhibition to synergistically potentiate the activity of the mTOR inhibitor everolimus to induce apoptosis and inhibit PanNET growth.
Methods
Human PanNET Patient Samples and Immunohistochemistry
PanNET samples were prospectively obtained from patients undergoing surgical tumor resection under an institutional review board–approved study through the University of Florida Clinical and Translational Science Institute Biorepository. Although PanNET patient samples were difficult to obtain, we were able to analyze several good-quality samples of matched adjacent normal pancreas, tumor, and metastasis. Additional details are provided in the Supplementary Methods (available online). All patients provided written informed consent prior to surgical resection.
Cell Lines and Culture Conditions
BON cells, derived from a serotonin-secreting PanNET (32), were a gift from Kirk Ives (University of Texas Medical Branch, Galveston, TX). QGP-1 cells, derived from a somatostatin-secreting PanNET (33), were purchased from the Japan Health Sciences Foundation Health Science Research Resources Bank. CM cells, derived from an insulin-secreting PanNET (34), were a gift from Aldo Scarpa (University of Verona, Italy). Additional details are provided in the Supplementary Methods (available online).
Reagents and Antibodies
The FAK inhibitor PF-04554878 was provided by Pfizer (New York, NY) via MTA. Phospho-FAK (Y397) antibody was purchased from Cell Signaling Technology (Beverly, MA). FAK clone 4.47 antibody was purchased from EMD Millipore (Merck KGaA, Darmstadt, Germany). Additional antibodies used are described in the Supplementary Methods (available online).
Immunoblot Analysis
Cells were lysed in culture plates in 1% IPEGAL CA-630 (Octylphenoxypolyethoxyethanol) (Sigma). Patient samples were homogenized and lysed with RIPA Lysis Buffer System (Santa Cruz Biotechnology, TX). Additional details are provided in the Supplementary Methods (available online).
Viability, Clonogenicity, and Proliferation Assays
Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT, Sigma-Aldrich) assay as recommended by the manufacturer. Additional details are provided in the Supplementary Methods (available online).
Determination of Apoptosis
To assess apoptosis, we used the In-Situ Cell Death Detection Kit (Roche) to visualize DNA strand breaks as recommended by manufacturer. Additional details are provided in the Supplementary Methods (available online).
In Vivo Experiments
Five-week-old male and female NOD.CB17-Prkdc scid/NcrCrl (NOD/SCID) mice were injected intraperitoneally with 5x105 CM cells stably expressing firefly luciferase (CM-Luc). For the patient-derived xenograft experiment, a fresh biopsy from a human PanNET lymph node metastasis with moderate differentiation was established and passaged exclusively in immunodeficient mice never cultured on plastic in vitro. The tumor was isolated, lentivirally transduced with firefly luciferase while mincing into 3 x 3 x 3mm pieces, and implanted subcutaneously into NOD.Cg-Prkdc scid Il2rg tm1Wjl/SzJ (NSG) mice. Treatment details are provided in the Supplementary Methods (available online). All animal experiments were performed under an approved University of Florida Institutional Animal Care and Use Committee protocol.
Combination Experiments and Determination of Synergy
To assess synergistic effects on cell growth, individual drugs alone and in combination were analyzed using CalcuSyn software (BIOSOFT, Cambridge, UK) according to the Chou and Talalay method (35). Additional details and descriptions of fixed and varying molar ratio combinations are provided in the Supplementary Methods (available online).
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA). When comparing means of two groups, an unpaired two-tailed Student’s t test was used. When comparing tumor growth curves, a repeated measures two-way analysis of variance was performed using GraphPad Prism software to assess the effect of treatment intervention in comparison to control. Error bars reflect the standard deviation of the mean. Additional details are provided in the Supplementary Methods (available online). All statistical tests were two-sided and a P value of less than .05 was considered statistically significant. Additional statistical methods are described in the Supplementary Methods (available online).
Results
FAK Expression in Pancreatic Neuroendocrine Tumors
Because FAK signaling promotes normal islet cells to evade apoptosis (12,13), we first sought to determine whether FAK is expressed in human PanNETs. Using primary human PanNET specimens and matched adjacent normal pancreas, we evaluated the protein expression of FAK by immunoblot analysis using a pan-FAK antibody. FAK activation was determined using a phospho-FAK–specific antibody (Y397). We found that FAK is overexpressed (1.9- to 8.7-fold) and hyperphosphorylated (1.2- to 6.4-fold) in all PanNET tumor samples tested, which included one metastasis, as compared with matched adjacent normal pancreas (Figure 1A). We also tested the human PanNET cell lines BON, QGP-1, and CM (32–34) to determine FAK expression in PanNET cells. We found that FAK was expressed and autophosphorylated in all three PanNET cell lines tested (Figure 1B).
Figure 1.
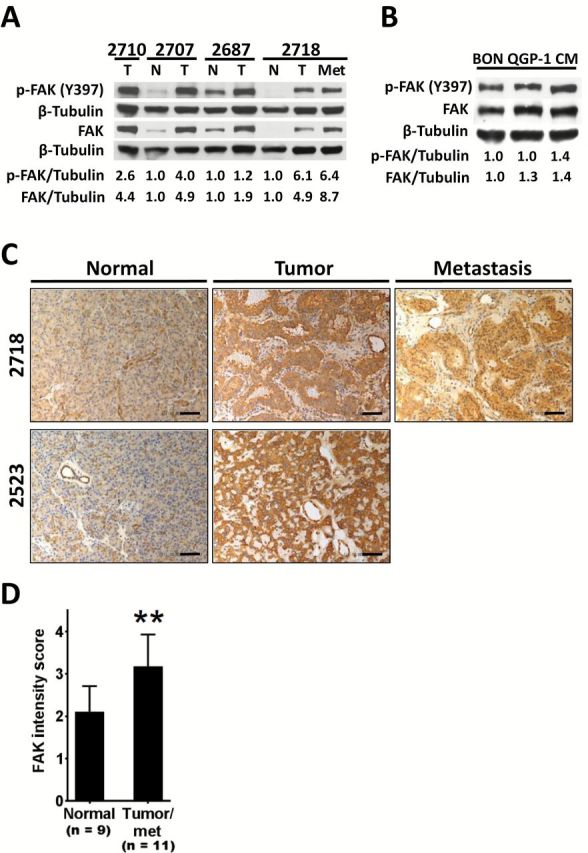
Focal adhesion kinase (FAK) expression in pancreatic neuroendocrine tumors (PanNETs). A) Immunoblot analysis of Y397 phospho- and total FAK in PanNET patient tumors (T) and/or matched metastatic sample (Met) as compared with matched normal pancreas (N). Numbers (2710, etc.) indicate samples obtained from the same patient. For densitometric analysis, results were normalized to tubulin and are expressed as fold induction over matched normal sample. For the unmatched tumor sample, results are expressed as fold induction over the mean of the other three normal samples. B) Immunoblot analysis of phospho- (Y397) and total FAK in human PanNET cell lines. Tubulin is shown as a loading control, and densitometric analyses are expressed as fold induction over BON cells. C) Representative images of FAK immunohistochemistry staining in human PanNET samples (scale bar = 200 µm). D) Analysis of FAK immunohistochemistry staining intensity in nine normal samples and 11 matched PanNET tumor samples (including one unmatched tumor and one matched metastasis sample) as determined by a masked pathologist using a two-tailed Student’s t test (**P < .01). Error bars represent standard deviation of the mean. FAK = focal adhesion kinase.
Because immunoblot analysis does not allow for discernment of which cell types express FAK, we performed immunohistochemical analysis on 20 matched normal and tumor samples, which included one metastasis. We found that tumor samples displayed increased FAK staining in tumor cells while adjacent stromal cells and matched normal samples exhibited basally low FAK staining (Figure 1C). Furthermore, analysis by a masked pathologist revealed that tumor samples (n = 11) displayed statistically significantly higher FAK staining intensity as compared with matched normal samples (n = 9) (normal: 2.111±0.601; tumor: 3.182±0.751, P = .002) (Figure 1D).
Effect of PF-04554878, an ATP-Competitive Kinase Inhibitor of FAK, on Proliferation and Clonogenicity of PanNET Cells
Because we detected increased FAK activation in human PanNET patient samples and cell lines, we next sought to determine the effect of FAK kinase inhibition in PanNET cell lines. Using the ATP-competitive FAK inhibitor PF-04554878, we evaluated the effect of FAK kinase inhibition on clonogenic colony formation and cell growth. PF-04554878 inhibited PanNET clonogenicity in a dose-dependent manner in all three cell lines tested (Figure 2A), with CM cells being five- and 10-fold more sensitive as compared with BON and QGP-1 cells, respectively (CM, 50% growth inhibition (GI50) 0.34 μM; QGP-1, GI50 3.34 μM; BON, GI50 1.56 μM) (Figure 2A; Supplementary Table 1, available online). We also found that 72 hours of PF-04554878 treatment inhibited PanNET viability in a dose-dependent manner, with CM cells showing a three-fold decrease in GI50 as compared with QGP-1 and BON cells (CM, GI50 1.77 μM; QGP-1, GI50 4.90 μM; BON, GI50 5.42 μM) (Figure 2B; Supplementary Table 1, available online). To determine the effect of FAK kinase inhibition on proliferation, we counted live cells daily following treatment with GI25 and GI50 concentrations of PF-04554878. We found that PF-04554878 markedly inhibited PanNET proliferation over five days in BON, QGP-1, and CM cells (Figure 2C).
Figure 2.
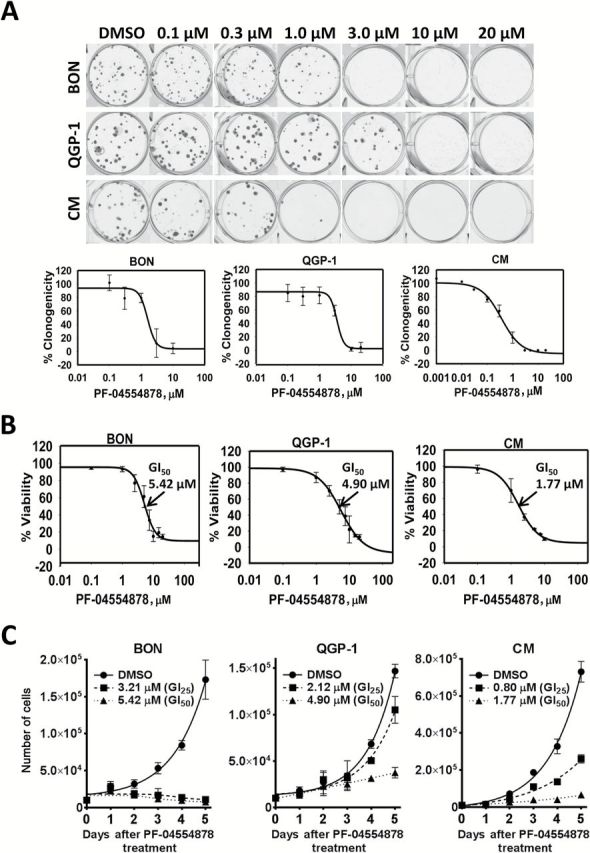
Effect of PF-04554878, a novel inhibitor of focal adhesion kinase (FAK) kinase activity, on growth of human pancreatic neuroendocrine tumor (PanNET) cells. A) Dose-response effect of PF-04554878 on colony formation of BON, QGP-1, and CM cells at low cell density after two- to four-week treatment as determined by the clonogenicity assay, and quantification of colonies for BON, QGP-1, and CM cells as determined by mechanical tally counter. Percent clonogenicity was calculated by normalizing colony counts to DMSO control. Error bars represent standard deviation of the mean. B) Viability of BON, QGP-1, and CM cells treated for 72 hours with increasing doses of PF-04554878 as determined by MTT assay. Percent viability was calculated by normalizing absorbance to DMSO control. Error bars represent standard deviation of the mean. C) Proliferation growth curves of BON, QGP-1, and CM cells treated for five days with DMSO control or PF-04554878 at GI25 or GI50 concentrations. Error bars represent standard deviation of the mean. GI50 = 50% growth inhibition, GI25 = 25% growth inhibition.
Effect of PF-04554878 on FAK Phosphorylation and Oncogenic Signaling in PanNET Cells
To confirm selectivity of PF-04554878 for inhibition of FAK phosphorylation, we performed immunoblot analysis of phosphorylated and total FAK in human PanNET cell lines following treatment with PF-04554878. After one hour of PF-04554878 treatment, we observed a marked inhibition of Y397 FAK autophosphorylation, the most proximal indicator of FAK activation (36) (Figure 3A). Densitometric analysis revealed that 50% inhibition of FAK autophosphorylation occured between 50 and 100nM in BON cells, and between 10 and 50nM in QGP-1 and CM.
Figure 3.
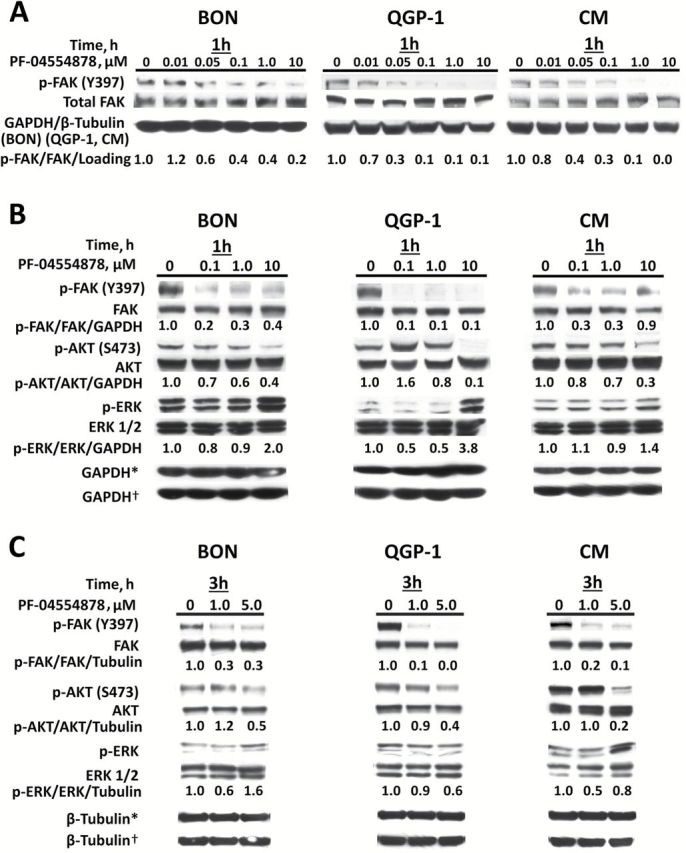
Effect of PF-04554878 on phosphorylation of focal adhesion kinase (FAK), AKT, and ERK. A) Immunoblot analysis of phosphorylated (Y397) and total FAK protein levels after one hour of treatment with increasing doses of PF-04554878 in human pancreatic neuroendocrine tumor cell lines. B) Effect of one hour or (C) three hours of PF-04554878 treatment on FAK, AKT, and ERK1/2 activation as determined by immunoblot analysis of phosphorylated FAK (Y397), AKT (S473), and ERK1/2 (Thr202/204) in comparison with total protein levels. Tubulin or GAPDH were used as loading controls. *Loading control for phosphoproteins. †Loading control for total proteins. FAK = focal adhesion kinase.
To confirm that the nanomolar inhibition of FAK activation observed in PanNET cells was not because of global decreases in cellular phosphorylation and to explore the effect of FAK kinase inhibition on downstream oncogenic signaling proteins, we treated human PanNET cell lines with varying concentrations PF-04554878 for one hour and used immunoblot analysis to examine the phosphorylation status of FAK as well as AKT (Figure 3B), which is a key downstream signaling molecule (14–20). We also chose to evaluate ERK phosphorylation, as this is an important indicator of aberrant oncogenic proliferation, although it is not directly regulated by FAK (37). We found that while Y397 phosphorylation of FAK was markedly inhibited at 100nM in all three cell lines, inhibition of ERK phosphorylation was not observed, but rather appeared to increase at the 10 µM dose only, while AKT phosphorylation was markedly inhibited at this dose in all three cell lines (Figure 3B). Densitometric analysis revealed that PF-04554878 inhibited FAK phosphorylation by more than 50% at the 100nM dose at one hour in all cell lines, while this extent of inhibition of AKT phosphorylation was only observed at the 10 µM dose. To determine whether lower doses of PF-04554878 could also modulate AKT and ERK signaling, we examine the effects of 1 µM and 5 µM concentrations of PF-04554878 in PanNET cell lines after three hours of treatment. We found that AKT phosphorylation was inhibited at three hours at the 5 µM dose in BON, QGP-1, and CM cells (Figure 3C).
Effect of PF-04554878 on Apoptosis in PanNET Cells
Because inhibition of AKT signaling was observed following PF-04554878 treatment and because the AKT pathway is implicated in cellular evasion of apoptosis (38), we sought to determine whether PF-04554878 treatment is associated with induction of apoptosis in PanNETs. To identify the optimal time requirement for PF-04554878 to induce apoptosis, we performed a time course experiment, and harvested BON cells at 12, 24, 48, and 72 hours after treatment. We then performed immunoblot analysis to assess proteolytic PARP-1 cleavage as a marker of apoptosis (39), as well as caspases 3, 8, and 9. We observed maximum levels of cleaved PARP-1 and cleaved caspase 9 at 72 hours after treatment and chose this time point to determine the dose of PF-04554878 required for apoptosis induction (Figure 4A).
Figure 4.
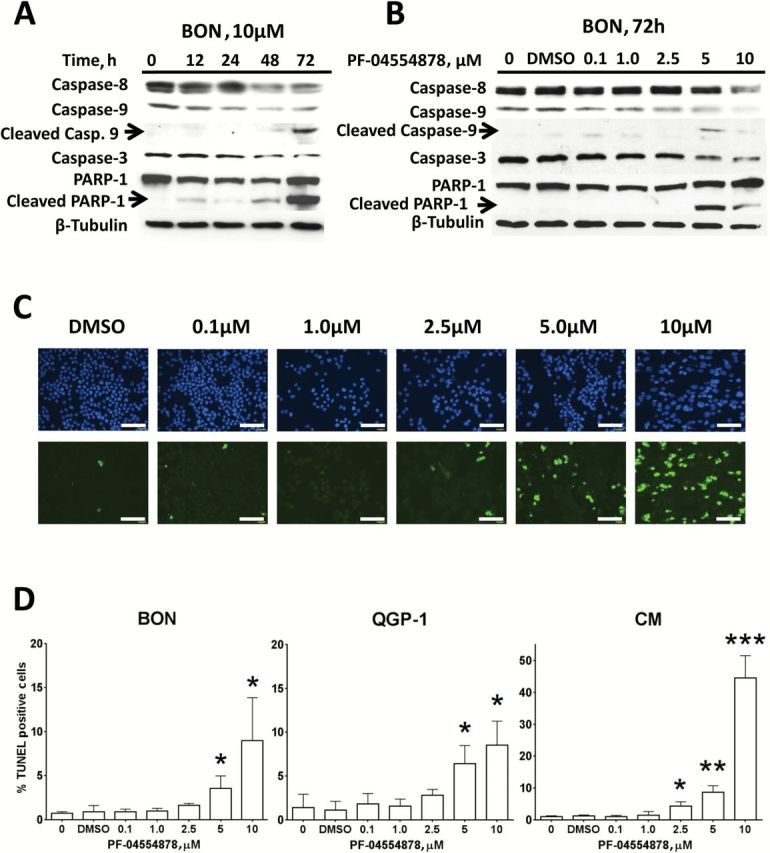
Effect of PF-04554878 on apoptosis in human pancreatic neuroendocrine tumor (PanNET) cells. A) Time course immunoblot analysis of the effects of 10 µM of PF-04554878 on induction of apoptosis as determined by caspase 3, 8, and 9 levels, as well as PARP-1 cleavage. B) Effect of increasing doses of PF-04554878 on caspases 3, 8, and 9 activation and PARP-1 cleavage in BON cells after 72 hours of treatment. C) Representative images of TUNEL staining for apoptosis in three independent dose-response experiments in CM cells following 72 hours of treatment with PF-04554878 (scale bar = 200 µm). D) Graphical quantification of PF-04554878 induction of apoptosis in human PanNET cell lines as determined by TUNEL staining. For each experiment, five images of each slide were analyzed, and the percentage of apoptotic cells was calculated by counting the number of TUNEL positive nuclei (green) in relation to total nuclei stained with DAPI (blue). Results are composite of three independent experiments. A two-tailed Student’s t test was used to compare each group with DMSO control (*P < .05, **P < .01, ***P < .001). Error bars represent standard deviation of the mean.
To determine whether apoptosis occurs in a dose-dependent manner, we performed a dose response experiment and immunoblot analysis of caspases 3, 8, and 9 as well as PARP-1. We found that PF-04554878 induced apoptosis at 5 μM to 10 μM with induction of PARP-1 cleavage and decreased full-length caspases 3, 8, and 9 observed at these doses (Figure 4B). Interestingly, while PARP-1 cleavage and decreases in full length caspases 3, 8, and 9 were consistently observed, cleaved caspase 9 was the only cleaved caspase detected in all three cell lines (Figure 4, A and B; Supplementary Figure 1, available online), suggesting that apoptosis induced by PF-04554878 in PanNET cells may be mitochondrial in nature, because caspase 9 is the most proximal caspase activated following mitochondrial cytochrome C release (40).
To confirm our findings that PF-04554878 can induce dose-dependent apoptosis, we next performed TUNEL staining for DNA strand breaks in BON, QGP-1, and CM PanNET cell lines. We found that PF-04554878 caused a dose-dependent induction of apoptosis in all cell lines tested (Figure 4, C and D). CM cells were the most sensitive to PF-04554878 induction of apoptosis, with statistical significance reached at the 2.5 μM dose, as compared with 5 μM in BON and QGP-1 cells (Figure 4D).
Effect of Oral Administration of PF-04554878 on Human PanNET Growth Inhibition In Vivo
Given the observed antitumor effects of PF-04554878 on PanNET cell growth, clonogenicity, AKT signaling, and induction of apoptosis, we next tested whether PF-04554878 could inhibit human PanNET cell growth in vivo. Since peritoneal carcinomatosis occurs in PanNETs (41,42), and is a negative prognostic marker in GEP-NETs (41,42), we performed intraperitoneal injection of the human PanNET cell line CM stably expressing luciferase (CM-Luc) in NOD/SCID mice as a model of human PanNET carcinomatosis. Animals were assigned to receive either vehicle or 50mg/kg of PF-04554878 by oral gavage once daily. Tumor burden was assessed every four days via bioluminescent imaging. We found that oral administration of PF-04554878 resulted in a statistically significant inhibition of carcinomatotic growth as compared with vehicle-treated mice (P = .03) (Figure 5, A and B).
Figure 5.

Effect of PF-04554878 on growth of human human pancreatic neuroendocrine tumors (PanNETs) in vivo. A) Effect of daily oral administration of PF-04554878 on growth of CM-Luc cells in NOD/SCID mice as measured by bioluminescent luciferase signal intensity in CM-Luciferase model of PanNET carcinomatosis. Fold induction bioluminescent signal intensity was calculated for each mouse by normalizing bioluminescent signal value to first measurement (Day 10 post-injection). Based on bioluminescent imaging, mice were assigned on Day 15 to vehicle control (n = 3) or 50mg/kg of PF-04554878 (n = 4). B) Bioluminescent imaging of mice on final day of study (Day 26). C) Immunohistochemical staining of FAK (Y397) and AKT (S473) phosphorylation in PanNET patient-derived xenograft tumor model (scale bar = 200 µm). D) Effect of daily oral administration of PF-04554878 on growth of human PanNET patient-derived xenograft transplant model in NSG mice. Animals were implanted with a 3 x 3 x 3mm piece of a human PanNET tumor sample 10 days prior to treatment assignment and commencement (Day 1) with vehicle control or 50mg/kg of PF-04554878. Tumors were measured when palpable using calipers, and tumor volume was calculated using the formula V = L2 x W x π/6. Statistical analysis of the effect of treatment intervention as compared with control was performed using a repeated measures two-tailed two-way analysis of variance. Error bars represent standard deviation of the mean.
Because a cell line xenograft model may not fully represent tumor responses observed in human patients, we next sought to confirm our findings in a patient-derived xenograft model. In this model, a primary human PanNET specimen freshly obtained following surgical resection, displaying high levels of FAK activation and moderate AKT activation (Figure 5C), was implanted subcutaneously into NSG mice. Mice were assigned to receive either vehicle or 50mg/kg of PF-04554878 orally, daily. We found that oral administration of PF-04554878 resulted in a statistically significant inhibition of tumor growth (P = .02) (Figure 5D, Day 24: Control: 1003.97±85.91mm3, PF-04554878: 429.35±53.75mm3, P = .001).
Effect of Dual Targeting of FAK and mTOR on PanNET Viability
Since maximal induction of apoptosis and inhibition of cell growth and clonogenicity were observed at relatively high doses of PF-04554878, and because solid tumors frequently acquire resistance to small molecule kinase inhibitors (43), we next sought to combine PF-04554878 with the mTOR inhibitor everolimus, which is currently an approved agent for the treatment of PanNETs. We chose to target mTOR because genes in this pathway are mutated in PanNETs (2). We found that dual targeting of FAK and mTOR by combining PF-04554878 and everolimus, respectively, in fixed molar ratio concentrations resulted in synergistic growth inhibition of QGP-1 and CM PanNET cells (Figure 6A). Synergy was confirmed by analysis of Combination Index (CI) values using CalcuSyn software. CI values were plotted against the fractional effect of each combination point (Figure 6B). According to the Chao and Talalay method, CI values below 1.0 indicate synergy (35). CI values are listed in Supplementary Tables 2 and 3 (available online). These data demonstrate that PF-04554878 synergizes with everolimus to inhibit cell growth.
Figure 6.
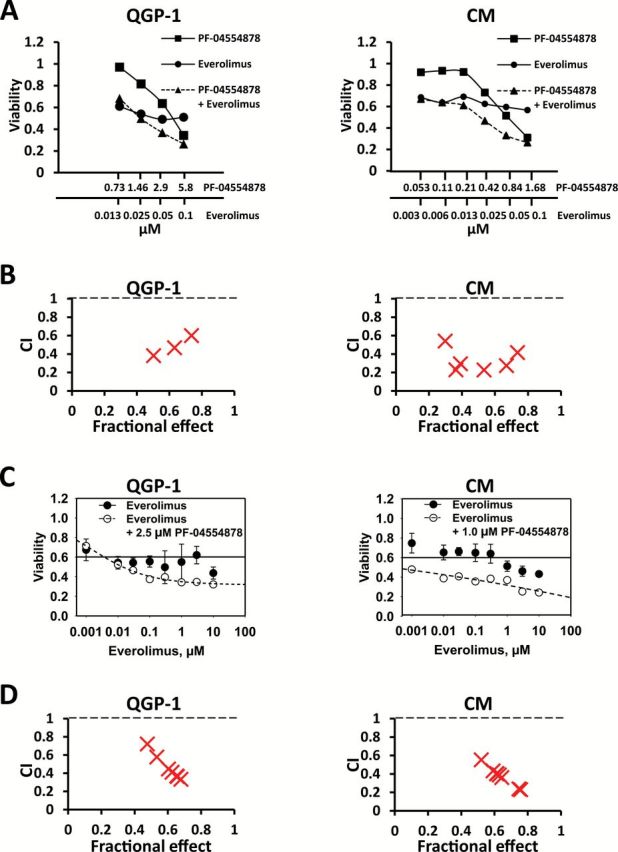
Effect of dual targeting of focal adhesion kinase (FAK) and mTOR on pancreatic neuroendocrine tumor (PanNET) cell growth. A) Effect of PF-0455878 and everolimus alone and in combination using the fixed molar ratio method in QGP1 and CM cells. PF-0455878 and everolimus were combined in a constant EC50 ratio, followed by serial one-half dilutions of this combination. B) CalcuSyn graph of combination index values (CI) and fraction of cells affected (Fa) from fixed molar ratio combination of PF-04554878 and everolimus in QGP-1 and CM cells. C) Effect of everolimus alone and in combination with a single dose of PF-04554878 using the varying molar ratio method in QGP1 and CM cells. A single nonapoptogenic dose of PF-04554878 was combined with increasing concentrations of everolimus. D) CalcuSyn graph of combination index values (CI) and fraction of cells affected (Fa) from varying molar ratio combination of PF-04554878 and everolimus in QGP-1 and CM cells.
To confirm that dual targeting of FAK and mTOR resulted in maximum inhibition of oncogenic signaling and induction of apoptosis, we performed immunoblot analysis at 72 hours for phosphorylation of FAK, AKT, and p70 S6K, and apoptotic cleavage of PARP-1. While PF-04554878 decreased phosphorylation of AKT, everolimus alone appeared to increase FAK and AKT phosphorylation (Supplementary Figure 2, available online). Importantly, the combination of PF-04554878 with everolimus resulted in a complete block of everolimus induced AKT phosphorylation. This abrogation was accompanied by maximal PARP-1 cleavage (Supplementary Figure 2, available online). These data demonstrate that PF-04554878 can abolish AKT-mediated evasion of everolimus-induced apoptosis in PanNET cells.
The largely cytostatic effect of mTOR inhibitors on human cancers is thought to be the reason why these agents rarely induce regression of solid tumors (7). Because we observed a potent cytostatic effect of everolimus in PanNET cells (Figure 6A), we next sought to determine whether adding a single fixed dose of PF-04554878 to increasing concentrations of everolimus—using a varying molar ratio method—would change the cytostatic profile of everolimus. We found that adding a single, nonapoptogenic dose of PF-04554878 (2.5 µM for QGP-1 cells and 1.0 µM for CM cells) to increasing concentrations of everolimus resulted in a cytotoxic effect profile in comparison with everolimus alone. In QGP-1 cells, the effect changed to resemble a classic sigmoidal dose-response curve, while in CM cells the combination effect appeared to be linearly cytotoxic (Figure 6C). Analysis of CI values plotted against fractional effect of combination points using CalcuSyn software indicated that the synergistic effect of this combination strategy increased as the fractional effect and dose of everolimus increased (Figure 6D). CI values are listed in Supplementary Tables 4 and 5 (available online). These data indicate that the addition of a single dose of PF-04554878 is sufficient to allow everolimus to drive further synergy in PanNET cells.
Discussion
We have demonstrated for the first time that FAK is hyperphosphorylated and overexpressed in PanNETs. FAK signaling has been implicated in the survival of normal pancreatic islets, and we now show that FAK expression and activation are increased upon malignant transformation of islet cells. In addition, it has been proposed that survival of normal islets requires FAK activation mediated by exposure to basement membrane proteins (12,13). Our findings of increased FAK expression and phosphorylation in PanNET metastasis and primary tumor samples as compared with matched normal pancreas support this hypothesis, suggesting that PanNET metastatic foci are seeded by tumor cells that can survive detachment from the double-layered islet basement membrane (44) by upregulating and activating FAK independently of basement membrane protein contact. Thus, our data predict that cells in the primary tumor with high levels of FAK would be selected during the metastatic process, in accord with the observation that FAK expression is increased in many different tumor types as they become invasive and metastatic (21,22).
We have discovered that an ATP-competitive FAK inhibitor displays antiproliferative and pro-apoptotic activity in PanNETs as a single-agent both in vitro and in vivo. Our findings of growth inhibition and apoptosis induction in vitro and in vivo following kinase inhibition of FAK highlight the importance of FAK activity and signaling for PanNET survival. These antitumor effects are likely facilitated by downregulation of AKT, because maximum apoptosis and inhibition of viability occurred at doses of PF-04554878 that decreased both FAK and AKT phosphorylation. The redundant nature of AKT signaling, which is critical to cancer cell evasion of apoptosis and survival (38), provides a strong rationale of simultaneous targeting of multiple oncoproteins in this pathway (45).
Although PF-04554878 was active as a single agent in our PanNET xenograft models, an important finding of this study is the discovery of a novel combination strategy using kinase inhibition of FAK to sensitize PanNETs to the mTOR inhibitor everolimus. Because PanNETs have been increasing in incidence for several decades (46–48) and treatment options for PanNETs have historically been limited (49), novel therapeutic strategies are needed for PanNET patients. In 2011, everolimus became the first drug approved by the FDA in nearly 30 years (followed by Sunitinib) on the basis of an increase in progression-free survival observed in everolimus treated patients in the RADIANT-3 trial (3). Despite this prolongation of progression-free survival, overall response rates were low, highlighting the need for a combination strategy that will maximize tumor regression in PanNET patients. One explanation for the lack of complete responses is the observation that mTOR inhibitors increase AKT activation and thus allow cells to evade apoptosis, limiting the efficacy of these rapalogs (50). Our results showing the cytostatic effect and increased FAK and AKT activation of everolimus in PanNETs as a single agent support this explanation, and also suggest that AKT activation by everolimus may be secondary to FAK activation.
Since FAK/AKT signaling has been shown to be critical for the survival of normal islets (12,13), and AKT is known to be aberrantly activated in the majority of PanNETs (8–10), we chose to test the effect of the combination of FAK and mTOR inhibition on the growth of PanNETs. We found that PF-04554878 synergized with everolimus and abolished its feedback activation of AKT. Importantly the addition of a fixed dose of PF-04554878 to everolimus was sufficient to overcome the cytostatic effect of everolimus in PanNET cells. Although this combination therapy is promising for PanNETs, some potential limitations of this study should be considered. These include the limited number of patient samples of this tumor type available for analysis and the number of commercially available PanNET cell lines accessible for research purposes. Nevertheless, it is likely that this strategy of targeting upstream of AKT will enhance mTOR inhibition by preventing feedback AKT activation. While Phase II clinical trials of a dual PI3K/mTOR inhibitor are already underway in PanNETs (ClinicalTrials.gov identifier: NCT01658436), our data propose a role for FAK/AKT signaling in resistance to chemotherapy. To this end, while our manuscript was in preparation we noted recent data showing that PF-04554878 can overcome AKT-mediated chemotherapeutic resistance in ovarian cancer (20). In summary, our findings warrant the clinical investigation of combined treatment with everolimus plus FAK inhibitors in PanNET patients.
Funding
This work was supported by the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine, by the National Cancer Institute T32 Training Grant and by the State Of Florida Bankhead-Coley Cancer Research Program.
Supplementary Material
The study sponsors had no role in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
References
- 1. Zhang J, Francois R, Iyer R, Seshadri M, Zajac-Kaye M, Hochwald SN. Current understanding of the molecular biology of pancreatic neuroendocrine tumors. J Natl Cancer Inst. 2013;105(14):1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oberstein PE, Saif MW. Safety and efficacy of everolimus in adult patients with neuroendocrine tumors. Clin Med Insights Oncol. 2012;6:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sankhala K, Giles FJ. Potential of mTOR inhibitors as therapeutic agents in hematological malignancies. Expert Rev Hematol. 2009;2(4):399–414. [DOI] [PubMed] [Google Scholar]
- 6. Easton JB, Houghton PJ. Therapeutic potential of target of rapamycin inhibitors. Expert Opin Ther Targets. 2004;8(6):551–564. [DOI] [PubMed] [Google Scholar]
- 7. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo SS, Wu X, Shimoide AT, Wong J, Moatamed F, Sawicki MP. Frequent overexpression of cyclin D1 in sporadic pancreatic endocrine tumours. J Endocrinol. 2003;179(1):73–79. [DOI] [PubMed] [Google Scholar]
- 9. Shah T, Hochhauser D, Frow R, Quaglia A, Dhillon AP, Caplin ME. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18(5):355–360. [DOI] [PubMed] [Google Scholar]
- 10. Ghayouri M, Boulware D, Nasir A, Strosberg J, Kvols L, Coppola D. Activation of the serine/theronine protein kinase akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010;30(12):5063–5067. [PubMed] [Google Scholar]
- 11. Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7(10):1133–1137. [DOI] [PubMed] [Google Scholar]
- 12. Miao G, Zhao Y, Li Y, et al. Basement membrane extract preserves islet viability and activity in vitro by up-regulating alpha3 integrin and its signal. Pancreas. 2013;42(6):971–976. [DOI] [PubMed] [Google Scholar]
- 13. Zhao Y, Xu J, Wei J, Li J, Cai J, Miao G. Preservation of islet survival by upregulating alpha3 integrin signaling: The importance of 3-dimensional islet culture in basement membrane extract. Transplant Proc. 2010;42(10):4638–4642. [DOI] [PubMed] [Google Scholar]
- 14. Fang Y, Wang L, Jin J, Zha X. Focal adhesion kinase affects the sensitivity of human hepatocellular carcinoma cell line SMMC-7721 to tumor necrosis factor-alpha/cycloheximide-induced apoptosis by regulating protein kinase B levels. Eur J Biochem. 2001;268(16):4513–4519. [DOI] [PubMed] [Google Scholar]
- 15. Schmitz KJ, Grabellus F, Callies R, et al. High expression of focal adhesion kinase (p125FAK) in node-negative breast cancer is related to overexpression of HER-2/neu and activated akt kinase but does not predict outcome. Breast Cancer Res. 2005;7(2):R194-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dasari VR, Kaur K, Velpula KK, et al. Downregulation of focal adhesion kinase (FAK) by cord blood stem cells inhibits angiogenesis in glioblastoma. Aging (Albany NY). 2010;2(11):791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crompton BD, Carlton AL, Thorner AR, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in ewing sarcoma. Cancer Res. 2013;73(9):2873–2883. [DOI] [PubMed] [Google Scholar]
- 18. Zheng Y, Gierut J, Wang Z, Miao J, Asara JM, Tyner AL. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene. 2013;32(36):4304–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee BY, Hochgrafe F, Lin HM, et al. Phosphoproteomic profiling identifies focal adhesion kinase as a mediator of docetaxel resistance in castrate-resistant prostate cancer. Mol Cancer Ther. 2014;13(1):190–201. [DOI] [PubMed] [Google Scholar]
- 20. Kang Y, Hu W, Ivan C, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105(19):1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Owens LV, Xu L, Craven RJ, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55(13):2752–2755. [PubMed] [Google Scholar]
- 22. Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28(1–2):35–49. [DOI] [PubMed] [Google Scholar]
- 23. Hochwald SN, Nyberg C, Zheng M, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chatzizacharias NA, Giaginis C, Zizi-Serbetzoglou D, Kouraklis GP, Karatzas G, Theocharis SE. Evaluation of the clinical significance of focal adhesion kinase and SRC expression in human pancreatic ductal adenocarcinoma. Pancreas. 2010;39(6):930–936. [DOI] [PubMed] [Google Scholar]
- 25. Kurenova E, Liao J, He DH, et al. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget. 2013;4(10):1632–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, He DH, Zajac-Kaye M, Hochwald SN. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle. 2014;13(19):3143–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim SJ, Park JW, Yoon JS, et al. Increased expression of focal adhesion kinase in thyroid cancer: immunohistochemical study. J Korean Med Sci. 2004;19(5):710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fujiwara Y, Monden M, Mori T, Nakamura Y, Emi M. Frequent multiplication of the long arm of chromosome 8 in hepatocellular carcinoma. Cancer Res. 1993;53(4):857–860. [PubMed] [Google Scholar]
- 29. Pan CC, Jong YJ, Chen YJ. Comparative genomic hybridization analysis of thymic neuroendocrine tumors. Mod Pathol. 2005;18(3):358–364. [DOI] [PubMed] [Google Scholar]
- 30. Park JH, Lee BL, Yoon J, et al. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum Pathol. 2010;41(12):1664–1673. [DOI] [PubMed] [Google Scholar]
- 31. Tonnies H, Toliat MR, Ramel C, et al. Analysis of sporadic neuroendocrine tumours of the enteropancreatic system by comparative genomic hybridisation. Gut. 2001;48(4):536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evers BM, Townsend CM, Jr, Upp JR, et al. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 1991;101(2):303–311. [DOI] [PubMed] [Google Scholar]
- 33. Kaku M, Nishiyama T, Yagawa K, Abe M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gann. 1980;71(5):596–601. [PubMed] [Google Scholar]
- 34. Baroni MG, Cavallo MG, Mark M, Monetini L, Stoehrer B, Pozzilli P. Beta-cell gene expression and functional characterisation of the human insulinoma cell line CM. J Endocrinol. 1999;161(1):59–68. [DOI] [PubMed] [Google Scholar]
- 35. Chou TC. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010;70(2):440–446. [DOI] [PubMed] [Google Scholar]
- 36. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cobb MH, Hepler JE, Cheng M, Robbins D. The mitogen-activated protein kinases, ERK1 and ERK2. Semin Cancer Biol. 1994;5(4):261–268. [PubMed] [Google Scholar]
- 38. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22(56):8983–8998. [DOI] [PubMed] [Google Scholar]
- 39. Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53(17):3976–3985. [PubMed] [Google Scholar]
- 40. Hakem R, Hakem A, Duncan GS, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94(3):339–352. [DOI] [PubMed] [Google Scholar]
- 41. Vasseur B, Cadiot G, Zins M, et al. Peritoneal carcinomatosis in patients with digestive endocrine tumors. Cancer. 1996;78(8):1686–1692. [PubMed] [Google Scholar]
- 42. Elias D, Sideris L, Liberale G, et al. Surgical treatment of peritoneal carcinomatosis from well-differentiated digestive endocrine carcinomas. Surgery. 2005;137(4):411–416. [DOI] [PubMed] [Google Scholar]
- 43. Cools J, Maertens C, Marynen P. Resistance to tyrosine kinase inhibitors: Calling on extra forces. Drug Resist Updat. 2005;8(3):119–129. [DOI] [PubMed] [Google Scholar]
- 44. Otonkoski T, Banerjee M, Korsgren O, Thornell LE, Virtanen I. Unique basement membrane structure of human pancreatic islets: implications for beta-cell growth and differentiation. Diabetes Obes Metab. 2008;10(Suppl 4):119–127. [DOI] [PubMed] [Google Scholar]
- 45. Aziz SA, Jilaveanu LB, Zito C, et al. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res. 2010;16(24):6029–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19(10):1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the united states. J Clin Oncol. 2008;26(18):3063–3072. [DOI] [PubMed] [Google Scholar]
- 48. Franko J, Feng W, Yip L, Genovese E, Moser AJ. Non-functional neuroendocrine carcinoma of the pancreas: Incidence, tumor biology, and outcomes in 2,158 patients. J Gastrointest Surg. 2010;14(3):541–548. [DOI] [PubMed] [Google Scholar]
- 49. Oberstein PE, Remotti H, Saif MW, Libutti SK. Pancreatic neuroendocrine tumors: Entering a new era. JOP. 2012;13(2):169–173. [PubMed] [Google Scholar]
- 50. O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.