

Abstract

High-temperature synthesized monodisperse superparamagnetic iron oxide nanoparticles are obtained with a strongly bound ligand shell of oleic acid and its decomposition products. Most applications require a stable presentation of a defined surface chemistry; therefore, the native shell has to be completely exchanged for dispersants with irreversible affinity to the nanoparticle surface. We evaluate by attenuated total reflectance−Fourier transform infrared spectroscopy (ATR−FTIR) and thermogravimetric analysis/differential scanning calorimetry (TGA/DSC) the limitations of commonly used approaches. A mechanism and multiple exchange scheme that attains the goal of complete and irreversible ligand replacement on monodisperse nanoparticles of various sizes is presented. The obtained hydrophobic nanoparticles are ideally suited for magnetically controlled drug delivery and membrane applications and for the investigation of fundamental interfacial properties of ultrasmall core–shell architectures.
Introduction
High-temperature synthesized superparamagnetic iron oxide nanoparticles (SPIONs) are well-defined biomedical materials with homogeneous physical properties in terms of diameter, polydispersity, surface area, and magnetic characteristics. SPIONs have an increasing number of applications, not the least in the biomedical field;1−3 they are used as magnetic resonance imaging (MRI) contrast agents in imaging diagnosis4 and in magnetic-field-assisted cancer therapy (hyperthermia)5 and have emerging use in magnetically addressable drug delivery systems6−8 as a result of their inherent low toxicity2,3 and suitable size.3 Clinical approval requires reproducible manufacture of highly uniform physicochemical characteristics, which explains the increasing demand for controlled synthesis and improved surface modification approaches.3 While remarkable progress has been achieved in the synthesis of monodisperse SPIONs,3,4,9−11 their application requires equally well-defined and stable ligand shells with tailored surface chemistries.3,9 Ultrapure interfaces of that kind allow for controlled assembly in amphiphilic superstructures,7,12 investigation of nanoparticle–membrane interactions,13,14 and internanoparticle interactions in membranes, and they aid in improving the underlying design principles for future generations of drug delivery systems.6,7,15 A prerequisite for these studies is exclusion of impurities, such as weakly bound dispersants or residual capping agents, that might obscure nanoparticle-induced effects. Grafting of such shells to monodisperse SPIONs has thus far received scant attention and remains a bottleneck. The challenge is that a strongly but reversibly bound ligand shell of, e.g., oleic acid (OA) and its decomposition products remains on the surface after high-temperature synthesis of monocrystalline SPIONs.
Attention has been given recently to ligand replacement of OA for hydrophilic dispersants providing steric stabilization to SPIONs, which has demonstrated difficulties in terms of lower achieved grafting density than required for strong colloidal stability.16 The likely cause is incomplete replacement of OA, as shown by isotope labeling,17 and the contradicting requirements for exchanging residual OA and densely grafting bulky polar dispersants in its place.
Ligand replacement of hydrophobic dispersants has received even less attention. Although the as-synthesized OA shell on monodisperse SPION is hydrophobic and strongly bound, it has been shown that these ligands dissolve in, e.g., membranes and cause undefined interactions and unstable aggregates.8 We elucidate the extent of OA on SPIONs after various purification steps and present a protocol that enables complete ligand replacement and stable hydrophobic shells to be grafted.
Experimental Section
Materials
All reagents were purchased from Sigma-Aldrich and used as received (see the Supporting Information for details). To ensure maximal reproducibility, all nanoparticles and ligands originated from the same batches.
Synthesis of Superparamagnetic Iron Oxide Nanoparticles
Monodisperse, spherical SPIONs were synthesized via high-temperature thermal decomposition of an organometallic precursor in a high-boiling surfactant solution following a route adapted from Park et al.10 Size control was achieved by altering the surfactant to organometallic precursor ratio (see SI 2.1 of the Supporting Information).11
Synthesis of 4-Nitrocatechol-Derived Ligands
6-nitrodopamine-hemisulfate (NDA-HSO4) was synthesized according to the literature, with slight modifications (see SI 1.1 of the Supporting Information).18N-Palmityl-6-nitrodopamide (P-NDA) and its alkyl-perdeuterated analogue N-(d31-palmityl)-6-nitrodopamide (d31P-NDA) were prepared by COMU-mediated peptide-coupling reactions (see SI 1.2 of the Supporting Information).19
Surface Modification of Magnetite Nanoparticles
Typically 50 mg of P-NDA (or NDA) was dissolved in 3 mL of dimethylformamide (DMF) under sonication and added to 200 mg of as-synthesized OA–SPIONs dissolved in 6 mL of chloroform (CHCl3). The SPION–ligand mixture was combined with 9 mL of methanol (MeOH) and sonicated for 3 h under N2. CHCl3 was evaporated from the coating mix, and the resulting SPIONs were collected by magnetic precipitation from excess MeOH (40 mL) and purified by 3 rounds of washing and magnetic separation from cold MeOH (20 mL each) to remove excess nitrocatechol ligands.
Purification Methods of Coated Magnetite Nanoparticles
A chart of all investigated purification pathways and their results can be found in SI 3.1 of the Supporting Information.
(a) As-Synthesized SPIONs
The crude SPION reaction mixture was purified from reaction solvent and byproducts by five precipitations from minimal toluene (tol) into excess ethanol (EtOH) (300 mL each) and collected by magnetic separation.
(b) OA (1 mM) in Hot MeOH Pre-extraction
A total of 200 mg of as-synthesized OA–SPIONs (a) was washed 4 times each with 20 mL of hot (heated with heat gun) 1 mM OA in MeOH and once with pure hot MeOH.
All purification methods on newly capped SPIONs have been performed after cold MeOH washings of the coating mixtures. SPIONs were routinely collected by magnetic separation, unless otherwise stated.
(c) Hot MeOH Extraction
A total of 200 mg of OA–SPIONs (a or b) were subjected to the standard coating exchange and subsequently washed 3 times with 30 mL of hot MeOH each.
(d) Standard Cold MeOH Extraction
Newly capped SPIONs were purified according to the standard protocol by 3-fold extraction with 20 mL of cold MeOH each.
(e) Syringe Filtering
A total of 200 mg of washed, precipitated mixed-dispersant SPIONs were dissolved in 3 mL of CHCl3 and purified by a single pass through commercially available filter units (0.2 μm cutoff).
(f) Addition of Surfactants
Surfactants were added in 100 mg quantities to 200 mg of mixed dispersant SPIONs. The mixes were sonicated in hot ethanol (EtOH) for 5 min. SPIONs were collected by magnetic separation.
(g) Column Chromatography
A total of 5 g of SiO2 60 was loaded in a 2 × 30 cm column, and 200 mg of mixed dispersant SPIONs dissolved in the mobile phase were eluted with tetrahydrofuran (THF)/MeOH (4:1).
(h) Ligand-Saturated Column Chromatography
A total of 200 mg of P-NDA in DMF was run twice over 5 g of SiO2 60, and the stationary phase was purged with 2 column volumes of THF/MeOH (4:1) prior to loading of the coated SPIONs.
(i) Recrystallization from MeOH/n-Hexane
A total of 200 mg mixed dispersant SPIONs was refluxed in 20 mL of a N2-saturated MeOH/n-hexane (1:1) solvent mixture under an inert atmosphere. Recrystallization was repeated 3 times.
(j) Base (2,6-Lutidine) as Solvent
A total of 200 mg of as-synthesized SPIONs and P-NDA was dissolved in 5 mL of 2,6-lutidine and sonicated under N2 for 3 h. Lutidine was evaporated under reduced pressure, and SPIONs were purified with three cold MeOH extractions (20 mL each).
(k) P-NDA Post-coating
A total of 200 mg of mixed dispersant SPIONs freed from physisorbed OA [prepared via pre-extraction (a) or cetyltrimethylammoniumbromid (CTAB) addition (f)] was subjected to an additional round of coating with 100 mg of P-NDA in a minimal amount of N2-saturated 2,6-lutidine at 50 °C for 48 h under an inert atmosphere and magnetic stirring. Lutidine was completely evaporated under reduced pressure, and the post-coating mix was washed 3 times with hot MeOH (30 mL each).
(l) d31P-NDA Post-coating
Pre-extracted OA-SPIONs (b) were ligand-exchanged with d31P-NDA and purified via hot MeOH extractions as in step c. Post-coating with d31P-NDA was analogous to step k.
(m) NDA Post-coating
Mixed dispersant NDA-HSO4-coated SPIONs obtained via the standard coating protocol were isolated by evaporating the coating mix to the DMF fraction, adding 10 volumes of n-hexane and collecting the SPIONs via magnetic separation. The particles were post-coated by adding 100 mg of NDA-HSO4 and decocting the dispersion under an inert atmosphere 3 times with each hot n-hexane, hot Me2CO, and hot MeOH (30 mL per wash). NDA–SPIONs were collected by magnetic separation and finally lyophilized from ultrapure water.
Characterization
Transmission Electron Microscopy (TEM)
TEM studies were performed on a FEI Tecnai G2 20 transmission electron microscope operating at 120 kV or at 200 kV for high-resolution imaging. Samples were prepared by drop-casting toluene dispersions onto 300-mesh carbon-coated copper grids and evaporating the solvent in air. Size distributions were evaluated using PEBBLES.20
Thermogravimetric Analysis/Differential Scanning Calorimetry (TGA/DSC) Measurements
Thermograms were recorded on a Mettler-Toledo TGA/DSC 1 STAR system in the temperature range of 25–650 °C with a ramp of 10 K/min under 80 mL/min synthetic air gas flow. Typically, 0.5–2 mg of sample was used per run, and the rest of the mass was evaluated at 500 °C by horizontal setting.
1H and 13C Nuclear Magnetic Resonance (NMR) Measurements
1H and 13C solution spectra were collected on a Bruker DPX operating at 300 MHz using tetramethylsilane (TMS) as an internal standard.
Electrospray Ionization–Mass Spectrometry (ESI–MS) Measurements
Mass spectra were collected using a Q-Tof Ultima ESI (Waters, Milford, MA) mass spectrometer in positive-ion mode. Samples were diluted with NH4+HCOO– buffer (65 mM, pH 3) or MeOH to a final concentration of 5–500 μg/mL.
Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy (ATR–FTIR) Measurements
Mid-infrared (IR) powder spectra of the lyophilized samples were collected using a Bruker Tensor 37 FTIR spectrometer with Bruker Platinum Diamond single-reflection ATR equipment at a resolution of 4 cm–1 by averaging 32 scans.
Ultraviolet–Visible (UV–Vis) Measurements
UV–vis absorption spectra were collected at a scan speed of 400 nm/min on a Hitachi UV-2900 spectrophotometer.
Results and Discussion
Figure 1 shows TEM micrographs of the as-synthesized, OA-capped magnetite nanoparticles with narrow size distributions of 3.5 ± 0.4, 5.0 ± 0.4, and 8.3 ± 0.4 nm.
Figure 1.

TEM of spherical, monodisperse magnetite nanoparticles with narrow size distributions of (a) 3.5 ± 0.4 nm, (b) 5.0 ± 0.4 nm, and (c) 8.3 ± 0.4 nm and (d) high-resolution (HR)-TEM of the 8.3 ± 0.4 nm SPIONs in panel c.
No difference was observed between sonicating and stirring OA-capped SPIONs in P-NDA ligand solution at an elevated temperature to perform the exchange step. Sonication was chosen because of the shorter time of preparation.
A large variety of purification and post-coating methods were tested for their potential to purify P-NDA-coated SPIONs from residual OA. Their efficiencies were evaluated by ATR–FTIR and TGA/DSC.
Figure 2 shows the IR spectra of 3.5 nm SPIONs after purification with various methods enumerated as c−m. The purification steps c−m are briefly described with motivations for why they were chosen in the following. Repeated pre-extraction with hot MeOH containing 1 mM OA as a stabilizer (b) to prevent aggregation,21 (c) hot MeOH extractions, (d) cold MeOH extractions, and (e) syringe filtering [polytetrafluoroethylene (PTFE)] were used as common, quick, and low-cost methods to remove excess unbound organics and aggregates.1 In step b, oleate monolayer-covered SPIONs are subjected to a single exchange step, while newly capped SPIONs derived from as-synthesized particles were purified in step c. The addition of surfactants (f), such as cationic cetyltrimethylammoniumbromid (CTAB), was tested because OA forms complexes with countercharged surfactants that might impede re-adsorption to the particle surface and prevent aggregation during the exchange of the coating.21 The influence of the charge of the surfactant on the removal of OA was studied by additionally using anionic sodium dodecyl sulfate (SDS) and neutral 1-octanol (OctOH) (see SI 3.4 of the Supporting Information). Column chromatography over SiO2 with (h) and without (g) P-NDA adsorbed to the stationary phase in THF/MeOH (4:1) was tested as a ubiquitous purification process for surface-active species that could capture OA but leave P-NDA as a result of its lower affinity to SiO2.22 Recrystallization (i) from MeOH/n-hexane (1:1) relies on dissolving the less tightly bound capping agent from the particle surface. The base 2,6-lutidine was similarly tested as solvent (j), which was inspired by the fact that weak hydrogen-bond acceptors significantly speed up the rate of nitrocatechol surface complexation.23 Lutidine was chosen because it is a sterically hindered, moderate, medium-polar base serving multiple purposes as it acts as a solvent for OA–SPIONs, sufficiently deprotonates P-NDA, and at the same time, offers the possibility to form ion pairs with OA without complexing the SPION surface strongly. Finally, post-coating strategies (k–m) were devised that emphasize the role of competitive binding to the particle surface; i.e., a high-affinity ligand (in our case, a strongly adsorbed OA-related species) can be displaced by a large excess of other ligands if it has a non-negligible desorption rate (koff). Post-coating with P-NDA (k) and d31P-NDA (l) involved exchange of mixed dispersant particles at 50 °C in 2,6-lutidine containing excess P-NDA for 48 h under an inert atmosphere, while NDA post-coated particles (m) were prepared by selectively desorbing oleate in the presence of excess nitrocatechol ligand, which could adsorb at emerging surface sites.
Figure 2.

ATR–FTIR spectra of various 3.5 nm magnetite nanoparticle preparations: (a) as-synthesized SPION containing excess physisorbed OA, (b) purified SPION with a monolayer of chemisorbed oleate, (c–j) mixed dispersant OA/P-NDA–SPION and (l–m) post-coated, pure nitrocatechol-ligand-capped SPIONs. SPIONs were purified by the following methods: (b) pre-extraction in hot MeOH containing 1 mM OA as a stabilizer, (c) hot MeOH extraction, (d) cold MeOH extraction, (e) syringe filtration (PTFE), (f) surfactant addition (CTAB), (g) silica column chromatography (THF/MeOH = 4:1), (h) ligand saturated chromatography, (i) repeated MeOH/n-hexane (1:1) recrystallization, (j) 2,6-lutidine as a solvent, (k) post-coated P-NDA–SPION, (l) post-coated d31P-NDA–SPION, (m) post-coated NDA–SPION, and (n) SPION with combusted shell (post-TGA) with 10× scaled inset of residual absorptions. Peaks corresponding to physisorbed and chemisorbed OA are indicated by circles and asterisks, respectively, while crosses depict bands related to nitrocatechol ligands. The post-coated P-NDA (k), d31P-NDA (l), and NDA particles (m) are the only particles that demonstrate an absence of the characteristic OA bands and, therefore, complete ligand replacement.
Association of OA with the primary surfactant layer on the particle surface to quasi-bilayer structures was reported to occur though intercalation of alkyl chains in combination with hydrogen bonding between the headgroups.15 The presence of physisorbed OA was, therefore, determined from absorptions at 1705 cm–1 in FTIR spectra assigned to the C=O stretch of hydrogen-bonded (dimeric) alkyl carboxylic acids and from other bands characteristic of free acid groups, such as the OH in-plane and out-of-plane bending vibrations at 1430 and 965 cm–1 (see SI 2.2 of the Supporting Information for further assignments).
Oleate complexed to the particle surface was evaluated at 1605, 1520, and 1410 cm–1, corresponding to two asymmetric and one symmetric stretching vibrations of the carboxylate headgroup.24 The respective positions and the split between νas,s(COO–) are indicative of the binding mode of the carboxylate headgroup to Fe ions.24,25 For perdeuterated samples, oleate exchange was concomitantly monitored by comparing νas,s(CH2) integral ratios of the newly capped SPIONs to the respective oleate monolayer-covered particles. Fe–O lattice vibrations of magnetite are found at 580 and 385 cm–1 corresponding to T1u optical phonon modes of the crystal lattice.26,27
For half of the tested purification methods, there is a substantial amount of unbound or physisorbed OA left (entry 1 in Table 1). Even extracting the coating mix 3 times with cold MeOH was insufficient to remove physisorbed OA (Figure 2d). This is in good agreement with the fact that dissociation of OA dimers is vital for complete removal of physisorbed OA and only occurs above 70 °C (curves b and c in Figure 2).24
Table 1. TGA Results for Various SPION Preparations.
| dcore (nm) | preparation method | TOCa (%, w/w) | ρgrafta (nm–2) | ρchain endsa (nm–2) |
|---|---|---|---|---|
| 3.5 | a, d, e, g, h, and j | 35–78 | nd | nd |
| 3.5 | b | 28–29 | 2.2–2.3 | 0.35–0.37 |
| c, f, and i | 28–30 | nd | nd | |
| 3.5 | 38–39 | 2.6–2.7 | 0.35–0.4 | |
| 5 | k | 32–35 | 2.8–3.2 | 0.6–0.7 |
| 8.3 | 22–23 | 2.8–3.1 | 1.0–1.1 | |
| 3.5 | 26–28 | 2.2–2.9b | 0.9–1.2b | |
| 5 | mb | 24–26 | 2.8–3.5b | 1.4–1.8b |
| 8.3 | 14–15 | 2.4–2.9b | 1.6–1.9b |
Average of three measurements evaluated in the temperature interval (100–200 °C to 500 °C).
Expanded range of values arises from ambiguous composition of the shell (see SI 4.2 of the Supporting Information).
Intercalated OA is also present in samples run over the SiO2 column, despite the fact that the samples passed as single fractions (curves g–h in Figure 2). Filtering either did not succeed in removing associated OA at all [regenerated cellulose (RC/polyethylene terephthalate (PET)/polyvinylidene difluoride (PVDF) filter material] or introduced additional bands, which originate from dissolvable material of the filter (PTFE/nylon) (Figure 2e and SI 3.3 of the Supporting Information).
TGA combined with DSC was used to access the total organic content (TOC) associated with the purified SPIONs (Table 1 and SI 3.2 of the Supporting Information). The organic fractions of the samples were evaluated from the rest of the mass of the TGA curve according to Benoit et al.28 The determination of the exact mass ratio of organic ligand to inorganic core is challenging to obtain for fatty acids, because they do not fully combust under an inert atmosphere. Instead, carbonaceous graphitic residues are formed, which cause significant reduction of the inorganic core to wüstite and ferrite above 760 °C.29 Burning in air was reported to only result in moderate residues (mainly COx surface complexes) and oxidation of the iron oxide core to hematite at around 520 °C.25 Thus, burning in synthetic air up to 500 °C was used to determine grafting densities of as-synthesized and purified core–shell SPIONs. Samples with an incomplete purification from excess OA yielded unreasonably high TOC values (entry 1 in Table 1); the grafting densities were thus not evaluated.
The purification methods in entry 2 in Table 1, e.g., through the addition of the surfactant, yielded particles that are free of physisorbed OA but still display peaks attributed to surface-complexed oleate. The TOC is strongly reduced and corresponds to the amount that could be expected for a mixed OA/P-NDA monolayer coverage, although an exact grafting density cannot be calculated because the composition of the shell is not known as a result of the remaining complexed OA–. Interestingly, the improved removal of OA was almost independent of surfactant charge (CTAB, SDS, OctOH; see SI 3.4 of the Supporting Information). Surfactant bands were not detected after the washing steps. CTAB led to least losses during collection of the coated particles.
Residual complexed oleate might be strongly integrated into the shell and possess a low desorption rate. Finding a procedure for complete replacement of the ligand is necessary to ensure uniform SPION shell properties and a stronger binding through nitrocatechol anchors. In a final approach, removal of oleate was, therefore, tested by adding a post-coating step in excess ligand at an elevated temperature. Particles (b or e) were stirred at 50 °C in 2,6-lutidine containing excess P-NDA for 48 h under an inert atmosphere.
FTIR spectra recorded after the post-coating step show markedly decreased absorptions at 1605, 1520, and 1410 cm–1, concomitant with a narrowing and blue shift of the 1645 cm–1 band and a slight decrease in the overall νas,s(CH2) and δs(CH2) intensities (see SI 4.3 of the Supporting Information). This is attributed to removal of surface-bound oleate. Complete exchange was additionally verified by post-coating with isotope-labeled d31P-NDA. Perdeuteration of the alkyl chain leads to a decrease in vibrational frequency and, hence, results in spectral separation of new dispersant and native ligand signals. Noteworthy, the νas,s(CH2) integral area of the post-coated sample, representing persistent surface-bound oleate, dropped by 22 times compared to its initial value on OA monolayer-covered particles (see SI 4.3 of the Supporting Information). This corresponds to a 96% reduction in the TGA-derived number of initial oleate molecules (see SI 4.2 of the Supporting Information) and roughly 4 remaining oleate molecules per particle. Similarly, OA exchange for NDA-HSO4 exhibits only marginal CH2 intensities, which closely correspond to the signal of the pure ligand. Thus, efficient stripping and replacement of the original OA stabilizer is achieved in both post-coating procedures.
The loss of intensities at 1605, 1520, and 1410 cm–1 is ascribed to two individual νas(COO–) and a common νs(COO–) mode arising from two different complexation patterns of the carboxylate headgroup to the metal ions on the mixed dispersant particle (see SI 2.2 and SI 4.3 of the Supporting Information).24 A splitting of νas(COO–) into two separate parts largely accounts for our observations of unequal intensities between the asymmetric and symmetric carboxylate stretching vibrations and their concurrent disappearance upon post-coating. This is reasoned from the fact that νs(COO–) is less affected by the coordination pattern than its asymmetric counterpart; therefore, it mostly retains its spectral position and, hence, exhibits twice the intensity of each of the νas(COO–) modes. The 110 cm–1 (1520–1410 cm–1) splitting hints at a bidentate, mononuclear complexation, while the second splitting of 195 cm–1 (1605–1410 cm–1) could be due to contributions of either a unidentate or binuclear, bridged OA– complex.24 Unidentate coordination is rarely found and not supported by our data because we have no indication of an uncomplexed carbonyl around 1710 cm–1. Induced binuclear binding is an appealing explanation for the necessity of a second coating step, in which more strongly bound oleate is replaced on the surface and agrees better with our model of two different complexation patterns, where the νs(COO–) vibrations merge. A slight change in the overall CH2 intensities upon post-coating presumably originates from alterations in alkyl chain packing caused by different headgroup areas of the capping agents.
It is worth noting that the as-synthesized SPION spectra do not exhibit a pronounced mixture of two different chemisorbed oleate species (Δ = 110 cm–1 ∼ 90%) nor does washing with hot MeOH during the purification alter the observed 50:50 ratio on the mixed dispersant SPIONs. This highlights the disruption of oleate packing along with a rearrangement of the carboxylate binding induced by intercalation of nitrocatechol ligands. The presence of two oleate species with different complexation strengths has previously been noted as an explanation for differences in precursor reactivity of iron–oleate complexes in nanoparticle synthesis.24 We can further demonstrate that P-NDA only fully displaces both species of surface-bound oleate through competitive binding once excess OA has first been removed, which requires two coating steps (curves k–m in Figure. 2). Competitive binding could also explain why partial surface dissolution occurs when no alcohol is added during the coating procedure; this is presumably due to competition of two strongly complexing agents (P-NDA– and OA–) for surface sites, whereas protonation of the displaced species lowers their affinity.
TGA shows that the grafting density of the pure P-NDA shell is high (Table 1 and SI 4.2 of the Supporting Information). The grafting density was compared for different nitrocatechol-coated SPIONs with size varied from 3.5 to 8.3 nm (Table 1 and SI 4.2 of the Supporting Information) and found to be almost independent of surface curvature and hydrophobic substitution. Slight deviations regarding higher grafting densities for larger particles could originate from relative differences in iron oxide phase content,4,30 in line with preferential binding of nitrocatechols to Fe3O423 and/or improved crystallinity of the lattice of the larger nanoparticles. However, the observed trend is within the experimental uncertainty and curvature therefore seems to not affect the grafting density in the case of low-molecular-weight ligands. An approximately constant grafting density suggests that the limiting factor for adsorbing densely packed low-molecular-weight dispersants to the substrate is given by either the number of surface iron sites (3.5–5.9 nm–2 depending upon crystal plane and termination)31 or the spatial requirements of the nitrocatechol anchor. The latter is substantial as approximated by the molecular areas of palmitic acid (23 Å2) and N-stearoyldopamine (42 Å2),32 which suggest a maximum achievable density of 2.4 nm–2. This correlates with the theoretically expected maximum coverage for dopamine (2.4 nm–2) based on an estimate from the molar volume. At similar grafting density, the curvature strongly affects the density of the shell and, thereby, the specific interaction volume with the hydrophobic ligands; the latter increases with decreasing radius, as exemplified by the number of chain ends per outer surface area unit (ρchain ends in Table 1 and SI 4.2 of the Supporting Information). Thus, small-sized core–shell SPIONs interact more strongly with surrounding solutes, whereas larger particles feature compact core–shell architectures.
Multistep TGA profiles observed below 400 °C have previously been attributed alternatively to vaporization of physisorbed OA,15 to chemisorbed oleate species with different binding strengths,33 or to partial cleavage of the capping agent.29 In our experiments, a significant second step in TGA was only observed when physisorbed OA or other impurities were present (Figure 3 and SI 3.2 of the Supporting Information). Also, by comparison of the FTIR spectra of the tested particles to the TGA/DSC data, the presence of minor amounts of residual-free OA (around 5% w/w) could not be reliably detected with TGA/DSC (Table 1, Figures 2 and 3, and SI 3.2 of the Supporting Information) using a difference in the decomposition profile as the basis for detection.
Figure 3.
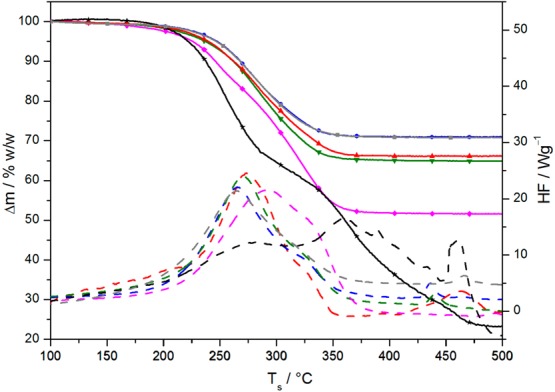
TGA (solid lines) and DSC curves (dashed lines) of representative 3.5 nm SPION preparations measured in synthetic air: as-synthesized OA–SPION containing excess physisorbed oleic acid (black, stars), purified OA–SPION with an oleate monolayer (gray, squares), column-chromatographed SPION with physisorbed impurities (magenta, diamonds), cold MeOH-extracted mixed dispersant SPION containing around 5% w/w physisorbed OA (green, triangles), CTAB-treated mixed dispersant SPION free of physisorbed OA (blue, circles), and spectroscopically clean P-NDA post-coated SPION (red, triangles).
The differently chemisorbed oleate species observed in FTIR on mixed ligand SPIONs seem to be too close in their dissociation energies to be resolved by TGA; co-adsorbed P-NDA and chemisorbed OA also give rise to similar curves (see Figure 3). A closer inspection of the DSC curves of the main mass loss shows two distinct heat peaks corresponding to one discernible step in TGA. The two peaks are, however, also observed when only P-NDA is present on the SPIONs, and the relative peak areas vary with the preparation method. The lower T peak is increasingly dominant toward more complete exchange of OA for P-NDA.
Only traces of organics were left after heat treatment of the mixed dispersant SPIONs to 500 °C, but clearly shifted nanoparticle absorptions became evident at 520 and 430 cm–1 (Figure 2n). This is also seen by eye as transformation to a purple inorganic residue. FTIR spectra of these SPIONs showed that all bands attributed to the nitro group have disappeared. Minimal residues of decomposed ligand were detected through bands at 2984 cm–1 [ν(Csp2–H)], 2896 cm–1 [ν(Csp3–H)], 2651 and 2350–2200 cm–1 (surface COx), 1650–1400 cm–1 [ν(C=C) and δ(Csp3–H)], 1059 cm–1 [ν(C–O)], and 906 cm–1 [δ(Csp2–H)]. The persistence of minute residual ligand on the particle supports the conclusion that vaporization of loosely associated molecules is the primary cause for multistep profiles in TGA rather than partial ligand decomposition or desorption of diverse chemisorbed agents.
Conclusion
We have demonstrated that standard methods to functionalize the surface of hydrophobic, monodisperse SPIONs through ligand replacement result in substantial residual OA. Removal of physisorbed OA requires repeated extraction in boiling solvents to promote dissociation of carboxylic acid dimers or the addition of countercharged surfactants to impede re-absorption via ion pair formation. Even extensive purification of SPIONs prepared by a single-exchange step results in mixed dispersant particles that are characterized by a large remaining fraction of two different chemisorbed oleate species coexisting with P-NDA on the particle surface. Intercalation of nitrocatechol anchors during the initial coating exchange triggers a rearrangement of the carboxylate binding mode, which ultimately results in a mixture of mononuclear, bidentate and binuclear, bridged oleate complexes. Complete ligand exchange to an irreversibly grafted dispersant shell was only achieved after post-coating of already purified, mixed ligand nanoparticle dispersions in high excess of nitrocatechol-derived capping agents. Grafting densities of 2.5–3 molecules/nm2 were obtained regardless of SPION size. Thus, we have demonstrated that monodisperse SPIONs can be equipped with an equally monodisperse, irreversibly grafted hydrophobic shell at maximal ligand density that makes them perfectly suited for further structural studies, drug delivery, and membrane applications. In further work, we will report on the highly improved performance of these particles for the creation of magnetically responsive membrane structures.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.5b01833.
Detailed synthetic and purification protocols as well as additional characterization of reagents and samples referred to in the text (PDF)
Author Contributions
Oliver Bixner designed, conducted, and evaluated all experiments. SPIONs were synthesized by Andrea Lassenberger. Dieter Baurecht and Erik Reimhult provided guidance. The manuscript was written by Oliver Bixner and Erik Reimhult.
The research leading to these results has received funding from the BMWFW IGS BioNanoTech and the European Research Council under the European Union’s Seventh Framework Program (FP/2007–2013)/ERC Grant Agreement 310034.
The authors declare no competing financial interest.
Supplementary Material
References
- Amstad E.; Textor M.; Reimhult E. Stabilization and Functionalization of Iron Oxide Nanoparticles for Biomedical Applications. Nanoscale 2011, 372819–2843 10.1039/c1nr10173k. [DOI] [PubMed] [Google Scholar]
- Liu G.; Gao J.; Ai H.; Chen X. Applications and Potential Toxicity of Magnetic Iron Oxide Nanoparticles. Small 2013, 99–101533–1545 10.1002/smll.201201531. [DOI] [PubMed] [Google Scholar]
- Ling D.; Hyeon T. Iron Oxide Nanoparticles: Chemical Design of Biocompatible Iron Oxide Nanoparticles for Medical Applications (Small 9–10/2013). Small 2013, 99–101449–1449 10.1002/smll.201370057. [DOI] [PubMed] [Google Scholar]
- Kim B. H.; Lee N.; Kim H.; An K.; Park Y. I.; Choi Y.; Shin K.; Lee Y.; Kwon S. G.; Na H. B.; et al. Large-Scale Synthesis of Uniform and Extremely Small-Sized Iron Oxide Nanoparticles for High-Resolution T1Magnetic Resonance Imaging Contrast Agents. J. Am. Chem. Soc. 2011, 1333212624–12631 10.1021/ja203340u. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Dutz S.; Häfeli U. O.; Mahmoudi M. Magnetic Fluid Hyperthermia: Focus on Superparamagnetic Iron Oxide Nanoparticles. Adv. Colloid Interface Sci. 2011, 1661–28–23 10.1016/j.cis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Bose A.; Bothun G. D. Controlled Release from Bilayer-Decorated Magnetoliposomes via Electromagnetic Heating. ACS Nano 2010, 463215–3221 10.1021/nn100274v. [DOI] [PubMed] [Google Scholar]
- Preiss M. R.; Bothun G. D. Stimuli-Responsive Liposome-Nanoparticle Assemblies. Expert Opin. Drug Delivery 2011, 881025–1040 10.1517/17425247.2011.584868. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Kohlbrecher J.; Müller E.; Schweizer T.; Textor M.; Reimhult E. Triggered Release from Liposomes through Magnetic Actuation of Iron Oxide Nanoparticle Containing Membranes. Nano Lett. 2011, 1141664–1670 10.1021/nl2001499. [DOI] [PubMed] [Google Scholar]
- Lu A.-H.; Salabas E. L.; Schüth F. Magnetic Nanoparticles: Synthesis, Protection, Functionalization, and Application. Angew. Chem., Int. Ed. 2007, 4681222–1244 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee E.; Hwang N.-M.; Kang M.; Kim S. C.; Hwang Y.; Park J.-G.; Noh H.-J.; Kim J.-Y.; Park J.-H.; et al. One-Nanometer-Scale Size-Controlled Synthesis of Monodisperse Magnetic Iron Oxide Nanoparticles. Angew. Chem., Int. Ed. 2005, 44192872–2877 10.1002/anie.200461665. [DOI] [PubMed] [Google Scholar]
- Hyeon T.; Lee S. S.; Park J.; Chung Y.; Na H. B. Synthesis of Highly Crystalline and Monodisperse Maghemite Nanocrystallites without a Size-Selection Process. J. Am. Chem. Soc. 2001, 1235112798–12801 10.1021/ja016812s. [DOI] [PubMed] [Google Scholar]
- Hickey R. J.; Haynes A. S.; Kikkawa J. M.; Park S.-J. Controlling the Self-Assembly Structure of Magnetic Nanoparticles and Amphiphilic Block-Copolymers: From Micelles to Vesicles. J. Am. Chem. Soc. 2011, 13351517–1525 10.1021/ja1090113. [DOI] [PubMed] [Google Scholar]
- Verma A.; Stellacci F. Effect of Surface Properties on Nanoparticle–Cell Interactions. Small 2010, 6112–21 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- Nel A. E.; Madler L.; Velegol D.; Xia T.; Hoek E. M. V.; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano-Bio Interface. Nat. Mater. 2009, 87543–557 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Sahoo Y.; Pizem H.; Fried T.; Golodnitsky D.; Burstein L.; Sukenik C. N.; Markovich G. Alkyl Phosphonate/Phosphate Coating on Magnetite Nanoparticles: A Comparison with Fatty Acids. Langmuir 2001, 17257907–7911 10.1021/la010703+. [DOI] [Google Scholar]
- Gillich T.; Acikgöz C.; Isa L.; Schlüter A. D.; Spencer N. D.; Textor M. PEG-Stabilized Core–Shell Nanoparticles: Impact of Linear versus Dendritic Polymer Shell Architecture on Colloidal Properties and the Reversibility of Temperature-Induced Aggregation. ACS Nano 2013, 71316–329 10.1021/nn304045q. [DOI] [PubMed] [Google Scholar]
- Davis K.; Qi B.; Witmer M.; Kitchens C. L.; Powell B. A.; Mefford O. T. Quantitative Measurement of Ligand Exchange on Iron Oxides via Radiolabeled Oleic Acid. Langmuir 2014, 303610918–10925 10.1021/la502204g. [DOI] [PubMed] [Google Scholar]
- Napolitano A.; d’Ischia M.; Costantini C.; Prota G. A New Oxidation Pathway of the Neurotoxin 6-Aminodopamine. Isolation and Characterisation of a Dimer with a tetrahydro[3,4a]iminoethanophenoxazine Ring System. Tetrahedron 1992, 48398515–8522 10.1016/S0040-4020(01)86599-6. [DOI] [Google Scholar]
- El-Faham A.; Funosas R. S.; Prohens R.; Albericio F. COMU: A Safer and More Effective Replacement for Benzotriazole-Based Uronium Coupling Reagents. Chem. - Eur. J. 2009, 15379404–9416 10.1002/chem.200900615. [DOI] [PubMed] [Google Scholar]
- Mondini S.; Ferretti A. M.; Puglisi A.; Ponti A. Pebbles and PebbleJuggler: Software for Accurate, Unbiased, and Fast Measurement and Analysis of Nanoparticle Morphology from Transmission Electron Microscopy (TEM) Micrographs. Nanoscale 2012, 4175356–5372 10.1039/c2nr31276j. [DOI] [PubMed] [Google Scholar]
- Klokkenburg M.; Hilhorst J.; Erné B. H. Surface Analysis of Magnetite Nanoparticles in Cyclohexane Solutions of Oleic Acid and Oleylamine. Vib. Spectrosc. 2007, 431243–248 10.1016/j.vibspec.2006.09.008. [DOI] [Google Scholar]
- Spycher P. R.; Hall H.; Vogel V.; Reimhult E. Patterning of Supported Lipid Bilayers and Proteins Using Material Selective Nitrodopamine-mPEG. Biomater. Sci. 2015, 3194–102 10.1039/C4BM00090K. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Gehring A. U.; Fischer H.; Nagaiyanallur V. V.; Hähner G.; Textor M.; Reimhult E. Influence of Electronegative Substituents on the Binding Affinity of Catechol-Derived Anchors to Fe3O4 Nanoparticles. J. Phys. Chem. C 2011, 1153683–691 10.1021/jp1109306. [DOI] [Google Scholar]
- Bronstein L. M.; Huang X.; Retrum J.; Schmucker A.; Pink M.; Stein B. D.; Dragnea B. Influence of Iron Oleate Complex Structure on Iron Oxide Nanoparticle Formation. Chem. Mater. 2007, 19153624–3632 10.1021/cm062948j. [DOI] [Google Scholar]
- Roonasi P.; Holmgren A. A Fourier Transform Infrared (FTIR) and Thermogravimetric Analysis (TGA) Study of Oleate Adsorbed on Magnetite Nano-Particle Surface. Appl. Surf. Sci. 2009, 255115891–5895 10.1016/j.apsusc.2009.01.031. [DOI] [Google Scholar]
- Nasrazadani S.; Raman A. The Application of Infrared Spectroscopy to the Study of Rust systems—II. Study of Cation Deficiency in Magnetite (Fe3O4) Produced during Its Transformation to Maghemite (γ-Fe2O3) and Hematite (α-Fe2O3). Corros. Sci. 1993, 3481355–1365 10.1016/0010-938X(93)90092-U. [DOI] [Google Scholar]
- Jubb A. M.; Allen H. C. Vibrational Spectroscopic Characterization of Hematite, Maghemite, and Magnetite Thin Films Produced by Vapor Deposition. ACS Appl. Mater. Interfaces 2010, 2102804–2812 10.1021/am1004943. [DOI] [Google Scholar]
- Benoit D. N.; Zhu H.; Lilierose M. H.; Verm R. A.; Ali N.; Morrison A. N.; Fortner J. D.; Avendano C.; Colvin V. L. Measuring the Grafting Density of Nanoparticles in Solution by Analytical Ultracentrifugation and Total Organic Carbon Analysis. Anal. Chem. 2012, 84219238–9245 10.1021/ac301980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph M.; Erler J.; Peuker U. A. A TGA–FTIR Perspective of Fatty Acid Adsorbed on Magnetite nanoparticles–Decomposition Steps and Magnetite Reduction. Colloids Surf., A 2012, 397016–23 10.1016/j.colsurfa.2012.01.020. [DOI] [Google Scholar]
- Martínez-Boubeta C.; Simeonidis K.; Angelakeris M.; Pazos-Pérez N.; Giersig M.; Delimitis D.; Nalbandian L.; Alexandrakis V.; Niarchos D. Critical Radius for Exchange Bias in Naturally Oxidized Fe Nanoparticles. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 74, 054430. 10.1103/PhysRevB.74.054430. [DOI] [Google Scholar]
- Santos-Carballal D.; Roldan A.; Grau-Crespo R.; de Leeuw N. H. A DFT Study of the Structures, Stabilities and Redox Behaviour of the Major Surfaces of Magnetite Fe3O4. Phys. Chem. Chem. Phys. 2014, 163921082–21097 10.1039/C4CP00529E. [DOI] [PubMed] [Google Scholar]
- Ri̅bena D.; Alekseev A.; van Asselen O.; Mannie G. J. A.; Hendrix M. M. R. M.; van der Ven L. G. J.; Sommerdijk N. A. J. M.; de With G. Significance of the Amide Functionality on DOPA-Based Monolayers on Gold. Langmuir 2012, 284916900–16908 10.1021/la303308m. [DOI] [PubMed] [Google Scholar]
- Zhang L.; He R.; Gu H.-C. Oleic Acid Coating on the Monodisperse Magnetite Nanoparticles. Appl. Surf. Sci. 2006, 25352611–2617 10.1016/j.apsusc.2006.05.023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.