

Abstract
Purpose.
Inherent instability of the P23H mutant opsin accounts for approximately 10% of autosomal dominant retinitis pigmentosa cases. Our purpose was to develop an overall set of reliable screening strategies to assess if either stabilization or enhanced degradation of mutant rhodopsin could rescue rod photoreceptors expressing this mutant protein. These strategies promise to reveal active compounds and clarify molecular mechanisms of biologically important processes, such as inhibition of target degradation or enhanced target folding.
Methods.
Cell-based bioluminescence reporter assays were developed and validated for high-throughput screening (HTS) of compounds that promote either stabilization or degradation of P23H mutant opsin. Such assays were further complemented by immunoblotting and image-based analyses.
Results.
Two stabilization assays of P23H mutant opsin were developed and validated, one based on β-galactosidase complementarity and a second assay involving bioluminescence resonance energy transfer (BRET) technology. Moreover, two additional assays evaluating mutant protein degradation also were employed, one based on the disappearance of luminescence and another employing the ALPHA immunoassay. Imaging of cells revealed the cellular localization of mutant rhodopsin, whereas immunoblots identified changes in the aggregation and glycosylation of P23H mutant opsin.
Conclusions.
Our findings indicate that these initial HTS and following assays can identify active therapeutic compounds, even for difficult targets such as mutant rhodopsin. The assays are readily scalable and their function has been proven with model compounds. High-throughput screening, supported by automated imaging and classic immunoassays, can further characterize multiple steps and pathways in the biosynthesis and degradation of this essential visual system protein.
Keywords: rhodopsin, rod photoreceptors, phototransduction, misfolded protein, GPCR, retinal degeneration, ocular pharmacology
An outline for high-throughput drug screening strategy of P23H mutant opsin is described that employs several complimentary techniques to identify active hit compounds. This broadly applicable approach can be readily extended to other pathological mutations in opsins or other membrane proteins.
Cell- and tissue culture–based high-throughput screening (HTS) represents a mainstream approach for drug discovery used either to identify biologically active small molecules or genes associated with a given phenotype in genome-wide loss-of-function screens.1–4 Frequently, HTSs are integrated with computational methods and selected signaling pathways.1–3
Cell-based chemical compound screening typically is employed to identify hit compounds that bind to a receptor or an enzyme. Improvement of hit efficacy is then achieved through massive scale testing of compounds with similar chemical structures.4 Further improvements in selectivity and potency are accomplished by structure–activity relationship studies that allow fine tuning of the active compounds through medicinal chemistry.5 Once this step is accomplished, testing in animal models, with and without genetic modifications, is the next step. Here, dose-response and toxicology studies provide critical information as to whether a compound is a viable candidate for further preclinical trials and eventually clinical trials in humans.
Developing drugs for retinal degenerative diseases presents unique challenges. The eye compartment housing postmitotic neuronal cells of the retina and the adjoining retinal pigmented epithelium (RPE) is relatively isolated, making it difficult to achieve safe and effective concentrations of a systemically administered drug candidate at this site. Yet, for treatment or prevention of retinal pathology, orally administered drugs provide the option that is most preferred, cost-effective, and amenable to patient compliance. Moreover, this can be readily discontinued if adverse events occur or better treatment options become available.
Mutations in opsin, an essential protein molecule for dim light vision,6,7 cause autosomal dominant retinitis pigmentosa (adRP). Among affected individuals, the P23H mutation opsin is the most frequent.6,8–10 Our proof-of-concept studies have focused on this mutation to understand its pathogenesis and develop pharmacological approaches for its treatment.11–21 The inherent instability of P23H opsin mutant leads to its enhanced degradation, but small amounts of the mutant opsin can still disrupt rod outer segment (ROS) disc organization.11,21 We considered two approaches that could combat the negative effect of this mutation. One is to stabilize the mutant protein with a small ligand that in turn would protect rod photoreceptor cells. The second is to destroy the mutant variant, leaving sufficient functional wild-type (WT) rhodopsin to carry out dim light vision and prevent retinal degeneration. A small molecule that selectively enhances degradation would be highly desirable. This idea is viable because heterozygote opsin knockout mice exhibit fully functional, albeit smaller rod photoreceptor cells.22 An additional benefit of this work could be the identification of other cellular factors that stabilize the mutant protein by, for example, enhancing the expression of chaperone proteins or modifying disulfide isomerase or proline cis-trans isomerase activities.
A major challenge for discovering ligands/pharmacological chaperones for membrane proteins arises from their compartmentalization into different cellular organelles in experimental cell lines, often due to use of a strong promoter. Other experimental artifacts include drug-dependent direct alteration of luminescence/fluorescence output, and conjugated reporter-mediated disruption of normal target protein folding, localization, or activity.23,24 Such problems can be resolved by modern high-content imaging techniques.
Here, we provide an outline for HTS of P23H mutant opsin that employs several complimentary techniques to identify active hit compounds and eliminate false positives. This broadly applicable approach can be readily extended to combat the effect of other pathological mutations in opsins or other mutated membrane proteins in the eye (Fig. 1; Table).
Figure 1.

High-throughput screening and orthogonal assays for the translocation and clearance of P23H mutant opsin in mammalian cells. (A) The β-Gal fragment complementation assay applied to a HTS for P23H mutant opsin translocation. Expressed in a U2OS cell line, the large subunit of β-Gal (EA, green tag) was fused with a plasma membrane anchored peptide, PLC (orange), whereas the small subunit of β-Gal (PK, pink tag) was fused on the C-terminal of P23H mutant opsin (blue). Only when P23H mutant opsin is transported to the PM upon treatment with an active compound will β-Gal be reconstituted and its restored activity quantified by its luminescence. (B) Diagram of the BRET assay for confirmation of P23H mutant opsin translocation. Briefly, the translocation of P23H mutant opsin was read by a BRET signal (dual emission ratio 530/480 nm) from a double stable cell line (HEK293) coexpressing mouse P23H mutant opsin (blue) fused with Renilla luciferase (Rluc, green) as an energy donor and membrane associated Kras peptide (orange) conjugated with fluorescent protein Venus (pink) as an acceptor. Increased PM transport of P23H-Rluc will increase the BRET signal. (C) The HTS assay for clearance of P23H mutant opsin. P23H mutant opsin was fused with Rluc and expressed in HEK293 cells to generate a stable cell line for the HTS assay. Treatment with an active compound will decrease P23H-Rluc levels noted as a decrease in luminescence from a microplate reader. (D) The amount of P23H mutant opsin expressed in a NIH3T3 stable cell line was quantified by the ALPHA immunoassay. Briefly, cells were fixed and lysed for exposure of epitopes of P23H mutant opsin to be captured by the biotinylated B6-30 anti-rhodopsin antibody and the 1D4 anti-rhodopsin antibody conjugated with acceptor beads. Upon addition of streptavidin-conjugated donor beads that bind to the biotinylated B6-30 antibody, the donor and acceptor beads are brought in close proximity. The laser excited donor (680 nm) bead then releases a singlet oxygen molecule and initiates an energy transfer to the nearby acceptor bead, which emits a red luminescence at 615 nm. Treated with active compounds, the reduced amount of P23H mutant opsin is represented by a lowered ALPHA signal.
Table.
Screening Assays for Compounds Affecting the Stabilization or Degradation of P23H Opsin
Methods
Stable Cell Lines
Mouse sequences were the source of all opsin constructs.
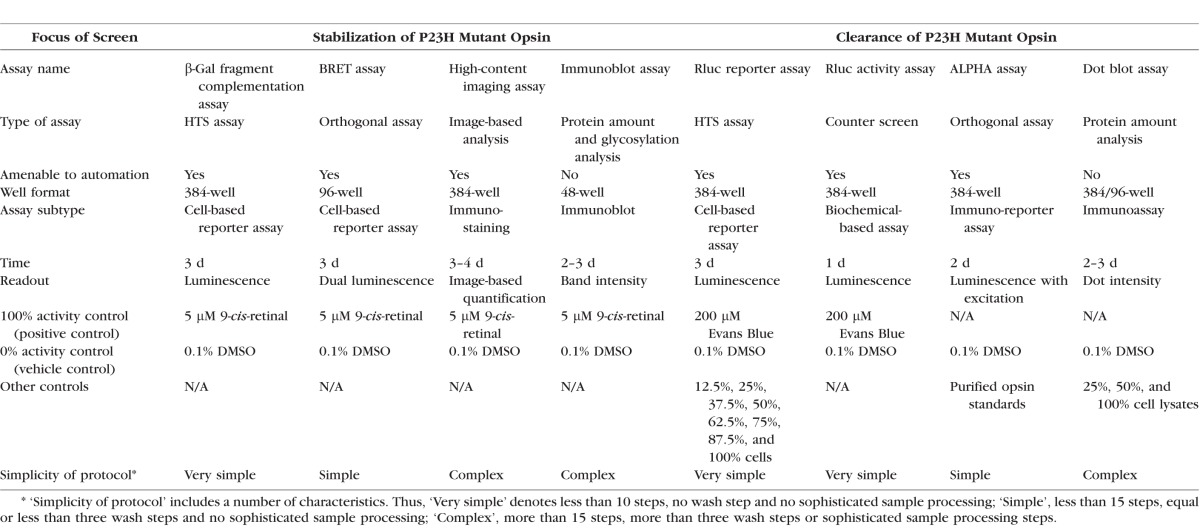
For the β-Galactosidase (β-Gal) fragment complementation assay (HTS of P23H-opsin translocation), the U2OS (PLC-EA and P23H-opsin-PK) cell line was generated by DiscoveRx, Inc. (Fremont, CA, USA). Two fusion proteins were expressed in the U2OS (PLC-EA and P23H-opsin-PK) cell line: the pleckstrin homology (PH) domain of phospholipase C-δ (PLC) was fused with the large subunit (EA) of β-Gal named PLC-EA; and the small subunit (PK) of β-Gal was fused on the C-terminus of the mouse P23H-opsin mutant named P23H-opsin-PK. Expression of PLC-EA and P23H-opsin-PK was confirmed by immunostaining (Fig. 2A).
Figure 2.
The β-Gal fragment complementation assay for a HTS of P23H mutant opsin translocation (top left). (A) Immunostaining of U2OS (PLC-EA and P23H-opsin-PK) stable cells treated with 0.1% DMSO (left) and 5 μM 9-cis-retinal (right). PLC-EA is the PH domain of phospholipase C δ fused with the large subunit of β-Gal; and P23H-opsin-PK is the P23H mutant opsin fused with the small subunit of β-Gal. Yellow, P23H-opsin-PK; blue, DAPI. (B) Dose-response curve of 9-cis-retinal assessed by the β-Gal fragment complementation assay. Concentrations of 9-cis-retinal tested were 0.001, 0.005, 0.01, 0.05, 0.10, 0.25, 0.5, 0.75, 1.0, 2.5, 5, 7.5, 10, 15, and 20 μM. Error bars, SDs of triplicate determinations. The HTS quality control parameters (S/B ratio and Z' described in Methods and the EC50 of 9-cis-retinal are listed in the inset.
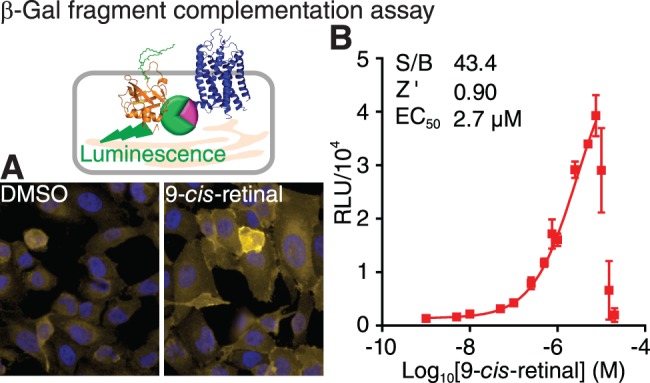
For the bioluminescence resonance energy transfer (BRET) assay25 (confirmation of increased translocation of P23H-opsin), an HEK293 (P23H-opsin-Rluc and Venus-Kras) cell line was generated by incorporating the P23H-opsin-Rluc pcDNA3.1 Zeo and Venus-Kras pcDNA3.1 constructs in sequence. These cells then stably expressed the two fusion proteins, namely a mouse P23H-opsin tagged with Renilla luciferase 8 (P23H-opsin-Rluc), and a 25 amino acid plasma membrane (PM) targeting peptide of the mouse V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (Kras) GTPase conjugated with the Venus fluorescence protein (Venus-Kras). Coexpression of these two fusion proteins was confirmed by immunostaining of P23H opsin and Venus fluorescence under a fluorescence microscope (Fig. 3A). Positive clones were tested in the BRET assay and one clone was selected as noted previously.26
Figure 3.
The BRET assay for confirming active compounds that facilitate P23H mutant opsin translocation (top left). (A) Immunostaining of HEK293 stable cells illustrates the localization of P23H-opsin-Rluc and Venus-Kras fusion proteins. Red, mP23H mutant opsin; green, Venus-Kras. An overlay of the two stains is shown in the right panel. (B) Dose-response curve of 9-cis-retinal obtained with the BRET assay. Concentrations of 9-cis-retinal tested were 0.0025, 0.005, 0.01, 0.039, 0.078, 0.16, 0.31, 0.63, 1.3, 2.5, 10, and 20 μM. Error bars, SDs of triplicate determinations.
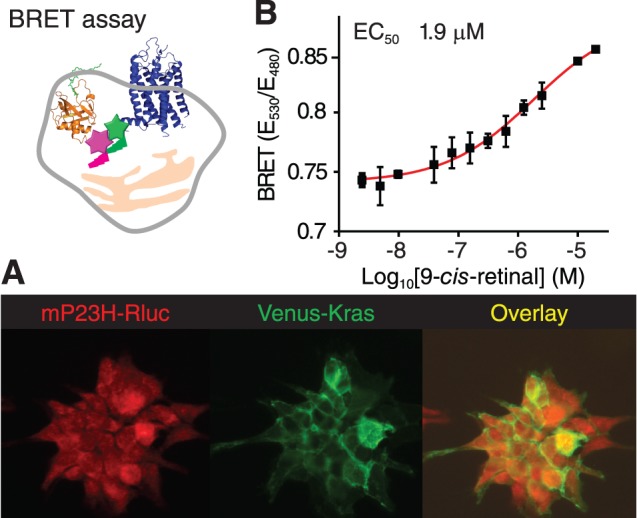
For the immunoblot, immunostaining, and ALPHA immunoassays, NIH3T3 (P23H-opsin/green fluorescent protein [GFP]), and NIH3T3 (WT-opsin/GFP) stable cells were generated by incorporating the pMiLRO 23 and pMiLRO DNA constructs, respectively (the backbone vector pMXs-IG was kindly provided by Toshio Kitamura from the University of Tokyo). Fluorescent GFP was expressed separately with the opsin protein, which was used for positive clone selection. Expression of the P23H mutant or WT opsin was confirmed by immunoblots and immunostaining (Figs. 4A, 5A).
Figure 4.
Image-based analysis of P23H mutant opsin localization. (A) Immunostaining of NIH3T3 stable cells expressing the WT opsin (left), the P23H mutant opsin treated with 9-cis-retinal (middle), or DMSO (right). Yellow, opsin; blue, DAPI; green, GFP. (B) Quantification of P23H mutant opsin localization and cytotoxicity after treatment with 9-cis-retinal. Left chart: The dose response curve of 9-cis-retinal plotted by the intensity (INT) ratio of P23H mutant opsin staining on the PM to that in the ER, as a function of the Log10 [9-cis-retinal]; right chart: The change in nucleus numbers after treatment with 9-cis-retinal as a function of the log of its concentration. Concentrations of 9-cis-retinal in both charts were 0.16, 0.31, 0.63, 1.3, 2.5, 5, 10, and 20 μM. Error bars, SDs of experiments carried out in triplicate.
Figure 5.

Immunoblot analyses of P23H mutant opsin. (A) Immunoblot of NIH stable cells expressing P23H mutant opsin (left panel) or WT opsin (right panel) without treatment (None), or treated with 0.1% DMSO alone (DMSO) or 9-cis-retinal at different concentrations. Thirty micrograms of P23H mutant opsin lysate or 10 μg WT opsin lysate were loaded onto each lane. Internal control, β-tubulin. Arrows: 1, opsin dimer with mature glycosylation; 2, opsin dimer with immature glycosylation; 3, opsin monomer with mature glycosylation; 4, opsin monomer with immature glycosylation. (B) Quantification of band intensities from the immunoblots shown in (A). Left: Chart of the total intensities of opsin normalized by β-tubulin immunoblot intensity. Right: Chart of the intensity ratios of opsin with mature glycosylation (bands 1 + 3) to total opsin. Error bars, SDs from measurements of two individual immunoblots. (C) Immunoblots of NIH stable cells expressing P23H mutant opsin (left panel) or WT opsin (right panel) treated with 0.1% DMSO or 5 μM 9-cis-retinal. Cell lysates were treated with or without PNGaseF for 1 hour at room temperature for deglycosylation before being loaded onto the SDS-PAGE gels. Double arrows: 5, deglycosylated opsin dimer, 6, deglycosylated opsin monomer.
For the luciferase reporter assay (HTS of P23H-opsin clearance and a counterscreen for the effect on WT-opsin), either HEK293 (P23H-opsin-Rluc) or HEK293 (WT-opsin-Rluc) cells were generated by incorporating the P23H-opsin-Rluc pcDNA3.1 Zeo or WT-opsin-Rluc pcDNA3.1 Zeo constructs, respectively. Expression of P23H-opsin-Rluc or WT-opsin-Rluc was confirmed by a luciferase assay as well as by immunoblotting (Fig. 6, Supplementary Fig. S2A).
Figure 6.
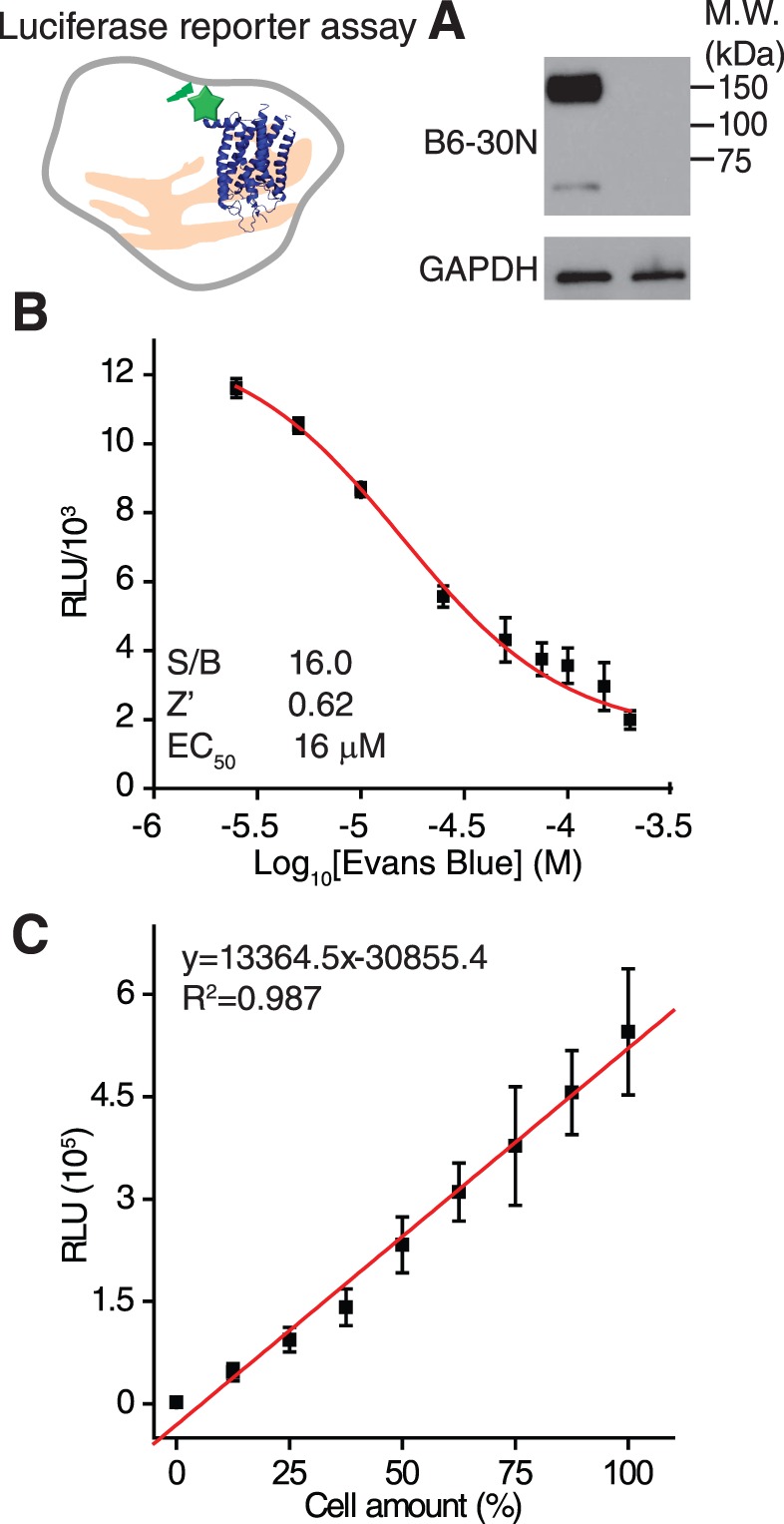
Luciferase reporter assay for a HTS of P23H mutant opsin clearance (top left). (A) Immunoblot (right panel) of HEK293 stable cells expressing the P23H-Rluc fusion protein (left lane) and the HEK293 cells without transfection (right lane). The top of this panel was blotted with B6-30 anti-rhodopsin antibody, and the bottom was blotted with the anti-GAPDH antibody as an internal control. (B) A dose-response curve for the positive control, Evans Blue, in the luciferase reporter assay. Concentrations of Evans Blue were 2.5, 5.0, 10, 25, 50, 75, 100, 150, and 200 μM. Quality control parameters (S/B ratios and Z') and EC50 of Evans Blue are listed in the inset. Error bars are SDs from experiments performed in triplicate. (C) A chart of luciferase activity related to cell seeding number in the P23H-Rluc reporter assay. Cells were seeded at 12.5%, 25%, 37.5%, 50%, 62.5%, 75%, 87.5%, and 100% per well in triplicate with the 100% cell amount equaling 8000 cells/well. A linear fit is shown as a red line. The slope, intercept, and R2 value of the fitted curve are listed in the inset.
Cell Cultures and Media
For all stable cell lines, cells were cultured in cell growth medium containing Dulbecco's modified Eagle's medium (DMEM; Hyclone, Logan, UT, USA) with 10% fetal bovine serum (FBS; Hyclone), 5 μg/mL plasmocin (InvivoGen, San Diego, CA, USA) at 37°C in 5% CO2. Cells were subcultured as per instructions in the ATCC Animal Cell Culture Guide (in the public domain, www.atcc.org). When cells were seeded for assays in 96- or 384-well plates, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2.92 μg/mL L-glutamine (Hyclone) were added to the cell growth media (designated as assay medium).
Chemicals and Reagents
The β-Gal fragment complementation assay buffer and substrate were from the Gal-Screen System (Applied Biosystems, Bedford, MA, USA). PNGaseF was purchased from NEB (Ipswich, MA, USA). Coelenterazine h (NanoLight, Pinetop, AZ, USA) was dissolved in ethanol at 1 mg/mL (2.5 mM) as a stock solution and stored at −80°C. DAPI (4′,6-diamidino-2-phenylindole; Life Technologies, Grand Island, NY, USA) was dissolved in ethanol at 14.3 mM and stored at −80°C until use. ViviRen (Promega, Madison, WI, USA) was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich Corp., St. Louis, MO, USA) to achieve a 60 mM stock solution that also was stored at −80°C. 9-cis-Retinal (Sigma-Aldrich Corp.) was dissolved in DMSO at 20 mM in the dark and covered with foil before storage at −80°C. Evans Blue (Sigma-Aldrich Corp.) was solubilized in H2O at 25 mM and kept at room temperature. Recombinant Rluc (RayBiotech, Norcross, GA, USA) was dissolved in PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 137 mM NaCl, and 2.7 mM KCl) at 50 μg/mL and maintained at 4°C for use within 2 weeks. Bovine opsin was obtained from bleached rhodopsin purified from bovine retina as described previously.27 Mouse monoclonal B6-30 and 1D4 anti-rhodopsin antibodies were purified from hybridoma cells also as noted before.27 Biotinylated B6-30 anti-rhodopsin antibody was generated and purified as listed in the instructions for EZ-Link NHS-LC-Biotin reagent (Thermo Scientific/Pierce, Rockford, IL, USA). The immunoassay buffer, cell lysis buffer, streptavidin-conjugated donor beads, and unconjugated acceptor beads for the ALPHA immunoassay were purchased from PerkinElmer (Waltham, MA, USA). Anti-rhodopsin antibody 1D4 conjugated acceptor beads were generated as described in the ALPHA immunoassay guide (PerkinElmer).
β-Gal Fragment Complementation Assay
On day 1, cultured U2OS (PLC-EA and P23H-opsin-PK) cells were detached, resuspended and diluted to 3 × 105 cells/mL with assay medium. The cell suspension was dispensed at 20 μL/well into a 384-well, white-walled, clear-bottom, tissue culture-treated plate (Greiner Bio-one, Monroe, NC, USA) with a Multidrop dispenser (Thermo Scientific, Waltham, MA, USA). The 384-well plate was centrifuged at 300g for 30 seconds, and then incubated at 37°C in 5% CO2. On day 2 of incubation, a dosage series of 9-cis-retinal working solution (5 times concentrated in the assay medium) was prepared. Dimethyl sulfoxide in the assay medium (at final concentration in the assay of 0.1%) was also provided as a negative control. Five microliters/well of either 9-cis-retinal working solution or 0.5% DMSO was dispensed into each well of a 384-well plate containing the cell cultures. Each dose of 9-cis-retinal was examined in triplicate, whereas the DMSO control was repeated 16 times. The plate was centrifuged at 300g for 30 seconds and incubated at 37°C in 5% CO2. On day 3, the reaction buffer was prepared as described in the instructions for the Gal-Screen System (Applied Biosystems), and dispensed into the prepared 384-well plate at 25 μL/well with the Multidrop dispenser. The plate was covered with foil and incubated at room temperature for 1 hour. The plate was read for luminescence with the EnVisionTM plate reader (PerkinElmer) with an integration time of 0.1 s/well. The averages and standard deviations of the relative luminescence unit (RLU) readouts from repeats of each condition are listed in Supplementary Table S1, and plotted as the y-values and error bars in the dose-response chart with the corresponding log10[9-cis-retinal] concentrations shown as the x-values in Figure 2B. The dose-response curve for 9-cis-retinal was fitted by Origin8.1 software (OriginLab, Northampton, MA, USA), and the data range was selected to rule out data points at high dosages which reflected cell death with a subsequent decrease in luminescence. An observed EC50 was obtained at the concentration of 9-cis-retinal, which achieved 50% of the maximal experimental luminescence signal. The seeding density, and β-Gal reaction time were optimized by this protocol, ensuring a high Z' value. Dimethyl sulfoxide was tested from 0.01% to 1%, which confirmed that addition of up to 0.5% DMSO did not affect the assay readout. The 100% value was measured at 5 μM 9-cis-retinal, and the 0% value in 0.1% DMSO. Quality control (QC) of the assay was tested with the 100% and 0% values, each treated with 16 repeats in a 384-well plate. The QC parameters, the signal-to-background ratio (S/B ratio) and Z' values were calculated as S/B ratio = Mean100% control/Mean0% control; and Z' = 1–3 × (SD0% control + SD100% control)/|Mean100% control − Mean0% control|, with Mean as the average of each control readout.28 In the QC experiment, an S/B ratio greater than 30 and Z' greater than 0.70, suggested that the established protocol was reliable for scaling up the HTS (Fig. 2B, inset).
BRET Assay
On day 1, cultured HEK293 (P23H-opsin-Rluc and Venus-Kras) cells were detached, resuspended in culture medium and diluted to 1 × 106 cells/mL. The cell suspension was dispensed into a 96-well tissue culture plate (BD Falcon, San Jose, CA, USA) at 200 μL/well. The plate was centrifuged at 300g for 30 seconds and then incubated at 37°C in 5% CO2. On day 2, the 9-cis-retinal doses were diluted with assay medium to five times their final tested concentrations under dim light. The DMSO sample also was diluted to five times its final concentration of 0.1% with assay medium. Then 50 μL 9-cis-retinal or DMSO diluent was added to each well of the 96-well plate under dim light. The plate was covered in foil and incubated at 37°C in 5% CO2. On Day 3 under dim light, the medium in the 96-well plate was aspirated and the treated cells were resuspended in 200 μL PBS per well with a multichannel pipette. Cell suspensions then were transferred into a white opaque 96-well plate (Corning, Tewksbury, MA, USA). Coelenterazine h was diluted to 25 μM with PBS and each well of the white 96-well plate was injected with 50 μL of this coelenterazine h solution, followed by a dual-emission reading at 480 and 530 nm after a 5-second delay. Readings were taken with a SpectraMax L plate reader featuring two MDT detectors installed with a BRET1 filter set (Molecular Devices, Sunnyvale, CA, USA), and the BRET signal was calculated as the ratio of emission at 530 nm to that at 480 nm. The average and SD of BRET assays repeated in triplicate for each dose of 9-cis-retinal are listed in Supplementary Table S2 and plotted in Figure 3B. The EC50 of 9-cis-retinal was then calculated as described in the protocol for the β-Gal assay. This protocol was finalized after optimization of plate type, cell seeding density, substrate concentration, and reaction time.
Imaging the Localization of P23H Mutant Opsin
In this assay, microplate liquid handling was performed with an EL406 Washer Dispenser (BioTek, Winooski, VT, USA). On day 1, cultured NIH3T3 (P23H-opsin/GFP) or NIH3T3 (WT opsin/GFP) cells were seeded at 5000 cells/well in a 384-well cell carrier plate (PerkinElmer) as described for the β-Gal fragment complementation assay. The plate was incubated at 37°C in 5% CO2 for 2 hours until cells were attached to the bottom of the plate. Then cells were treated as described for the β-Gal fragment complementation assay. Day 2, the assay medium was aspirated from the 384-well plate and cells were fixed in freshly prepared 4% paraformaldehyde at 20 μL/well for 20 minutes. Cell membranes were permeabilized with 0.1% Triton X-100 in PBS three times at 50 μL/well for 5 minutes each, blocked by 10% goat serum at 20 μL/well for 1 hour, and then incubated with 20 μg/mL mouse monoclonal 1D4 anti-rhodopsin antibody at 20 μL/well at 4°C overnight. After three 0.1% Tween 20 washes in PBS, 20 μL/well Cy3-conjugated goat anti-mouse IgG (1:200 dilution; Life Technologies) was added to the plated cell samples, which were incubated for 1 hour under dim light. Following three more 0.1% Tween 20 washes in PBS, 20 μL/well 300 nM DAPI solution was added to each well for a 5 minute incubation after which the plate was washed a final three times with 0.1% Tween 20 in PBS. Cells immersed in the last wash solution were then used for imaging. Fluorescence images were obtained with the Operetta High Content Imaging System (PerkinElmer) according to the user manual. Five fields were taken from each well, and three channels were taken for DAPI (50 ms), GFP (100 ms), and Cy3 (100 ms) fluorescence. Images were analyzed with Acapella Software from the Columbus data storage and analysis system (PerkinElmer). An image analysis is illustrated in Supplementary Figure S1. Nuclei were defined by DAPI staining and the edge line of each DAPI-stained circle was defined as the 50% line of the cell. The cytoplasm was defined by GFP fluorescence and the outer border of GFP fluorescence was set as the 0% line of the cell. Incomplete cell images were omitted from the final analyses. The PM region was defined within ± 5% of the cell border, and endoplasmic reticulum (ER) was the perinuclear space defined as the 25% to 50% region. Intensities were measured from the Cy3 channel cell by cell. The intensity ratios of Cy3 on the PM region to that in ER region also were calculated cell by cell. Resulting intensity ratios were averaged among the population of intact cells in each well (INT ratio PM-to-ER). The mean and SD of PM-to-ER intensity ratios from three repeats of each treatment condition are listed in Supplementary Table S3 and plotted in Figure 4B. This assay was optimized by cell seeding number, primary and secondary antibody concentration, plate type, starting position of autofocusing, excitation intensity, and scanning time for each channel of fluorescence. The algorithm for quantification of P23H mutant opsin translocation was finalized after comparison with alternative calculations, such as total Cy3 intensity per cell and PM-to-total Cy3 INT ratio per cell. The PM-to-ER INT ratio provided the greatest difference between 5 μM 9-cis-retinal and 0.1% DMSO treatments.
Immunoblot Analyses
NIH3T3 (P23H-opsin/GFP) or NIH3T3 (WT-opsin/GFP) cells were seeded at 8 × 105/mL in 400 μL/well of a 48-well plate (Corning Bio-one). Two hours after cells were attached, 100 μL of either a five times concentrated final dose of 9-cis-retinal or DMSO at 0.5% in assay medium was added to each well under dim light. The treated plate was covered with foil and incubated at 37°C in 5% CO2 overnight. Next day the cells were lysed with 100 μL/well 0.1% TritonX-100 in PBS and complete protease inhibitor cocktail (Roche, Basel, Switzerland) followed by a 3-s/well sonication. Protein concentrations of cell lysates were determined by OD 280 readings in a Nanodrop spectrometer (Thermo Scientific). For PNGase F catalyzed deglycosylation, 3 μL (1500 units) PNGase F (NEB) was added to the cell lysate and incubated at room temperature for 1 hour before immunoblotting (Fig. 5C). For the NIH3T3 (P23H-opsin/GFP) lysate, 30 μg of total protein was loaded for each treatment condition. But for the NIH3T3 (WT-opsin/GFP) lysate, 10 μg total protein was loaded per well for immunoblotting performed as previously described.29 Two immunoblots were used to assess each treatment condition. The working concentration of 1D4-anti-rhodopsin antibody was 0.5 μg/mL and the horseradish peroxidase (HRP) conjugated goat anti-mouse IgG secondary antibody (Life Technologies) was employed at a 1:10,000 dilution. Band intensities were measured with ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA) and opsin amounts were normalized first by the intensity ratios of immunoblot bands obtained from opsin to those from β-tubulin, and then to the DMSO-treated condition. Ratios of maturely glycosylated-to-total P23H mutant opsin ratio were calculated as (INTband1 + INTband3)/(INTband1 + INTband2 + INTband3 + INTband4). These P23H mutant opsin bands are illustrated in Figure 5A. These values were averaged and SDs were calculated from experiments performed in duplicate, and the data are listed in Supplementary Table S4 and plotted as columns and error bars in Figure 5B.
The Luciferase Reporter Assay
On day 1, HEK293 (P23H-opsin-Rluc) cells were seeded at 8000 cells/well (32 μL/well from a 2.5 × 105/mL cell suspension) into a 384-well white-walled clear-bottom tissue culture treated plate (Greiner Bio-one), and then incubated at 37°C in 5% CO2. On day 2, cells were treated as described in the β-Gal fragment complementation assay, with 8 μL/well containing five times the final concentrations of Evans Blue or 0.5% DMSO, respectively. Each Evans Blue dose or DMSO control was performed in triplicate. On day 3, cells were treated with 5 μL/well 0.2% n-dodecyl-β-D-maltoside (DDM; Affemetrix, Santa Clara, CA, USA) for 5 minutes at room temperature. The modified Rluc substrate ViviREN was added to achieve a final concentration of 5 μM (5 μL/well 50 μM ViviREN working solution). The plate was shaken at 100 rpm for 5 seconds, covered by foil and incubated at room temperature for 60 minutes. Luminescence then was read with an Envision plate reader (PerkinElmer). Averages and SDs of readouts at each dose are listed in Supplementary Table S5 and plotted as y-values and error bars, with corresponding log10[Evans Blue] x-values shown in Figure 6B. The dose-response curve fitting was done as described for the β-Gal fragment complementation assay. The QC experiments were each carried out 16 times with 0.1% DMSO and 200 μM Evans Blue used as 0% and 100% controls. These QC experiments provided an S/B ratio greater than 10 and a Z' greater than 0.5, suggesting that the luciferase reporter assay is reliable for HTS (Fig. 6, inset). This protocol was finalized by optimization of cell seeding density, substrate stability, detergent conditions, and reaction times to ensure the HTS quality of the assay.
To calibrate the luciferase reporter activity with amount of P23H mutant opsin, on days 1, 4, 8, 12, 16, 20, 24, and 28, 32 μL/well cell suspension (2.5 × 105 cells/mL) was added to columns 3 to 4, 5 to 6, 7 to 8, 9 to 10, 11 to 12, 13 to 14, 15 to 16, and 17 to 18, respectively, in a 384-well Greiner plate. Then 40, 36, 32, 28, 24, 20, 16, 12, 8, and 4 μL/well assay medium was added to columns 1 to 2, 3 to 4, 5 to 6, 7 to 8, 9 to 10, 11 to 12, 13 to 14, and 15 to 16, respectively, to achieve a total of 40 μL solution per well. On day 3, this calibration plate was treated and read as described above. Averages and SDs of readouts from each cell number were plotted as y-values and error bars, whereas cell amounts in percentages were plotted as x-values (Fig. 6C). A linear curve fitting was performed for calibration of the P23H mutant opsin amount (%) to the luminescence readout.
This same protocol was repeated in HEK293 cells expressing the WT-opsin-Rluc fusion protein (Supplementary Fig. S2).
The Rluc Activity Assay
Recombinant Rluc stock solution was diluted to 50 μg/mL and dispensed into a 384-well white-walled clear-bottom plate (Greiner Bio-one) at 16 μL/well (800 ng/well). Evans Blue or DMSO were prepared at five times of their final concentrations (200 μM for Evans Blue or 0.5% for DMSO) and dispensed at 4 μL/well. The plate was incubated at 37°C for 15 minutes. Coelenterazine h stock solution diluted to a final concentration of 25 μM in assay medium was then dispensed into the plate at 20 μL/well. Luminescence from each well was read 2.5 seconds after substrate injection in a well-by-well sequence. Each control condition was carried out 16 times, and the averages and SDs of readouts from these replicates were calculated and plotted as columns with error bars (Fig. 7). The S/B ratio was higher than 10, suggesting that this assay could reliably detect Rluc inhibitors as false positives from the HTS of P23H mutant opsin clearance.
Figure 7.
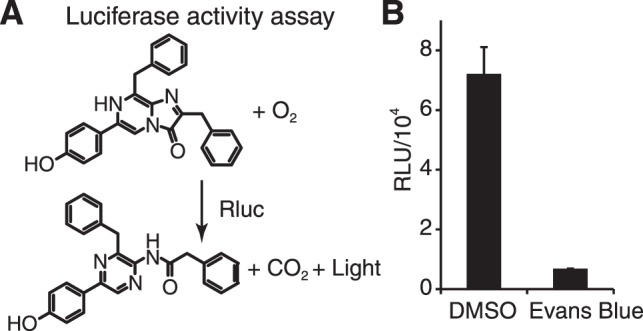
Luciferase activity assay for the counter screen to identify false positives from the HTS of P23H mutant opsin clearance. (A) Coelenterazine h, catalyzed by recombinant Rluc, reacts with oxygen and generates coelenteramide h, CO2 and light that is read as luminescence. Inhibition of Rluc by a model compound causes a significant reduction of this luminescence. (B) The activity of Rluc treated with 0.1% DMSO or 200 μM Evans Blue. Error bars, SDs from 16 experiments.
The ALPHA Immunoassay
Liquid handling for this assay was accomplished with the EL406 Washer Dispenser (BioTek). On day 1, NIH3T3 (P23H-opsin/GFP) cells were seeded at 10,000, 7500, 5000, and 2500 cells/well (for quantification of 100%, 75%, 50%, and 25% of cell lysate) in a 384-well white-walled clear-bottom plate (Greiner Bio-one) leaving three columns blank. After 2 hours of incubation at 37°C in 5% CO2, cells were treated with 0.1% DMSO and reincubated at 37°C with 5% CO2. On day 2, the assay medium was aspirated and cells were fixed with freshly prepared 4% PFA, followed by three washes each with 0.1% Triton X-100 in PBS for 5 minutes. Fixed cells were lysed by 2.5 μL/well lysis buffer (PerkinElmer) at room temperature for 10 minutes. Purified bovine opsin standard diluted from 100 ng/mL to 3 pg/mL was added at 2.5 μL/well in three replicates to the three columns of blank wells of the 384-well plate. Each well then received 5 μL 0.625 μg/mL biotinylated B6-30 antibody and 7.5 μL 33.33 μg/mL 1D4-conjugated acceptor beads. The plate was centrifuged at 300g for 30 seconds, and incubated at room temperature for 1 hour. Under dim light, each well of the plate then received 10 μL 100 μg/mL streptavidin-conjugated donor beads and the plate was covered by foil and incubated at room temperature for 30 minutes. The ALPHA signal was read with a PerkinElmer Enspire plate reader. Each condition was assayed in triplicate. The averages and SDs obtained for each experimental condition are listed in Supplementary Table S6. The standard curve of ALPHA signals versus opsin amounts was plotted and fitted by the Hill function, y = a × xn/(bn + xn) with Origin8.1 software (Fig. 8A). P23H mutant opsin amounts were calculated by calibrating ALPHA signals from NIH3T3 (P23H-opsin/GFP) cell lysates to a standard curve derived from purified bovine opsin (Supplementary Table S7; Fig. 8B). The linear correlation between calculated opsin amounts with cell lysate amounts suggests reliability of the ALPHA assay in confirming P23H mutant opsin degradation.
Figure 8.
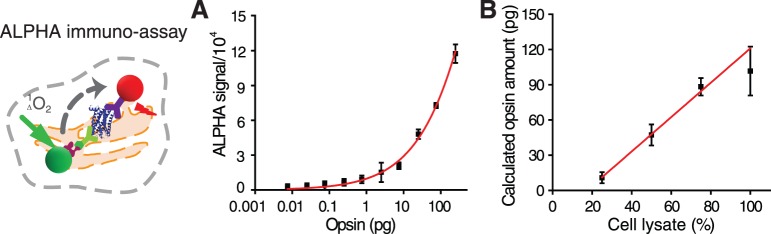
The ALPHA immunoassay for confirmation of P23H mutant opsin clearance (left). (A) A standard curve of purified opsin fitted by the Hill function. Opsin standards were 0.0075, 0.025, 0.075, 0.25, 0.75, 2.5, 7.5, 25, 75, and 250 pg. (B) Quantification of P23H mutant opsin in lysates containing different numbers of cells. The value of 100% for a cell lysate was obtained from 10,000 cells cultured in a single well of a standard 384-well plate for 18 hours. Error bars represent SDs from experiments performed in triplicate.
The Dot Blot Assay
On day 1, NIH3T3 (P23H-opsin-GFP) cells were seeded in a 384-well tissue culture-treated plate and treated as described for the ALPHA immunoassay (five replicates for each experimental condition). On day 2, the assay medium was aspirated and cells were lysed over 5 minutes by 20 μL/well 0.2% SDS in PBS with complete protease inhibitor cocktail (Roche). Each well of cell lysate was subjected to sonication for 3 seconds at low energy to break down the DNA. Then 5 μL/well cell lysate was blotted onto a nitrocellulose membrane (Thermo Scientific) with a multichannel pipette. The nitrocellulose membrane was air-dried for 30 minutes and the immunoblot procedure was performed as described above in ‘Immunoblot analyses.' Dot intensities were measured with ImageJ software. The averages and SDs of measured dot intensities from the five replicates of each condition were calculated and plotted as columns with error bars as shown in Figure 9B.
Figure 9.
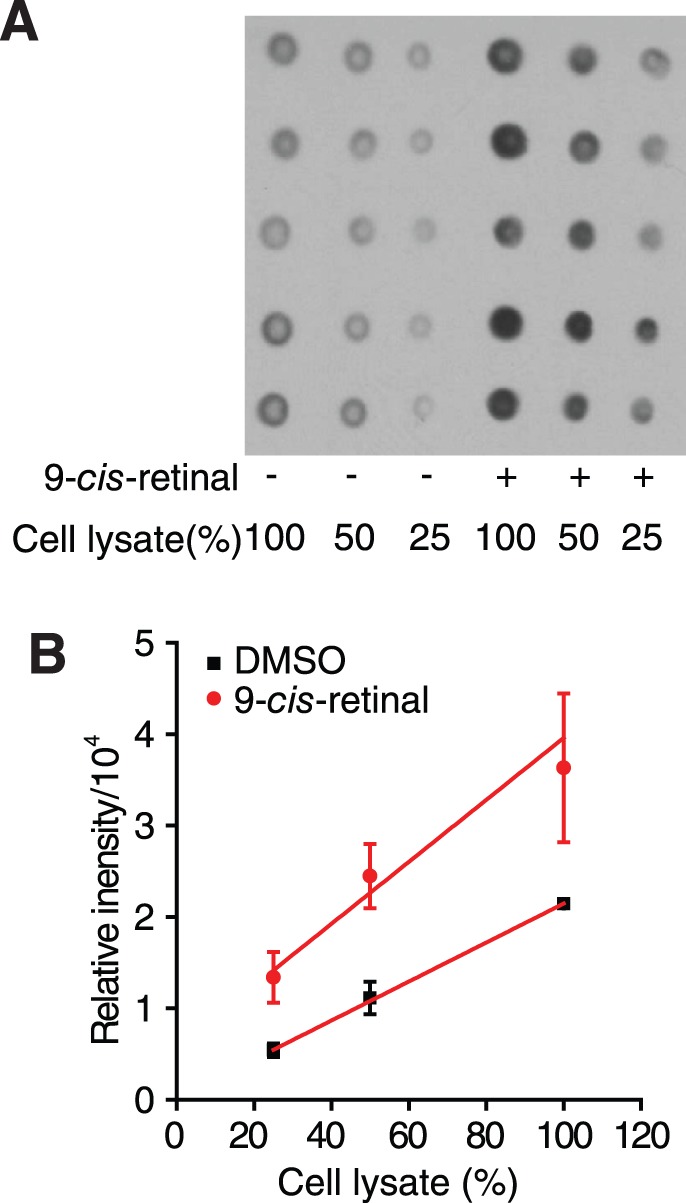
Dot blot assay for confirmation of P23H mutant opsin clearance. (A) A dot blot of P23H mutant opsin expressed by NIH3T3 stable cells imaged with the 1D4 anti-rhodopsin antibody. Each dot was blotted from cells incubated in one well of a standard 384-well plate for 18 hours. The 100% cell lysate was obtained from 2500 cells. (B) Chart of the relationship between dot intensities and the amounts of cell lysate in DMSO and 9-cis-retinal–treated samples. Error bars represent SDs from experiments performed five times.
Results
To identify active hit compounds for treatment of P23H mutant opsin associated adRP and identify potential pathways that affect its homeostasis, we developed a set of cell-based assays to identify compounds that either stabilize misfolded P23H mutant opsin or increase its degradation (Fig. 1; Table). Image-based and immunoblot assays were optimized for further characterization of target localization, target amounts and posttranslational modifications.
9-cis-Retinal, an analog of the natural chromophore 11-cis-retinal, is a pharmacological chaperone that can stabilize the folding of P23H mutant opsin by binding to the ligand pocket of this mutant opsin and forming an isorhodopsin pigment.13,20,30,31 Thus, 9-cis-retinal was used as a model compound to validate assays used to screen for compounds that stabilize the P23H mutant opsin. The solubility of 9-cis-retinal in DMEM is higher than 200 μM, because of the albumin present in the serum, and thus no precipitation was observed at that concentration.
We did not have available compounds known to enhance the degradation of P23H mutant opsin. So instead for the luciferase reporter assays, we used the reported luciferase inhibitor, Evans Blue,32 as a model compound to validate the luciferase screening assays. Assays for P23H mutant opsin degradation were also quantified by titrations of differing numbers of cells and amounts of purified opsin (Figs. 6C, 8B, 9).
The β-Gal Fragment Complementation Assay for HTS of P23H Mutant Opsin Translocation
We adapted a cell-based HTS assay to quantify the translocation of P23H mutant opsin from the ER to the PM, a marker for folded P23H mutant opsin that has escaped the ER quality control system of the cell.20,30,31 Using the complementary effect of two separated subunits of β-Gal,33 one fused with the P23H mutant opsin and the other with a plasma membrane anchored peptide (PLC), the translocation of P23H mutant opsin was quantified as the recovered β-Gal activity read by luminescence (Fig. 1A). Upon treatment with the vehicle control (DMSO), P23H-opsin-PK still accumulated in the ER, such that the basal readout of β-Gal activity was as low as that of the blank. But upon treatment with 9-cis-retinal, the folded P23H isorhodopsin translocated from the ER to the PM, resulting in a significant increase in the readout signal for the β-Gal assay in a 384-well format (Fig. 2). The simple protocol for this assay ensured its adaptation to automation. The dose response curve for 9-cis-retinal also indicated that the β-Gal assay could detect subtle increases in P23H mutant opsin translocation because the maximum effect of 9-cis-retinal produced a greater than 40-fold increase in the signal with only small error bars. This result allows a reliable quantification of P23H mutant opsin translocation for a HTS assay (Fig. 2B). The Z' value, calculated as 1–3 × (SD0% control + SD100% control)/(Mean100% control − Mean0% control),28 is a critical quality control parameter for a HTS assay, taking both the S/B ratio and assay variations into account. It is generally agreed that a good HTS assay should have Z' greater than or equal to 0.5. The Z' value for the β-Gal fragment complementation assay was 0.90, indicating this assay had a high reliability for HTS.
The BRET Assay for Confirmation of P23H Mutant Opsin Translocation
A HTS of a 25,000 Structure Diversity Set from a compound library could yield up to 200 “hit” compounds with desired activity in a primary HTS assay.34 In contrast, only a couple of candidate compounds usually emerge for further medicinal chemical optimization and in vivo studies. Therefore, an orthogonal assay is immediately required to distinguish ‘true' hit compounds from false positives. Using the BRET effect, we developed a cell-based assay to confirm the effect of P23H mutant opsin translocation after treatment with hits identified from the preliminary HTS (Fig. 1B). A stable cell line was generated expressing the P23H-opsin-Rluc (BRET donor) and Venus-Kras (BRET acceptor) fusion proteins (Fig. 3A). With the vehicle control (DMSO), the BRET donor P23H-opsin-Rluc aggregated in the ER, whereas the BRET acceptor Venus-Kras remained in the plasma membrane, providing a low basal BRET signal. Treatment with 9-cis-retinal, a positive control, evidenced a dose-dependent increase of the BRET signal, due to colocalization of the BRET donor and acceptor on the PM (Fig. 3B). This assay followed a simple protocol in a 96-well format with the capability of testing 200 compounds with reliable quality. False positives such as activators of β-Gal activity and autoluminescent compounds were easily detected and ruled out with 0.1% DMSO and 5 μM 9-cis-retinal used, respectively, as 0% and 100% controls. Compounds can increase the signal from BRET or β-Gal complementary assays by (1) increasing the total amount of P23H mutant opsin, (2) shifting P23H mutant opsin from ER to PM, and (3) increasing the cleavage of BRET donor or β-Gal subunit into the cytosol. The model compound 9-cis-retinal both increases the translocation of P23H mutant opsin and increases its total mount in cell culture. Either mechanism could effectively rescue photoreceptors and need to be tested further. However, other undesired effects such as cleavage of tag proteins or an increased fraction of P23H mutant opsin in the ER should be identified as artifacts by image-based analysis described below.
Image-Based Analysis to Quantify the P23H Mutant Opsin by its Location
More careful characterization can accomplished with a reduced number of active compounds for further investigation. Immunostaining is a common method for analyzing protein localization in cultured cells. However, conventional fluorescence microscopy cannot consistently differentiate subtle fluorescence intensity changes among a large group of samples. In contrast, image-based high-content screening systems coupled to advanced image processing software can provide the capability of scanning and analyzing a large number of samples.35,36 High-content imaging analysis was applied to quantify the translocation of G protein–coupled receptors.37–39 In a 384-well plate, we immunostained a stable NIH3T3 cell line separately expressing the P23H mutant opsin and GFP. As shown in Figure 4A, the automated imaging system provided high quality fluorescent images. Each experimental condition was applied to three wells and each well was scanned over five fields with images of 500 to 4000 cells. Both DAPI and GFP fluorescence were recorded to define the nucleus and cytoplasm of each cell (Supplementary Fig. S1). Based on these defined cellular localizations, immunostaining intensities of P23H mutant opsin at different regions of each cell were then measured. The boundary of cytoplasmic region was defined as PM and the perinuclear region was defined as the ER (Supplementary Fig. S1). The intensity ratios of PM-to-ER staining of P23H mutant opsin were measured and calculated as described in Methods, representing the mean values per cell in each well. We also tested the quality of image analysis with different cell seeding numbers (250–10,000 cells/well), and decided to seed 5000 cells/well which reached approximately 60% confluence (Fig. 4A). This cell number ensured that enough cells could be analyzed in the five image fields per well, identifying up to 2000 to 4000 cells to be analyzed per well when treated by DMSO. Also critical to assay quality, these cells were not tightly crowded so that their cell borders and shapes were better defined. Given sufficient numbers of cells in each measurement, these image-based analyses became much more reliable compared with conventional imaging analysis of immunostained cells on cover glasses. This fact was demonstrated by a marked dose-dependent increase in the PM-to-ER INT ratio of the P23H mutant opsin stain in cells treated with 9-cis-retinal (Fig. 4B, left chart). In agreement with previous studies, 9-cis-retinal also increased the average P23H mutant opsin stain of the entire cell, and decreased the ER-to-total P23H opsin stain ratio (data not shown). In addition, the cytotoxicity (CC50) of 9-cis-retinal was obtained by the plot of nuclei numbers, suggesting that the concentration of this model compound needed to be in the 10−6 to 10−7 M concentration range to reduce its cytotoxicity (Fig. 4B, right chart). Therefore, this image-based assay allows a dual analysis of P23H mutant opsin localization as well as cytotoxicity. Compounds with undesired effects, such as those with high cytotoxicity or causing an increased ER fraction of P23H mutant opsin (decrease of PM-to-ER INT ratio), can be ruled out with this assay. Other possible effects of tested compounds can be identified as well, such as: (1) a decrease of P23H mutant opsin aggregating in ER, (2) an increase in the total amount of mutant opsin, and (3) a shift of the mutant opsin from the ER to the PM. Because this assay features nontagged mutant opsin, it would also eliminate artifacts due to the addition of tags in the β-Gal fragment complementation and BRET assays.
Immunoblot Analysis of P23H Mutant Opsin
Due to its ER retention, misfolded P23H mutant opsin features N-linked glycosylation that is susceptible to cleavage by endoglycosidase H.21,40 Differences in glycosylation content are also manifested by molecular masses that are lower than that of WT opsin when expressed in NIH3T3 cells. Compared with that of WT opsin, the total intensity of P23H mutant opsin was significantly reduced and was seen primarily as a dimer band (Fig. 5A). After treatment with 9-cis-retinal, the total intensity of the P23H mutant opsin bands was increased; the monomer band of the mutant opsin became visible (left, Fig. 5A), suggesting that the mutant opsin was stabilized. In addition, the molecular masses of the P23H mutant opsin monomer and dimer shifted to sizes similar to those of WT opsin, suggesting an increased amount of the mutant opsin with mature glycosylation (Fig. 5A). Deglycosylation by PNGaseF led to new bands with lower molecular mass at 30 and 60 kDa from lysates of NIH3T3 cells expressing P23H mutant or WT opsin treated with DMSO or 5 μM 9-cis-retinal, confirming (1) the difference of molecular mass between P23H mutant and WT opsin is due to a difference in glycosylation, and (2) the shift of molecular mass of P23H mutant opsin monomer and dimer bands by treatment of 9-cis-retinal is also due to a change of glycosylation. With measurements of band intensities from repeated immunoblots, these effects also could be quantified in a dose-dependent fashion (Fig. 5B). To summarize, this immunoblot analysis could reveal the effects of an active compound on both the amount of protein and its posttranslational modifications.
The first drug discovery strategy was developed based on the hypothesis that stabilizing the P23H mutant opsin could rescue rod photoreceptors. Alternatively, we also hypothesized that specific clearance of the P23H mutant opsin could protect rod photoreceptors, while the copy of normal opsin sustains the structure and function of retina. The following assays were developed to identify novel compounds, which specifically increase the degradation of P23H mutant opsin.
Luciferase Reporter Assay for a HTS of P23H Mutant Opsin Clearance
With Rluc used as a reporter in a HEK293 stable cell line, a HTS assay was developed to identify compounds that decrease the total amount of P23H mutant opsin, and thereby reduce its cytotoxicity (Fig. 1C). Due to its misfolding and instability, the P23H mutant opsin is already heavily degraded in cell culture, reducing its basal amount significantly lower than that of WT opsin. Fusion of Rluc to P23H mutant opsin generated a luminescence signal for quantification of the mutant opsin, providing a high S/B ratio (S/B = 16.0) for the HTS (Z' = 0.62, with Evans Blue used as the 100% control and DMSO as the 0% control; Figs. 6A, 6B). Because no active compounds were available that increase the clearance of P23H mutant opsin, Evans Blue, a reported Rluc inhibitor,32 was used instead as the control model compound. Potential false positive compounds, such as dyes and Rluc inhibitors, were ruled out by using the counterscreen and confirmation assays described below. In addition, amounts of P23H mutant opsin were determined by titrating different numbers of cells expressing P23H-opsin-Rluc (Fig. 6C). The linear correlation of the luminescence readout with cell amounts suggests the readout of this assay can be used to quantify P23H mutant opsin, if treatment with the compound does not affect the number of cells. Compounds that decrease cell numbers can be identified by the image-based assay described above that will count cell numbers and reveal changes in cell shape.
To identify active compounds that specifically decrease the amount of P23H mutant opsin, we generated HEK293 stable cells expressing WT-opsin-Rluc fusion protein. The same luciferase reporter assay was tested in this cell line (Supplementary Fig. S2). The amount of WT-opsin-Rluc can be determined by titration of cell numbers as described above. Compounds that selectively decrease the readout in cells expressing P23H-opsin-Rluc without changing the readout in cells expressing WT-opsin-Rluc would be selected as active compounds for further characterization.
Counter Screen Rluc Activity Assay to Identify False Positives From the HTS of P23H Mutant Opsin Clearance
Because the HTS assay for P23H mutant opsin clearance was designed to search for compounds that decrease the signal of the Rluc reporter, a counter screen assay was needed to distinguish compounds that directly affect Rluc enzymatic activity from true hit candidates that reduce the amount of P23H mutant opsin. Thus, we developed a biochemical assay to test the effect of hit compounds on the activity of a recombinant Rluc (Fig. 7A). As expected, Evans Blue (200 μM) provided a strong inhibition of Rluc activity (Fig. 7B), suggesting this assay could identify luciferase inhibitors and dyes that block the luminescence readout. Thus, only hit compounds that showed no effect on Rluc activity in this assay would qualify for further confirmation.
The ALPHA Immunoassay to Confirm P23H Mutant Opsin Clearance
The ALPHA assay was designed as an improved alternative to the ELISA because it provides a better quality and simpler protocol for quantification of certain proteins of interest.41,42 Here, we developed a modified ALPHA immunoassay for the quantification of P23H mutant opsin to confirm the effect of active compounds on its clearance (Fig. 1D). With purified bovine opsin used as a standard, the ALPHA signal was calibrated to detect picogram levels of opsin (Fig. 8A). Due to its instability after cell lysis, the P23H mutant opsin often aggregates resulting in low ALPHA readouts. Therefore, we fixed the NIH3T3 (P23H-opsin-GFP) cells before lysis, exposing their P23H mutant opsin epitopes to obtain reliable quantification. Due to lack of available active compounds before running the HTS, we used lysates from different numbers of cells seeded in the 384-well plate to represent different amounts of P23H mutant opsin in the ALPHA assay. The superior linear correlation of cell numbers and calculated P23H mutant opsin amounts from this ALPHA calibration suggest that this assay can reliably indicate the effect of tested compounds on the clearance of mutant opsin (Fig. 8B).
The Dot Blot Assay for Alternative Confirmation of P23H Mutant Opsin Clearance
The dot blot assay is a simplified alternative to the conventional immunoblot, allowing semiquantification of protein amounts under a large range of experimental conditions. We developed a dot blot assay with lysates from NIH3T3 (P23H-opsin/GFP) cells to confirm P23H mutant opsin clearance activity. To compensate for higher variability, each condition was tested in 5 wells of a 384-well format. As shown in Figure 9, the decreased cell amounts were correlated with decreases in dot intensity, whereas enhanced dot intensities reflected increases in dot blot intensities provided by greater amounts of P23H mutant opsin upon pretreatment with increasing concentrations of 9-cis-retinal. This assay, together with an immunoblot assay and high-content imaging analysis from immunostaining of P23H mutant and WT opsin should further confirm their changes in amount, localization, and posttranslational modification after treatment with active compounds. Cytotoxicity will also be detected by the image-based analyses.
Finally, only a handful of active compounds will be selected upon being fully characterized by these different pharmacological assays. After careful chemoinfomatic analysis of each compound,43,44 two to four compounds from each P23H mutant opsin stabilization or clearance study would qualify for further development and in vivo testing.
Discussion
To discover novel, small molecular compounds with desired activity for treating retinal diseases, initial compounds can be identified from a cell-based HTS with a high S/B ratio and low variability. Initially identified hit compounds from HTS should be further confirmed and characterized by multiple assays, so that only those compounds with the desired activity in all these assays are selected for further evaluation by medicinal chemical and in vivo studies. Here we established a strategic set of reliable cell-based and scalable protocols to search for active compounds that stabilize or increase the degradation of P23H mutant opsin. Active compounds identified from this methodology might also reveal novel cellular compensatory mechanisms and machinery that could be incorporated to enrich pharmacological approaches for the treatment of RP. Moreover, the general protocol schematic can readily be adapted to identify promising active compound precursors for the treatment of other retinal diseases. We purposely selected the misfolded P23H mutant opsin as one of the most difficult to study.
Retinitis pigmentosa actually consists of a set of hereditary retinal diseases that cause progressive rod and cone photoreceptor cell degeneration with visual impairment often culminating in blindness.45,46 Among RP patients, the rod opsin gene accounts for approximately 25% of adRP cases.1,10–12 More than 100 mutations in the opsin gene were noted to manifest wide variations in the severity of symptoms, among which distinct cellular pathways were hypothesized to be involved.21,45,47–51 The first reported opsin mutation, P23H, accounts for approximately 10% of all adRP cases. Specific signs and symptoms of this mutation in humans have been described in detail.21,52–54
Rhodopsin, a G protein–coupled receptor, has a special role in our vision.6,55 The visual pigment rhodopsin comprises the major protein component (>90%, 5 mM) in the pancake-like stacks of disk membranes located in the outer segments of rod photoreceptors.6,55 Approximately 10% of this structure is phagocytized and renewed daily,56 resulting in a high metabolic burden for rod cells because 50,000 rhodopsin molecules must be synthesized de novo every second. Rhodopsin expression is essential for the formation of ROS, which are absent from opsin−/− mice57,58 and humans with null mutations in the opsin gene.59 Thus, even subtle changes in the structure of rhodopsin can lead to retinal degeneration.
Experimental models with the P23H mutant opsin have been generated in cell cultures,40,48,60,61 mice,62–64 rats,65,66 pigs,67 Drosophila melanogaster,17,68 Caenorhabditis elegans,13 and frogs15,19,69–71 to advance our understanding of this disease. Biochemically, the P23H mutant opsin is unstable and misfolded; thus, it is located in the ER and features enriched N-glycosylation in cell culture.40 9-cis-Retinal binds in the ligand pocket of the mutant opsin and stabilizes its structure by forming an isorhodopsin pigment.13,72 However, due to its light sensitivity and cytotoxicity at high concentrations, other active compounds are needed to stabilize the P23H mutant opsin. We previously generated a P23H opsin knock-in mouse that is genetically equivalent to the human adRP condition21 and found that the P23H protein was both less abundant (1%–10% of WT rod opsin) and glycosylated. Moreover, this protein failed to accumulate in rod photoreceptor cell ER and instead disrupted rod photoreceptor disk morphology.11,12 Genetically engineered P23H mice lacking visual chromophore also showed accelerated photoreceptor cell degeneration. Recently, we found that only early activation of ER-associated protein degradation (ERAD) occurred in opsinP23H/+ knock-in mice, whereas late apoptotic ER-stress signals such as activation of the activating transcription factor 4 (ATF4) or C/EBP homologous protein (CHOP) were not observed.12 These results indicate that most P23H protein is degraded and the ER quality control system of the photoreceptor cells is competent. Therefore, photoreceptor cell death could be due to cytotoxicity of the residual P23H mutant opsin that escapes degradation as well as lack of sufficient 11-cis-retinal to stabilize this opsin. Thus, enhancing the degradation of P23H mutant opsin appears the most logical path forward.
Experimental efforts to combat RP based on current knowledge of this disease have yet to result in approved clinical treatments. Here, we list some possible successful strategies: (1) sequence-specific knockdown of mutant mRNA65,73–75; (2) stabilization of rod cells by increased expression of ER chaperone-like proteins18; (3) coexpression of WT opsin to increase the ratio of normal to mutant protein; (4) reduction of expression of both mutant and WT proteins76–78; (5) nutritional therapy based on vitamin A and other additives such as docosahexanoic acid79,80; (6) stabilization of mutant protein by native and artificial chromophores or other pharmacological agents20,31,72,81,82; (7) treatment with 17-allylamino-17-demethoxygeldanamycin (17-AAG), an ansamycin antibiotic that binds to the heat shock protein Hsp90, activating a heat shock response83,84; (8) increasing neurotrophic factors such as rod-derived viability factor85–87 or many others78; (9) increasing or decreasing levels of specific micro-RNAs that exert neuroprotective or neurotoxic effects88–90; (10) transplantation of photoreceptors and the RPE91; (11) restoring metabolic regulation and managing oxidative stress92; (12) manipulation with light exposure19,93; (13) reprogramming rod into cone cells94; and (14), expressing proinsulin as a neuroprotective molecule.95 These examples are by no means an exhaustive list of proposed approaches, and all possibilities must be considered and compared systematically to identify the best option for treating RP.
Our results hold promise to advance the discovery of optimal pharmacological approaches that slow or prevent retinal degeneration caused by the P23H opsin mutations in the opsin gene. Novel compounds from HTS, as well as previously reported compounds could be tested in the confirmation assays and image-based assays described in this study, such that their pharmacological activities (efficacy, potency, and cytotoxicity) could be compared in a consistent experimental format. Combinatorial treatment with different compounds targeting separate cellular pathways could be tested and optimized for safer and more effective outcomes. P23H knock-in mice11,21 in combination with HTS and high-resolution imaging should dramatically advance our understanding of RP pathophysiology and yield active compounds for future development.
Supplementary Material
Acknowledgments
The authors thank Leslie T. Webster Jr (Case Western Reserve University, CWRU) for his comments on the manuscript, Grzegorz Bereta for generating the NIH3T3 (P23H-opsin/GFP) and NIH3T3 (WT-opsin/GFP) stable cells, and Paul J. Tesar for his guidance in using the Operetta Imaging System (CWRU).
Supported by grants from the Case Western Reserve University School of Medicine (Cleveland, OH, USA) as well as Grants EY021126 and EY024992 from the National Institutes of Health (Bethesda, MD, USA). KP is the John H. Hord Professor of Pharmacology.
Disclosure: Y. Chen, None; H. Tang, None; W. Seibel, None; R. Papoian, None; X. Li, None; N.A. Lambert, None; K. Palczewski, None
Correspondence: Krzysztof Palczewski, Department of Pharmacology, Case Western Reserve University, 2109 Adelbert Road, Cleveland, OH 44106, USA; kxp65@case.edu.
References
- 1. Reddy AS,, Zhang S. Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol. 2013; 6: 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carrieri A,, Perez-Nueno VI,, Lentini G,, Ritchie DW. Recent trends and future prospects in computational GPCR drug discovery: from virtual screening to polypharmacology. Curr Top Med Chem. 2013; 13: 1069–1097. [DOI] [PubMed] [Google Scholar]
- 3. Zhao S,, Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol. 2012; 52: 505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kenakin TP. Cellular assays as portals to seven-transmembrane receptor-based drug discovery. Nat Rev Drug Discov. 2009; 8: 617–626. [DOI] [PubMed] [Google Scholar]
- 5. Vanni S,, Rothlisberger U. A closer look into G protein coupled receptor activation: x-ray crystallography and long-scale molecular dynamics simulations. Curr Med Chem. 2012; 19: 1135–1145. [DOI] [PubMed] [Google Scholar]
- 6. Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006; 75: 743–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kiser PD,, Golczak M,, Palczewski K. Chemistry of the retinoid (visual) cycle. Chem Rev. 2014; 114: 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ballesteros J,, Palczewski K. G protein-coupled receptor drug discovery: implications from the crystal structure of rhodopsin. Curr Opin Drug Discov Devel. 2001; 4: 561–574. [PMC free article] [PubMed] [Google Scholar]
- 9. Filipek S,, Stenkamp RE,, Teller DC,, Palczewski K. G protein-coupled receptor rhodopsin: a prospectus. Annu Rev Physiol. 2003; 65: 851–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palczewski K. Retinoids for treatment of retinal diseases. Trends Pharmacol Sci. 2010; 31: 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakami S,, Kolesnikov AV,, Kefalov VJ,, Palczewski K. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum Mol Genet. 2014; 23: 1723–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiang WC,, Kroeger H,, Sakami S,, et al. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration [published online ahead of print October 2, 2014]. Mol Neurobiol. doi:10.1007/s12035-014-8881-8. [DOI] [PMC free article] [PubMed]
- 13. Chen Y,, Jastrzebska B,, Cao P,, et al. Inherent instability of the retinitis pigmentosa P23H mutant opsin. J Biol Chem. 2014; 289: 9288–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguila M,, Bevilacqua D,, McCulley C,, et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum Mol Genet. 2014; 23: 2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee DC,, Vazquez-Chona FR,, Ferrell WD,, et al. Dysmorphic photoreceptors in a P23H mutant rhodopsin model of retinitis pigmentosa are metabolically active and capable of regenerating to reverse retinal degeneration. J Neurosci. 2012; 32: 2121–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorbatyuk MS,, Gorbatyuk OS,, LaVail MM,, Lin JH,, Hauswirth WW,, Lewin AS. Functional rescue of P23H rhodopsin photoreceptors by gene delivery. Adv Exp Med Biol. 2012; 723: 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griciuc A,, Aron L,, Roux MJ,, Klein R,, Giangrande A,, Ueffing M. Inactivation of VCP/ter94 suppresses retinal pathology caused by misfolded rhodopsin in Drosophila. PLoS Genet. 2010; 6. [DOI] [PMC free article] [PubMed]
- 18. Gorbatyuk MS,, Knox T,, LaVail MM,, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010; 107: 5961–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tam BM,, Moritz OL. Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: a chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J Neurosci. 2007; 27: 9043–9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noorwez SM,, Malhotra R,, McDowell JH,, Smith KA,, Krebs MP,, Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004; 279: 16278–16284. [DOI] [PubMed] [Google Scholar]
- 21. Sakami S,, Maeda T,, Bereta G,, et al. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011; 286: 10551–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang Y,, Fotiadis D,, Maeda T,, et al. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem. 2004; 279: 48189–48196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baell J,, Walters MA. Chemistry: chemical con artists foil drug discovery. Nature. 2014; 513: 481–483. [DOI] [PubMed] [Google Scholar]
- 24. Soares JG,, Fiorani M,, Araujo EA,, et al. Cone photopigment variations in Cebus apella monkeys evidenced by electroretinogram measurements and genetic analysis. Vision Res. 2010; 50: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lan TH,, Liu Q,, Li C,, Wu G,, Lambert NA. Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic. 2012; 13: 1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamdan FF,, Audet M,, Garneau P,, Pelletier J,, Bouvier M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen. 2005; 10: 463–475. [DOI] [PubMed] [Google Scholar]
- 27. Palczewski K,, Kumasaka T,, Hori T,, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000; 289: 739–745. [DOI] [PubMed] [Google Scholar]
- 28. Zhang JH,, Chung TD,, Oldenburg KRA. Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999; 4: 67–73. [DOI] [PubMed] [Google Scholar]
- 29. Alegria-Schaffer A. Western blotting using chemiluminescent substrates. Methods Enzymol. 2014; 541: 251–259. [DOI] [PubMed] [Google Scholar]
- 30. Saliba RS,, Munro PM,, Luthert PJ,, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002; 115: 2907–2918. [DOI] [PubMed] [Google Scholar]
- 31. Ohgane K,, Dodo K,, Hashimoto Y. Retinobenzaldehydes as proper-trafficking inducers of folding-defective P23H rhodopsin mutant responsible for retinitis pigmentosa. Bioorg Med Chem. 2010; 18: 7022–7028. [DOI] [PubMed] [Google Scholar]
- 32. Auld DS,, Southall NT,, Jadhav A,, et al. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008; 51: 2372–2386. [DOI] [PubMed] [Google Scholar]
- 33. Hammer MM,, Wehrman TS,, Blau HM. A novel enzyme complementation-based assay for monitoring G-protein-coupled receptor internalization. FASEB J. 2007; 21: 3827–3834. [DOI] [PubMed] [Google Scholar]
- 34. Chen Y,, Tang H,, Seibel W,, et al. Identification and characterization of novel inhibitors of mammalian aspartyl aminopeptidase. Mol Pharmacol. 2014; 86: 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J,, Xie X. Development of a quantitative cell-based, high-content screening assay for epidermal growth factor receptor modulators. Acta Pharmacol Sin. 2007; 28: 1698–1704. [DOI] [PubMed] [Google Scholar]
- 36. Singh S,, Carpenter AE,, Genovesio A. Increasing the content of high-content screening: an overview. J Biomol Screen. 2014; 19: 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen L,, Jin L,, Zhou N. An update of novel screening methods for GPCR in drug discovery. Expert Opin Drug Discov. 2012; 7: 791–806. [DOI] [PubMed] [Google Scholar]
- 38. Agler M,, Prack M,, Zhu Y,, et al. A high-content glucocorticoid receptor translocation assay for compound mechanism-of-action evaluation. J Biomol Screen. 2007; 12: 1029–1041. [DOI] [PubMed] [Google Scholar]
- 39. Garippa RJ,, Hoffman AF,, Gradl G,, Kirsch A. High-throughput confocal microscopy for beta-arrestin-green fluorescent protein translocation G protein-coupled receptor assays using the Evotec Opera. Methods Enzymol. 2006; 414: 99–120. [DOI] [PubMed] [Google Scholar]
- 40. Sung CH,, Schneider BG,, Agarwal N,, Papermaster DS,, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eglen RM,, Reisine T. Photoproteins: important new tools in drug discovery. Assay Drug Dev Technol. 2008; 6: 659–671. [DOI] [PubMed] [Google Scholar]
- 42. Eglen RM,, Gilchrist A,, Reisine T. The use of immortalized cell lines in GPCR screening: the good bad and ugly. Comb Chem High Throughput Screen. 2008; 11: 560–565. [DOI] [PubMed] [Google Scholar]
- 43. Baell JB,, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010; 53: 2719–2740. [DOI] [PubMed] [Google Scholar]
- 44. Hoelder S,, Clarke PA,, Workman P. Discovery of small molecule cancer drugs: successes challenges and opportunities. Mol Oncol. 2012; 6: 155–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hartong DT,, Berson EL,, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 46. Sahel J,, Bonnel S,, Mrejen S,, Paques M. Retinitis pigmentosa and other dystrophies. Dev Ophthalmol. 2010; 47: 160–167. [DOI] [PubMed] [Google Scholar]
- 47. Zhang N,, Kolesnikov AV,, Jastrzebska B,, et al. Autosomal recessive retinitis pigmentosa E150K opsin mice exhibit photoreceptor disorganization. J Clin Invest. 2013; 123: 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sung CH,, Davenport CM,, Hennessey JC,, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 6481–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jacobson SG,, Kemp CM,, Sung CH,, Nathans J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol. 1991; 112: 256–271. [DOI] [PubMed] [Google Scholar]
- 50. Mendes HF,, van der Spuy J,, Chapple JP,, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005; 11: 177–185. [DOI] [PubMed] [Google Scholar]
- 51. Nakao T,, Tsujikawa M,, Notomi S,, Ikeda Y,, Nishida K. The role of mislocalized phototransduction in photoreceptor cell death of retinitis pigmentosa. PLoS One. 2012; 7: e32472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993; 34: 1659–1676. [PubMed] [Google Scholar]
- 53. Berson EL. Retinitis pigmentosa: unfolding its mystery. Proc Natl Acad Sci U S A. 1996; 93: 4526–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berson EL,, Rosner B,, Sandberg MA,, Dryja TP. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His). Arch Ophthalmol. 1991; 109: 92–101. [DOI] [PubMed] [Google Scholar]
- 55. Palczewski K. Chemistry and biology of vision. J Biol Chem. 2012; 287: 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kevany BM,, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda). 2010; 25: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Humphries MM,, Rancourt D,, Farrar GJ,, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997; 15: 216–219. [DOI] [PubMed] [Google Scholar]
- 58. Lem J,, Krasnoperova NV,, Calvert PD,, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999; 96: 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rosenfeld PJ,, Cowley GS,, McGee TL,, Sandberg MA,, Berson EL,, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992; 1: 209–213. [DOI] [PubMed] [Google Scholar]
- 60. Oprian DD. Molecular determinants of spectral properties and signal transduction in the visual pigments. Curr Opin Neurobiol. 1992; 2: 428–432. [DOI] [PubMed] [Google Scholar]
- 61. Kaushal S,, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994; 33: 6121–6128. [DOI] [PubMed] [Google Scholar]
- 62. Olsson JE,, Gordon JW,, Pawlyk BS,, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992; 9: 815–830. [DOI] [PubMed] [Google Scholar]
- 63. Naash MI,, Hollyfield JG,, al-Ubaidi MR,, Baehr W. Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci U S A. 1993; 90: 5499–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roof DJ,, Adamian M,, Hayes A. Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest Ophthalmol Vis Sci. 1994; 35: 4049–4062. [PubMed] [Google Scholar]
- 65. Lewin AS,, Drenser KA,, Hauswirth WW,, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998; 4: 967–971. [DOI] [PubMed] [Google Scholar]
- 66. LaVail MM,, Yasumura D,, Matthes MT,, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A. 2000; 97: 11488–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Petters RM,, Alexander CA,, Wells KD,, et al. Genetically engineered large animal model for studying cone photoreceptor survival and degeneration in retinitis pigmentosa. Nat Biotechnol. 1997; 15: 965–970. [DOI] [PubMed] [Google Scholar]
- 68. Kosmaoglou M,, Schwarz N,, Bett JS,, Cheetham ME. Molecular chaperones and photoreceptor function. Prog Retin Eye Res. 2008; 27: 434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haeri M,, Knox BE. Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One. 2012; 7: e30101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tam BM,, Moritz OL. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006; 47: 3234–3241. [DOI] [PubMed] [Google Scholar]
- 71. Zhang R,, Oglesby E,, Marsh-Armstrong N. Xenopus laevis P23H rhodopsin transgene causes rod photoreceptor degeneration that is more severe in the ventral retina and is modulated by light. Exp Eye Res. 2008; 86: 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Noorwez SM,, Kuksa V,, Imanishi Y,, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003; 278: 14442–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Scherer LJ,, Rossi JJ. Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol. 2003; 21: 1457–1465. [DOI] [PubMed] [Google Scholar]
- 74. Drenser KA,, Timmers AM,, Hauswirth WW,, Lewin AS. Ribozyme-targeted destruction of RNA associated with autosomal-dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1998; 39: 681–689. [PubMed] [Google Scholar]
- 75. Mao H,, Gorbatyuk MS,, Rossmiller B,, Hauswirth WW,, Lewin AS. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum Gene Ther. 2012; 23: 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mao H,, James T Jr,, Schwein A,, et al. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther. 2011; 22: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Frederick JM,, Krasnoperova NV,, Hoffmann K,, et al. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci. 2001; 42: 826–833. [PubMed] [Google Scholar]
- 78. Rossmiller B,, Mao H,, Lewin AS. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol Vis. 2012; 18: 2479–2496. [PMC free article] [PubMed] [Google Scholar]
- 79. Berson EL,, Rosner B,, Sandberg MA,, et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol. 2004; 122: 1306–1314. [DOI] [PubMed] [Google Scholar]
- 80. Berson EL,, Rosner B,, Sandberg MA,, et al. Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol. 2004; 122: 1297–1305. [DOI] [PubMed] [Google Scholar]
- 81. Noorwez SM,, Ostrov DA,, McDowell JH,, Krebs MP,, Kaushal S. A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Invest Ophthalmol Vis Sci. 2008; 49: 3224–3230. [DOI] [PubMed] [Google Scholar]
- 82. Krebs MP,, Holden DC,, Joshi P,, Clark CL III,, Lee AH,, Kaushal S. Molecular mechanisms of rhodopsin retinitis pigmentosa and the efficacy of pharmacological rescue. J Mol Biol. 2010; 395: 1063–1078. [DOI] [PubMed] [Google Scholar]
- 83. Tam LC,, Kiang AS,, Campbell M,, et al. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90). Hum Mol Genet. 2010; 19: 4421–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mendes HF,, Cheetham ME. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum Mol Genet. 2008; 17: 3043–3054. [DOI] [PubMed] [Google Scholar]
- 85. Chalmel F,, Leveillard T,, Jaillard C,, et al. Rod-derived Cone Viability Factor-2 is a novel bifunctional-thioredoxin-like protein with therapeutic potential. BMC Mol Biol. 2007; 8: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Leveillard T,, Mohand-Said S,, Lorentz O,, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004; 36: 755–759. [DOI] [PubMed] [Google Scholar]
- 87. Yang Y,, Mohand-Said S,, Danan A,, et al. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009; 17: 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhu Q,, Sun W,, Okano K,, et al. Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J Biol Chem. 2011; 286: 31749–31760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Georgiadis A,, Tschernutter M,, Bainbridge JW,, et al. AAV-mediated knockdown of peripherin-2 in vivo using miRNA-based hairpins. Gene Ther. 2010; 17: 486–493. [DOI] [PubMed] [Google Scholar]
- 90. Zhang L,, Wang T,, Wright AF,, et al. A microdeletion in Xp11.3 accounts for co-segregation of retinitis pigmentosa and mental retardation in a large kindred. Am J Med Genet A. 2006; 140: 349–357. [DOI] [PubMed] [Google Scholar]
- 91. Yang Y,, Mohand-Said S,, Leveillard T,, Fontaine V,, Simonutti M,, Sahel JA. Transplantation of photoreceptor and total neural retina preserves cone function in P23H rhodopsin transgenic rat. PLoS One. 2010; 5: e13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Punzo C,, Xiong W,, Cepko CL. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J Biol Chem. 2012; 287: 1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tam BM,, Qazalbash A,, Lee HC,, Moritz OL. The dependence of retinal degeneration caused by the rhodopsin P23H mutation on light exposure and vitamin a deprivation. Invest Ophthalmol Vis Sci. 2010; 51: 1327–1334. [DOI] [PubMed] [Google Scholar]
- 94. Montana CL,, Kolesnikov AV,, Shen SQ,, Myers CA,, Kefalov VJ,, Corbo JC. Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc Natl Acad Sci U S A. 2013; 110: 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fernandez-Sanchez L,, Lax P,, Isiegas C,, et al. Proinsulin slows retinal degeneration and vision loss in the P23H rat model of retinitis pigmentosa. Hum Gene Ther. 2012; 23: 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.