

Abstract
Background
Myocardial infarction (MI) has likely contributed to the increased prevalence of heart failure (HF). As a result of reduced cardiac function, splanchnic blood flow decreases, causing ischemia in villi and damage to the intestinal barrier. The induction of heme oxygenase-1 (HO-1) could prevent, or lessen the effects of stress and inflammation. Thus, the effect and mechanism thereof of HO-1 on the intestines of rats with HF was investigated.
Methods
Male Wistar rats with heart failure through ligation of the left coronary artery were identified with an left ventricular ejection fraction of < 45% through echocardiography and then divided into various experimental groups based on the type of peritoneal injection they received [MI: saline; MI + Cobalt protoporphyrin (CoPP): CoPP solution; and MI + Tin mesoporphyrin IX dichloride (SnMP): SnMP solution]. The control group was comprised of rats without coronary ligation. Echocardiography was performed before ligation for a baseline and eight weeks after ligation in order to evaluate the cardiac function of the rats. The bacterial translocation (BT) incidence, mesenteric microcirculation, amount of endotoxins in the vein serum, ileum levels of HO-1, carbon oxide (CO), nitric oxide (NO), interleukin (IL)-10, tumour necrosis factor-α (TNF-α), and the ileum morphology were determined eight weeks after the operation.
Results
The rats receiving MI + CoPP injections exhibited a recovery in cardiac function, an amelioration of mesenteric microcirculation and change in morphology, a lower BT incidence, a reduction in serum and ileac NO and TNF-α levels, and an elevation in ileac HO-1, CO, and interleukin-10 (IL-10) levels compared to the MI group (P < 0.05). The rats that received the MI + SnMP injections exhibited results inverse to the MI (P < 0.05) group.
Conclusions
HO-1 exerted a protective effect on the intestines of rats with HF by inhibiting the inflammation and amelioration of microcirculation through the CO pathway. This protective effect could be independent from the recovery of cardiac function.
Keywords: Carbon monoxide, Heart failure, Heme oxygenase-1, Intestine
1. Introduction
Heart failure (HF) is one of the most common medical conditions, effecting an estimated 23 million people worldwide.[1] The Framingham Heart Study found no change in the incidence of HF in men and an approximately one-third decline in the incidence of HF in women in the past 50 years.[2] Furthermore, a recent study has shown that the overall mortality and hospital readmission rates of patients with HF remains unacceptably high.[3] Advances in the treatment of coronary artery disease and the reduction in mortality associated with myocardial infarction (MI) have likely contributed to the increased prevalence of HF.[1] The cardiac remodeling process after acute myocardial infarction (AMI) has been well documented in experimental and clinical investigations, and has been associated with the development of congestive HF.[4]
Splanchnic blood flow occupies 30% of all cardiac output of a person at rest.[5] A reduced splanchnic blood flow is believed to maintain brain and kidney perfusion during shock or severe stress, and a reduced cardiac output can induce a reduction in mesenteric arterial blood flow.[6] The lowest oxygen pressure at the top of a villus is determined by the structure of its blood supply; even a slight change in blood flow can cause ischemia.[7] Intestinal mucosa permeability increases with inadequate mucosal perfusion.[5] During HF, the gastrointestinal tract is impaired by anoxia or ischemia before other organs,[8] and the changes in intestinal vascularity can precede changes in heart rate or blood pressure,[9] suggesting that even discrete HF can lead to varying degrees of intestinal ischemia. The main influence of gut-derived inflammation is impaired microcirculation due to reduced cardiac output.[10]
The independent paracellular absorption of transfer proteins in the small intestine increases during HF, suggesting a rupture in the intestinal barrier.[11],[12] However, passive transfer mediated by transfer proteins decreases, suggesting an ATP deficit.[11],[13] The intestinal change of patients with HF was identified by a 2007 imaging examination.[11] Arutyunov, et al.[14] reported that, a thickening of the small intestine wall, an increase in collagen tissue, and an increase in the distance between the enterocyte basal membrane and the blood capillary were correlated with the severity of HF. These structural changes could worsen enterocyte nutrition, thereby causing abnormal intestinal absorption.
A study by Niebauer, et al.[15] demonstrated that patients with HF and edema exhibited an increase in plasma endotoxin concentration compared to those without edema; this concentration normalized after diuretic therapy. Lipopolysaccharides (LPS), also known as endotoxins, are abundant in intestinal lumens during physical homeostasis and are considered one of the most powerful inducers of pro-inflammatory substances by triggering cytokine release,[7] including the tumor necrosis factor-α (TNF-α). A pro-inflammatory cytokine, the importance of TNF-α was primarily described by Levine, et al.[16] in 1990. A minute, even pathophysiologically relevant amount of LPS has demonstrated effectiveness in inducing TNF release.[17],[18] TNF-α is not only a marker of HF severity,[19],[20] but also induces HF by exacerbating ventricular dysfunction and remodeling.[21],[22] In addition, an elevated concentration of TNF-α can cause changes in the intestinal epithelial barrier.[9],[23] Thus, the following hypothesis was formed: HF → reduced intestinal perfusion → change in the intestinal barrier and function → elevated serum LPS level and cytokine release → exacerbation of HF and intestinal barrier change.
Free heme, released from hemoproteins, has pro-inflammatory properties and increases in various pathologies.[24] Heme oxygenase-1 (HO-1), the inducible type of HO, breaks heme into biliverdin, carbon oxide (CO), and ferrous iron. HO-1 has been indicated to be cytoprotective, particularly in inflammatory conditions, and its induction can be an adaptive mechanism against stress.[24],[25] Previous studies have shown that HO-1 induction exerts protective effects in cells stimulated by inflammatory cytokines through the down-regulation of inflammatory mediators.[26],[27]
Based on this evidence, a hypothesis was made that the induction of HO-1 could ameliorate HF and/or intestinal change, while the inhibition of HO-1 activity could exert a contra-effect. We used Cobalt protoporphyrin (CoPP) to induce HO-1 over-expression, Tin mesoporphyrin IX dichloride (SnMP) to inhibit HO-1 activity, and investigate the resulting mechanism of HO-1 protection.
2. Methods
2.1. Animal preparation
The experimental procedures were conducted in adherence to the Guiding Principles in the Use and Care of Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised in 1996) and the Guide for the Care and Use of Laboratory Animals published by the Government of the People's Republic of China (issued on June 3rd, 2004); they were approved by the Institutional Animal Care and Use Committee of the PLA General Hospital in Beijing, China. All procedures and evaluations were performed in a blinded manner. Male Wistar rats with body weights ranging from 220−240 g were obtained from the Animal Care Center Laboratory at the PLA General Hospital in Beijing, China. All rats were maintained at a constant humidity (60% ± 5%), temperature (24 ± 1°C), and light cycle (7 a.m. to 7 p.m.) and were fed a standard rat pellet diet. All rats were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal or intramuscular) before euthanasia eight weeks following the operation.
2.2. Experimental and control groups
The experimental group was subjected to coronary ligation. After anesthesia by intraperitoneal injection with 3% pentobarbital sodium at 50 mg/kg rat body weight, the rats were fixed to a pad, and the chest was shaved and disinfected. The animals were then intubated and ventilated with a positive pressure artificial respirator (tidal volume 7−8 mL, respiratory rate 80 breaths/min). The normal electrocardiogram (ECG) was monitored and recorded. After draping the sterilization towel, a left thoracotomy was performed through the sternum third and fourth intercostal space. The ophthalmology forceps were used to gently tear the pericardium open to expose the heart. The heart was extruded, and a silk suture was used to ligate the left anterior descending coronary artery 2−3 mm underneath the root of left atrial appendage. The ligation was deemed successful when the myocardial colour turned shallow and pale, and the limb lead of ECG monitoring showed the increased amplitude of R wave and ST segment. The chest was closed with the suture, and 8 × 105 U of penicillin and 0.2 mL of lidocaine were subcutaneously injected. The rates were extubated once spontaneous breathing recovered. The rats were kept warm and given food and water four hours later. Some of the rats died as a result of ventricular tachycardia or HF postoperatively. Of the total of 96 rats subjected to coronary ligation, 67 (69.8% of 96) rats survived. All the 67 rats were tested using transthoracic echocardiography. Ultrasound measurements were carried out according to the diagnosis standards of HF in rats issued by American Society of Echocardiography. Left ventricular ejection fraction (LvEF) was ≤ 45%, suggesting that the ligation of the left anterior descending coronary artery in rats can successfully replicate the rat model of HF after MI.[28] Forty-eight (71.6% of 67) rats had LvEF, less than 45% were selected, but the rats with LvEF, above 45%, one day post-operation were excluded from the study. The rats with LvEF, less than 45% were randomized into three experimental groups; ten rats were used for bacterial translocation examination and four rats were used for each of the microcirculation examination groups. Of the microcirculation examination groups, the MI group consisted of rats given injections of saline, the MI + CoPP group consisted of rats given injections of CoPP solution, and the MI + SnMP group consisted of rats given injections of SnMP solution. Eighteen rats, which were not subjected to coronary ligation, were used as the control group. The Western blotting and Real-time PCR tests identified an over-expression of HO-1 by CoPP at both the mRNA level and the protein level. As an index of HO-1 activity, the intestinal carbon monoxide concentration, a product of the decomposition of heme by HO-1, was used to indicate HO-1 activity in the CoPP and SnMP groups.
2.3. Drug administration and protocol
The CoPP, a hemoxygenase-1 inducer, was bought from Aldrich (UT, USA). SnMP, a hemoxygenase-1 inhibitor, was bought from Frontier Scientific Inc. (Logan, UT, USA). Drug administration was performed as previously described by Cao, et al.[29] and Chen, et al.[30] Briefly, CoPP (0.5 mg/100 g of rat weight) and SnMP (0.2 mg/100 g of rat weight) were injected into rats twice a week for eight weeks. The rats were weighed before each injection. The drug was freshly dissolved in NaOH; then, the free bacteria were eradicated by filtrating through a sterilized 0.22 µm pore filtrum. The solution was adjusted to a pH of 7.4 with hydrochloric acid, and was further diluted with PBS to meet the required volume before intraperitoneal injection, and the same approximate volume PBS was given to non-treated rats.[31] The rats prepared for microcirculation examination received intramuscular injections rather than intraperitoneal injections two weeks before examination. The CoPP and SnMP were prepared in the dark and protected from light. All of the acids, bases, and PBSs were sterilized. Rats were weighed before each injection.
2.4. Echocardiography
Echocardiography of the 32 rats was performed and analyzed before and at eight weeks after the operation with a high-resolution (17.5 MHz) Vevo 770 transthoracic echocardiography system (Visual Sonic Inc, Canada). The wall depth and ventricular diameters were measured during three consecutive cardiac cycles with a short-axis M-mode image. LvEF was computed using the Vevo 770 Standard Measurement Package. Next, the arithmetic means of these values were calculated. Before and during the ultrasound scanning, the rats were anesthetized with a mixture of 1.5% isoflurane gas (2.5% isoflurane to induce anesthesia) and 1 L/min of oxygen then placed in the supine position on a platform.
2.5. Collection of tissue and blood samples
After discarding the distal 5 cm of the ileum adjacent to the cecum, the remaining lower 5 cm was collected for morphological examination or other tests. The tissues used for the Real-time PCR, Western blot, and enzyme-linked immunosorbent assay (ELISA) tests were snap frozen using liquid nitrogen and transferred for preservation at –80°C. Plasma extracted from portal vein blood and inferior vena cava (centrifuged at 3,000 r/min for 10 min) was used for endotoxin examination through the ELISA test. The serum obtained from abdominal aortic blood (centrifuged at 3,000 r/min for 10 min) was frozen to a temperature of –80°C until use.
2.6. Endotoxin examination
Plasma extracted from portal vein blood and inferior vena cava under endotoxin-free conditions were tested using an ELISA kit (CUSABIO BIOTECH CO., Ltd, Wuhan, China), following the manufacturer's instructions. The results were expressed in pg/mL (n = 10).
2.7. BT examination
Liver, spleen, and mesenteric lymph node (MLN) samples were homogenized in PBS before being placed on blood agar (Oxoid, UK) for aerobic incubation, or on anaerobic agar (Oxoid, UK) for anaerobic incubation immediately after collection. The samples were then incubated in corresponding anaerobic chambers and aerobic atmospheres for 48 h at 37°C. The sterile swab technique was used to culture the peritoneal surface. If bacteria were present, BT was indicated. The number of living bacteria was calculated and expressed in colony forming units per gram of organ tissue (cfu/g).
2.8. RNA extraction and real-time PCR
All RNA was extracted using Trizol reagent (Invitrogen) under the manufacturer's protocol. The Revert Aid First Strand cDNA Synthesis Kit (1622, Fermentas) was used to complete reverse transcription. Real-time PCR was then executed twice for each sample type using 2 × Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (600882, Agilent Technologies). The data for each sample was analyzed according to the comparative Ct method and normalized through glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. The primers of the polymerase chain reaction are shown in Table 1.
Table 1. Primers of the polymerase chain reaction.
| Gene | Sense primer (5′- 3′) | Antisense primer (3′- 5′) | Amplication (bp) |
| HO-1 | GAAACAAGCAGAACCCAGTC | AGAGGTCACCCAGGTAGCG | 225 |
| TNF-α | GCCACCACGCTCTTCTGTC | GCTACGGGCTTGTCACTCG | 362 |
| IL-10 | AGTCAGCCAGACCCACAT | GGCAACCCAAGTAACCCT | 140 |
| GAPDH | GCAAGTTCAACGGCACAG | GCCAGTAGACTCCACGACAT | 128 |
GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HO-1: heme oxygenase-1; IL-10: interleukin-10; TNF: tumor necrosis factor.
2.9. Measurement of ileac NO
Ileac NO levels were detected using commercially available kits for the colorimetric determination of NO (Nanjing Jiancheng Bioengineering Institute, China); these kits are based on the enzymatic induced conversion of nitrate to nitrite with nitrate reductase. The Griess reaction results in the colorimetric detection of nitrite as an azo dye product.
2.10. Measurement of ileac CO
The ileum samples were homogenized in a hemolytic agent (w/v = 1:9). A 10 µL supernatant, containing 10% of the ileum homogenate, was diluted to a 2.5 mL diluent in order to calculate the absolute derivative absorption of the Hb content at 540 nm. The 20 µL supernatant was diluted to a 2.5 mL diluent using a biuret reagent, and the absolute derivative absorption value of the diluent at 540 nm was compared with the standard solution and blank solution of the saturated tissue proteins. With 0.2 mL of the supernatant diluted to a 2.3 mL diluents (for HbCO measurement), the absolute derivative absorption value of diluent respectively at 568 nm and 581 nm was respectively compared with saturated HbCO samples (2.5 mL diluted supernatant was saturated by bubbling CO gas for 15 min and nitrogen for 20 min) and saturated HbO2 samples (2.5 mL diluted supernatant was saturated by bubbling O2 gas for 20 min) to get the percentage of saturated HbCO. CO content was calculated by this formula: CO (µmol/g) = HbCO% × Hb (g/L) × 106 × 4 / [64456 × protein content (g/L)]. The reagent kit was obtained from the Nanjing Jiancheng Bioengineering Institute in Nanjing, China.
2.11. Enzyme-linked immunosorbent assay
Ileac mucosa and underlying muscular tissue were homogenized with the RIPA buffer (1 × tris-buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.004% sodium azide, 10 µl/mL phenylmethanesulfonylfluoride, 10 µL/mL protease inhibitor cocktail, and 10 µL/mL sodium orthovanadate). After homogenization, the sample was centrifuged at 14,000 r/min for 10 min at 4°C, and the supernatant was analyzed using ELISA. Both IL-10 and TNF-α were determined according to the manufacturer's (Cusabio Biotech, CO., Ltd., Wuhan, China) recommendations.
2.12. Western blotting
The rat ileum samples were lysed in radioimmunoprecipitation assay buffer [50 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1% Triton X-100, 0.5% deoxycholic acid, and 0.1% SDS]. The protein concentration was measured using the Coomassie Plus Protein Assay Reagent (Nanjing Jiancheng Bioengineering Institute, China). The lysate proteins (30 µg/lane) were separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred by electroblotting to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membrane was blocked for one hour with 5% non-fat milk in PBST (phosphate-buffered saline containing 0.1% Tween-20), before blotting with anti-HO-1 or anti-β-actin (Beijing Biosynthesis Biotechnology Co., Ltd, Beijing, China; 1:1,000) antibodies at 4°C overnight. The membranes were washed in TBST and incubated for one hour with horseradish peroxidase-conjugated anti-rabbit IgG antibody (Zhongshan Goldernbridge Biotechnology Co., Ltd., Beijing, China; 1: 3,000). The membrane was visualized (Kodak, USA) and analyzed with the ultraviolet photometry (UvP) imaging analysis system (Thermo, USA). The result was presented as HO-1/β-actin.
2.13. Mesentery microcirculation and intestinal Doppler
All animals were anesthetized with sodium pentobarbital (50 mg/kg, intramuscular) then incised on the midline of the abdomen. Next, the intestine and corresponding mesentery were carefully extracted. The mesentery was placed on a transparent observation plate. The areas of the intestine that were not to be examined were covered with gauze and continuously superfused with, or dipped in 37 °C isotonic saline in order to prevent dehydration and exposure to ambient air. The diameters of the grade III stretching arterioles (A3), capillaries, and grade I stretching venules (V1) was observed using a video microscope (Olympus BH-2). The adherent leukocytes in the grade III stretching venules (V3) were observed within 60 s and were used to locate each vessel segment in order to prevent the leukocyte from moving, or detaching, from the endothelial lining during the observation period. The result was reported as the number of cells per mm of venule. Three video images were digitally stored and analyzed with Image-Pro image analyzing software (Media cybernetics LP) for each measurement.
A longitudinal incision 2 cm in length was made in the lateral side of the ileum, about 5 cm from the ileocecal junction. The intestinal contents were removed with saline wash. The microcirculatory blood flow intensity of the intestinal villi was measured using the Perimed Laser Doppler technique (Perimed AB, Sweden), a laser speckle contrast imaging technique. The images were recorded and analyzed using the PeriScan System. The microcirculation of the intestine at a 1 mm depth was evaluated. The working distance between the head of the scan and the measurement site was 12 cm and the dimension of the measurement sites were 3 × 3 mm. The results were reported as mean volts.
2.14. Morphology examination
The ileac tissue was fixed with 10% paraformaldehyde, embedded in paraffin, sectioned into 4-µm-thick slices, and stained with hematoxylin and eosin (HE) for histological evaluation using light microscopy. Twenty consecutive visual fields were observed. The results were scored as the following: grade 1 (score of 0) was used to indicate a normal histology; grade 2 (score of 1) was used to indicate minimal damage, hydropic degeneration and/or separation of the surface epithelial cells from the lamina propria; grade 3 (score of 2) was used to indicate mild damage and/or epithelial cell necrosis confined to the tips of the villi; grade 4 (score of 3) was used to indicate moderate damage and/or complete villus necrosis; or grade 5 (score of 4) was used to indicate severe damage and/or transmural necrosis.[32] The arithmetic mean of the 20 visual fields of the ileum was regarded as the morphology score for each rat.
2.15. Statistics
The data was presented as the mean ± SE unless stated otherwise. In order to compare the multiple groups, a 1-way ANOVA was used if there was one independent variable, and the post-hoc test was used to adjust for multiple comparisons. The Kruskal-Wallis test was also used when necessary. Chi-Square tests were used to compare the ratio differences among groups. A statistical significance value of P < 0.05 was used for analysis.
3. Results
3.1. Echocardiography
Before the operation, the LvEF% (n = 8) of rats was determined for all four groups (Control 65.440 ± 3.767, MI + CoPP 37.005 ± 6.322, MI 36.272 ± 7.713, MI + SnMP 36.564 ± 6.252). No significant difference was found among the three postoperative groups (MI, MI + CoPP, and MI + SnMP), and each group had a significantly lower EF% (P < 0.01) than the control group, as shown in Figure 1A. After 8 weeks of treatment, the MI (38.080 ± 13.786 and P < 0.01 vs. Control) and MI + SnMP (33.810 ± 8.919 and P < 0.01 vs. Control) groups had significantly lower LvEF% than that of the control group (66.254 ± 7.002). The MI + CoPP (53.833 ± 15.755) group exhibited a prominent elevation of LvEF% compared to the MI (P < 0.05) and MI + SnMP (P < 0.01) groups. However, such an LvEF% still presented a lesion on the left ventricular cardiac function compared to the control group (P < 0.05 for MI + CoPP vs. Control). The MI + SnMP group suffered a lower mean LvEF% than the MI group. However, this reduction did not exhibit as high a statistical difference as expected (P > 0.05), as shown in Figure 1B. Considering the prominent recovery in LvEF% in the MI + CoPP group compared to the MI and MI + SnMP groups, four rats with LvEF% less than 45%, as shown in Figure 1C, were chosen from each group in order to eliminate the difference in cardiac function of the microcirculation test (MI + CoPP 40.280 ± 9.168, MI 31.788 ± 11.397, MI + SnMP 2.212 ± 12.308, n = 4, P > 0.05).
Figure 1. LvEF% of rats at baseline (A, n = 8) and eight weeks postoperative (B, n = 8), and rats for microcirculation test (C, n = 4).

*P < 0.05, **P < 0.01vs. Control; #P < 0.05, ##P < 0.01 vs. MI. CoPP: cobalt protoporphyrin; LvEF: left ventricular ejection fraction; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
3.2. Bacterial translocation
In all tests, the MI group demonstrated a higher BT incidence and higher number of living bacteria than the control group (P < 0.01), whereas the MI + CoPP group had no difference to the control group (P > 0.05). The MI + CoPP group exhibited a reduction in both the total number of BT incidences and the number of living bacteria compared to the MI group (P < 0.01), and the MI + SnMP group had an inverse effect to the MI group (P < 0.05). The MI + CoPP group of the liver and spleen samples also exhibited a reduction in BT incidence when compared to the MI group (P < 0.05). The CoPP treatment of the MLN samples did not have a similar result to the MI group (P > 0.05), even though the positive incidence was only slightly higher than that of the liver and spleen. However, this “defect” was “repaired” with a 100% BT incidence in the MI + SnMP sample. This situation was also observed in the number of BT incidences. Of the 18 rats with BT incidences, three of the rats (one in the MI + CoPP group, and two in the MI + SnMP group) exhibited BT incidences in the lymph node only and no BT incidences were observed in the liver and spleen in the MLN. The results were listed in Table 2.
Table 2. Incidence of BT in different experimental groups (positive/n, n = 10).
| Treatment | Liver | Spleen | MLN | Totala | Numbers of rats of BT | Numbers of living bacteria |
| Control | 0/10 | 0/10 | 0/10 | 0/30 | 00/10 | 0,0 |
| MI + CoPP | 1/10# | 1/10# | 2/10 | 4/30## | 02/10 | 0,3# |
| MI | 6/10** | 6/10** | 6/10** | 18/30** | 06/10** | 0307.5,2270.0** |
| MI + SnMP | 8/10** | 8/10** | 10/10**,# | 26/30**,# | 10/10**,# | 4530.0,8680.0**,# |
aTotal = Liver + Spleen + MLN. *P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05 compared to the MI group,##P < 0.01 compared to the MI group. The BT incidence was examined using Chi-Square tests. The number of living bacteria, represented as “median, Q75 - Q25” was examined using the Kruskal-Wallis Test. BT: bacterial translocation; CoPP: cobalt protoporphyrin; MI: myocardial infarction; MLN: mesenteric lymph node; SnMP: Tin mesoporphyrin IX dichloride.
3.3. Endotoxin
Endotoxin levels of plasma in both portal vein and inferior vena cava eight weeks postoperative were Control 53.571 ± 21.710 pg/mL, MI + CoPP 61.790 ± 21.038 pg/mL, MI 107.126 ± 65.673 pg/mL, MI + SnMP 165.016 ± 40.751 pg/mL, n = 10, and Control 42.609 ± 12.662 pg/mL, MI + CoPP 49.310 ± 25.273 pg/mL, MI 78.383 ± 36.740 pg/mL, MI + SnMP 141.249 ± 40.772 pg/mL, n = 10, respectively. The rats in the MI group exhibited significantly higher endotoxin levels than the control group (P < 0.01 in the portal vein and P < 0.05 in inferior vena cava). The Copp treatment exhibited a reduction in endotoxin levels compared to the MI group (P < 0.05), and no difference compared to the control group (P > 0.05); however, the SnMP treatment exhibited a prominent increase in endotoxin levels compared to the MI group (P < 0.01). The plasma endotoxin levels of both the portal veins and inferior vena cava exhibited the same tendencies and statistical results among groups, as shown in Figure 2.
Figure 2. Endotoxin levels of plasma in both portal vein (A) and inferior vena cava (B) eight weeks postoperative.

N = 10 in each group. *P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. CoPP: cobalt protoporphyrin; LvEF: left ventricular ejection fraction; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
3.4. HO-1 expression and its activity determined by CO level
The expression of HO-1 in the ileum obtained with Real-time PCR (examined using the Kruskal-Wallis Test, n = 5) was Control 1.242 ± 0.383, MI + CoPP 44.120 ± 5.115, MI 3.968 ± 1.152, MI + SnMP 4.236 ± 1.696. Expression of HO-1 in ileum as determined by Western bloting (n = 5) and HO-1/β-actin was: Control 0.183 ± 0.052, MI + CoPP 0.359 ± 0.025, MI 0.254 ± 0.037, and MI + SnMP 0.258 ± 0.033. The MI group rats expressed a higher HO-1 level than the control group at both the mRNA level (P < 0.01) and the protein level (P < 0.05). The CoPP treatment significantly increased HO-1 expression both at the mRNA level (P < 0.01) and the protein level (P < 0.01 vs. Control, and P < 0.05 vs. MI). The SnMP treatment did not significantly affect HO-1 expression despite being an HO-1 inhibitor; the HO-1 expression of this treatment was approximately equal to that of the MI treatment. The HO-1 activity of the MI group determined by the ileac CO level (Control 0.962 ± 0.291 µmol/g, MI + CoPP 1.647 ± 0.373 µmol/g, MI 1.317 ± 0.287 µmol/g, MI + SnMP 0.906 ± 0.269 µmol/g, n = 8), was significantly greater than that of the control group (P < 0.05). The CO level was even greater in the MI + CoPP group compared to the MI group (P < 0.05). The ileac CO level was inhibited by SnMP; thus, the SnMP group exhibited a significant reduction in CO compared to the MI group (P < 0.05), and an even lower mean CO level than the control group despite the lack of statistical significance (P > 0.05). The comparison results of HO-1 among four groups were shown in Figure 3.
Figure 3. HO-1 expression level in ileum determined by Real-time PCR (A) and Western blotting (B&C), and CO level (D).

*P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. CO: carbon oxide; CoPP: cobalt protoporphyrin; HO-1: heme oxygenase-1; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
3.5. Ileac NO level
The ileac NO level, Control 43.07 ± 10.104 µmol/g, MI + CoPP 51.32 ± 7.745 µmol/g, MI 60.058 ± 15.211 µmol/g, MI + SnMP 71.942 ± 10.559 µmol/g, n = 5, was significantly higher in the MI group than the control group (P < 0.01), and was even higher after the SnMP treatment (P < 0.05 vs. MI and P < 0.01 vs. Control). The CoPP treatment successfully suppressed the NO level compared to the control group (P > 0.05). No difference was observed in either compared to the MI group (P > 0.05), as shown in Figure 4.
Figure 4. Ileac NO level.

*P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. Copp: cobalt protoporphyrin; MI: myocardial infarction; NO: nitric oxide; SnMP: Tin mesoporphyrin IX dichloride
3.6. IL-10 and TNF-α level in ileum determined by Real-time PCR and ELISA
The IL-10 in ileum by Real-time PCR was Control 2.035 ± 1.086, MI + CoPP 9.548 ± 1.793, MI 6.148 ± 3.174, MI + SnMP 2.374 ± 1.624, n = 5. IL-10 in ileum by ELISA was Control 25.991 ± 11.753, MI + CoPP 56.724 ± 16.413, MI 39.039 ± 13.633, MI + SnMP 22.341 ± 9.607, n = 8. TNF-α in ileum by Real-time PCR was Control 0.738 ± 0.340, MI + CoPP 0.826 ± 0.621, MI 2.002 ± 1.047, MI + SnMP 3.324 ± 0.931, n = 5. TNF-α in ileum by ELISA was Control 156.739 ± 40.699, MI + CoPP 196.034 ± 84.048, MI 272.877 ± 81.945, MI + SnMP 313.591 ± 77.674, n = 8. The MI group demonstrated a higher IL-10 (P < 0.01) value and lower TNF-α (P < 0.05) mRNA expression compared to the control group. The MI + CoPP group exhibited an elevated ileac IL-10 mRNA expression compared to the control group (P < 0.01), and MI group (P < 0.05). The MI + CoPP group also demonstrated a TNF-α mRNA expression level equivalent to that of the control group (P > 0.05) and lower than that of the MI group (P < 0.05). Due to its HO-1 inhibitory action, the MI + SnMP treatment achieved a significant reduction in IL-10 mRNA expression compared to the MI group (P < 0.05) and an elevation in TNF-α mRNA expression compared to the MI group (P < 0.05). A similar result was observed in the ELISA test, in which an increasing trend of the IL-10 level in the MI group compared to the control group (P = 0.056) and the TNF-α level in the MI + SnMP group compared to the MI group (P > 0.05) was observed, although insignificant. The comparison results among four groups about IL-10 and TNF-α were shown in Figure 5.
Figure 5. Ileac TNF-α and IL-10 expression levels determined by real-time PCR (A & C, n = 5) or ELISA (B & D, n = 8).

*P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. CoPP: cobalt protoporphyrin; IL-10: interleukin-10; MI: myocardial infarction; TNF: tumor necrosis factor; SnMP: Tin mesoporphyrin IX dichloride.
3.7. Mesentery microcirculation and intestine Doppler
Number of adherent leukocytes in grade III stretching venule (V3) of per millimeter vessel within a period of 60 s was: Control 0.250 ± 0.500, MI + CoPP 0.725 ± 0.506, MI 4.075 ± 1.576, MI + SnMP 8.900 ± 2.432, n = 4. Diameter of grade III stretching Arteriole was Control 30.575 ± 0.419 μm, MI + CoPP 25.100 ± 2.760 μm, MI 18.775 ± 1.517 μm, MI + SnMP 17.675 ± 1.533 μm, n = 4. Diameter of capillary was Control 18.600 ± 1.219 μm, MI + CoPP 17.200 ± 0.829 μm, MI 13.375 ± 0.330 μm, MI + SnMP 11.375 ± 0.472 μm, n = 4. Perfusion of villi in ileum determined by Doppler was MI + CoPP 2.490 ± 0.261, MI 2.085 ± 0.170, MI + SnMP 2.055 ± 0.210, n = 4. All four rats with an LvEF% less than 45% in the MI + CoPP group were chosen for the purposes of the microcirculation test. Four rats with similar LvEF% levels in each postoperative group (MI and MI + SnMP) were matched correspondingly, and the microcirculation measurements among these groups were compared. A mesentery microcirculation without the influence of other cardiac function was expected to be obtained (the LvEF% statistics are shown in Figure 1C, with P > 0.05). The MI group rats exhibited obvious contracted artery vessels of either grade compared to the control group rats (P < 0.01). The CoPP treatment (MI + CoPP) rats exhibited increased diameters in both the capillary (P < 0.01) and grade III stretching arterioles (P < 0.01) and improved intestinal villi perfusion (P < 0.05) compared to the MI group rats (P < 0.01). SnMP treatment (MI + SnMP) rats had exacerbated capillary contraction (P < 0.01) compared to the MI rats. The SnMP treatment (MI + SnMP) rats expressed a contraction tendency in the grade III stretching arterioles and a reduction in intestinal villi perfusion, but there was no statistical difference when compared to the MI group rats (P > 0.05), as shown in Figure 6.
Figure 6. Determination of mesenteric microcirculation (n = 4).

(A): Number of Adherent Leukocytes in grade III stretching venule (V3) of per millimeter vessel within a period of 60 s; (B): Diameter of grade III stretching Arteriole; (C): capillary diameter; and (D): Perfusion of villi. *P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. CoPP: cobalt protoporphyrin; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
3.8. Morphology
Score of morphological examination of ileum was Control 0.015 ± 0.034, MI + CoPP 0.070 ± 0.054, MI 0.145 ± 0.080, MI + SnMP 0.260 ± 0.077, n = 5. The MI group rats had obvious intestinal damage compared to the control group (P < 0.01). The CoPP group had significantly attenuated intestinal change that contributed to the HF (P < 0.05 vs. MI) and obtained the same score as the control group (P < 0.05 vs. Control). The inhibition of HO-1 activity by SnMP significantly increased the score reflecting intestinal deterioration (P < 0.01 vs. MI), as shown in Figure 7. Intestinal edema was observed in both groups, but obvious necrosis was only observed in the MI + SnMP group, indicating increased intestinal injury, as shown in Figure 8. In addition, lymphocyte infiltration was common in the stratum submucosum of the MI and MI + SnMP groups, but seldom in the control and MI + CoPP groups, suggesting intestinal inflammation in the MI and MI + SnMP groups.
Figure 7. Score of morphological examination of ileum.

*P < 0.05, **P < 0.01 compared to the Control group; #P < 0.05, ##P < 0.01 compared to the MI group. CoPP: cobalt protoporphyrin; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
Figure 8. Morphological examination of ileum.
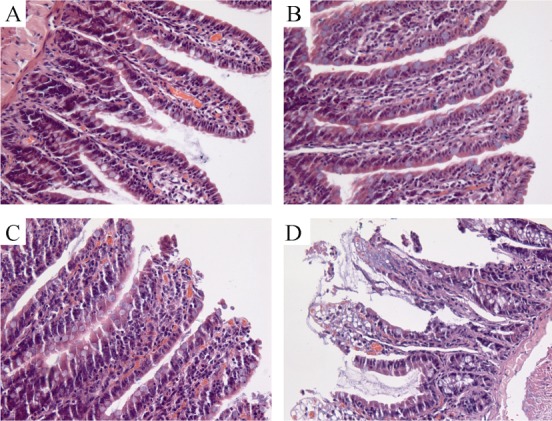
(A): Control, The normal morphology of the ileum; (B): MI + CoPP, CoPP protected the ileum from ischemia, and only mild edema was observed; (C): MI, The necrosis at the villus tip; (D): MI + SnMP, SnMP aggravated the necrosis. CoPP: cobalt protoporphyrin; MI: myocardial infarction; SnMP: Tin mesoporphyrin IX dichloride.
4. Discussion
In this paper, Real-time PCR tests identified an over-expression of HO-1 by CoPP at both the mRNA level and the protein level. Concentration of CO was used to indicate high and low HO-1 activity in the CoPP and SnMP groups, respectively. As in other experiments, the HO-1 induction treatment (MI + CoPP) assisted in the recovery of LvEF% (P < 0.05 vs. MI), with a mean greater than 50%.[33]–[35] Unfortunately, the MI + CoPP treatment did not completely abolish the cardiac function lesion (P < 0.05 vs. Control). In addition, the HO-1 inhibition treatment (MI + SnMP) did not reduce the LvEF% as expected (P > 0.05 vs. MI). In order to identify HF after the experimental MI surgery, the rats did not receive injection intervention until approximately six to eight hours after the operation. HO-1 is sensitive and reacts immediately to stress. According to the research of Zhang, et al.[33] the peak expression of HO-1 in rats with MI occurs within three days after MI. Thus, the “delayed” inhibition of HO-1 provided an opportunity for the ischemic myocardium to benefit from the decomposition of heme by HO-1. However, unexpected chance is accounted for in significance testing.
A reduction of LvEF is known to incur mesenteric arterial hypofusion.[6] In order to prevent the interference of varied LvEF, the four rats with the lowest EF% in MI + CoPP group were used in the microcirculation test, along with four rats with similar LvEF% from each of the other two groups. The result indicated an amelioration of the contraction of mesentery arteriole, capillary, and intestinal villi perfusion as well as leukocyte adhesion when subjected to the CoPP treatment. The SnMP treatment abolished the protective effect of HO-1, thereby exacerbating mesentery microcirculation and leukocyte adhesion. The importance of leukocyte adhesion in microcirculation has been observed to cause a reduction or lack of blood flow in villus microcirculation.[36] Thus, the accelerated leukocyte adhesion in the MI + SnMP group inferred impaired villi perfusion. The amelioration of oxidative injury by reducing leukocyte endothelial interactions was observed in microvascular endothelial cells with HO-1 over-expression.[37] Carbon monoxide causes smooth muscle relaxation and anti-aggregatory effects on plates in capillary pericytes, thereby improving blood flow and villi perfusion.[38] Thus, the following can be inferred from the small sample result: the protective effect of HO-1 on the intestine through the CO pathway could be independent from the amelioration of cardiac output in patients with HF. However, confirmation with more studies with larger sample sizes is necessary.
In addition to the ameliorated mesenteric microcirculation and villi perfusion, the intestinal villi edema was significantly attenuated in the MI + CoPP group. The MI and MI + SnMP groups exhibited more villi edema. Obvious necrosis was also observed in the MI + SnMP group. These findings suggested that edema was a characteristic change in the intestines of those with HF.[11],[14] The duration of intestinal damage could be a longer process. HO-1 exerted an important protective effect on the intestines of rats with HF. Due to the morphology of the mucosal barrier is the epithelial apical junctional complex,[12] it was inferred that the difference in apical tight junctions and adherence junctions might also be observed.
The importance of the integrity of the intestinal epithelial barrier as a defence mechanism against the intestinal lumen, with abundant germs and LPS, was evident. Inadequate mucosal perfusion increased the intestinal mucosal permeability.[5] In theory, the reduction of BT incidences in the MI + CoPP group should have contributed to the protection of the intestinal barrier. However, the MI + SnMP group had 100% BT in both MLN and all rats, and exhibited higher numbers of living bacteria and exacerbated intestinal morphology. A recent study, which unveiled the effect of HO-1 and CO on the augmented clearance of bacteria that breach the epithelial barrier, showed that bactericidal activity was not secondary to increased epithelial barrier integrity.[39] This could jointly affect the difference in the BT and intestinal morphology changes. In this study, three rats were observed with BT, and no BT was found in samples other than the MLN samples. During observation of these intestinal sections, numerous lymphocytes were found infiltrating the stratum submucosum in the MI and MI + SnMP groups, implying that the intestinal structure and function changes were probably combined with immunological abnormalities associated with HF. Thus, the immunological barrier could play an important role in the pathophysiological process, and the breach of gut-derived bacteria could precede the breach of morphological structure.
LPS, an ingredient of the wall of gram-negative germs, entered the circulation through an edematous, hypo-perfused bowel in increased amounts and activated monocytes and macrophages to release pro-inflammatory mediators, leading to an inflammatory state.[7],[17] Even minute amounts of LPS have been shown to be effective in the induction of TNF release.[17],[18] Subsequently, an elevated concentration of TNF-α could induce changes in the intestinal epithelial barrier and exacerbate HF.[9],[22],[23],[31] In vitro, HO-1 induction abolishes the induction of the pro-inflammatory cytokine TNF-α with IL-1β treatment, significantly decreasing HO-1 expression.[40] In this study, TNF-α was significantly suppressed with HO-1 elevation and increased HO-1 activity, where higher levels of IL-10 were detected. TNF-α accumulated as the HO-1 activity was inhibited by SnMP, as indicated by lower IL-10 levels. In addition, due to the restricted peripheral blood flow resulting from reduced cardiac output, micro-circulation was impaired, playing a crucial role in gut-derived inflammation.[10] LPS likely triggered catecholamine release in response to the exertion of additional unfavorable effects on intestinal perfusion by granulocytes and phagocytes.[41] A reduction in LPS level not only reduced pro-inflammation cytokine release, but could also attenuate the impaired microcirculation, thereby protecting the intestinal barrier and further reducing BT incidence.
NO is generated by three forms of NOS: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS). Both nNOS and eNOS are constitutively expressed in normal physiologies, but iNOS is expressed in response to inflammatory stimuli injury[42] and several immunological stimuli,[43] such as TNF-α and LPS.[44] Inflammation is generally believed to lead to a reduction of eNOS and eNOS-derived NO, while the excessive production of NO through iNOS expression is believed to further exacerbate the disease process and lead to tissue damage.[45],[46] In vitro, Lowry, et al.[47] observed that not only the NO-dependent reduction of migration in cytokine-treated endothelial cells, suggesting a reduction in wound healing, but also, eNOS activation generated by iNOS activity. However, a deleterious outcome of iNOS inhibitor was found in a clinical trial.[48] Under LPS priming, the inflammatory response was restricted through the induction of iNOS and inversely correlated to the inflammation through the NF-κB pathway[49],[50] but the iNOS derived NO did not affect the TNF-α production induced by LPS.[50] In addition, a recent study indicated the bacterial clearance is modulated by NO in sepsis.[51] In the early phases of sepsis, the contribution of nitric oxide production to the efficacy of microbial clearance with phagocytic cells was likely due to the antimicrobial activities of NO-derived mediators.[52] Despite of the dispute regarding NO, the over-expression of NO correlated with elevated TNF-α and LPS levels was confirmed in this study, indicating the severity of inflammation, and inverse correlation with IL-10. TNF-α prompted a dramatic and rapid global redistribution of chromatin activators to massive de novo clustered enhancer domains.[53] NO, a molecule reactive to inflammation and parallel to the severity of inflammation, when induced by LPS or other inflammatory stimuli, could exert a protective effect independent of TNF-α suppression.
In conclusion, the results of this study revealed the protective effect of HO-1 on the intestine of rats with HF through the CO pathway through the inhibition of inflammation and the amelioration of microcirculation. Furthermore, the induction of HO-1 and its mechanisms could strengthen the structural and immunological protection of the intestinal barrier. This protective effect could be independent of cardiac function recovery.
Acknowledgments
This work was supported by grants from the General Hospital of Nanjing Military Region Academician Li Jie-shou's “Intestinal Barrier” Special Fund (LJS-201110), Hainan Provincial Research Program (No. YJJC20130009) and Science and Technology Cooperation Fund Project of Hainan Province of China (No. KJHZ2015-36).
References
- 1.Costa MA, Pencina M, Nikolic S, et al. The PARACHUTE IV trial design and rationale: percutaneous ventricular restoration using the parachute device in patients with ischemic heart failure and dilated left ventricles. Am Heart J. 2013;165:531–536. doi: 10.1016/j.ahj.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Levy D, Kenchaiah S, Larson MG, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Normand SL, Wang Y, et al. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998–2008. JAMA. 2011;306:1669–1678. doi: 10.1001/jama.2011.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaudron P, Eilles C, Kugler I, et al. Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation. 1993;87:755–763. doi: 10.1161/01.cir.87.3.755. [DOI] [PubMed] [Google Scholar]
- 5.Takala J. Determinants of splanchnic blood flow. Br J Anaesth. 1996;77:50–58. doi: 10.1093/bja/77.1.50. [DOI] [PubMed] [Google Scholar]
- 6.Fiore G, Brienza N, Cicala P, et al. Superior mesenteric artery blood flow modifications during off-pump coronary surgery. Ann Thorac Surg. 2006;82:62–67. doi: 10.1016/j.athoracsur.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Anker SD, Egerer KR, Volk HD, et al. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol. 1997;79:1426–1430. doi: 10.1016/s0002-9149(97)00159-8. [DOI] [PubMed] [Google Scholar]
- 8.Secchi A, Ortanderl JM, Schmidt W, et al. Effect of endotoxemia on hepatic portal and sinusoidal blood flow in rats. J Surg Res. 2000;89:26–30. doi: 10.1006/jsre.1999.5811. [DOI] [PubMed] [Google Scholar]
- 9.Krack A, Sharma R, Figulla HR, et al. The importance of the gastrointestinal system in the pathogenesis of heart failure. Eur Heart J. 2005;26:2368–2374. doi: 10.1093/eurheartj/ehi389. [DOI] [PubMed] [Google Scholar]
- 10.Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105:2619–2624. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- 11.Sandek A, Bauditz J, Swidsinski A, et al. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol. 2007;50:1561–1569. doi: 10.1016/j.jacc.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Sandek A, Anker SD, von Haehling S. The gut and intestinal bacteria in chronic heart failure. Curr Drug Metab. 2009;10:22–28. doi: 10.2174/138920009787048374. [DOI] [PubMed] [Google Scholar]
- 13.Anker SD, Ponikowski P, Varney S, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–1053. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 14.Arutyunov GP, Kostyukevich OI, Serov RA, et al. Collagen accumulation and dysfunctional mucosal barrier of the small intestine in patients with chronic heart failure. Int J Cardiol. 2008;125:240–245. doi: 10.1016/j.ijcard.2007.11.103. [DOI] [PubMed] [Google Scholar]
- 15.Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–1842. doi: 10.1016/S0140-6736(98)09286-1. [DOI] [PubMed] [Google Scholar]
- 16.Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 17.Genth-Zotz S, von Haehling S, Bolger AP, et al. Pathophysiologic quantities of endotoxin-induced tumor necrosis factor-alpha release in whole blood from patients with chronic heart failure. Am J Cardiol. 2002;90:1226–1230. doi: 10.1016/s0002-9149(02)02839-4. [DOI] [PubMed] [Google Scholar]
- 18.Sharma R, Bolger AP, Rauchhaus M, et al. Cellular endotoxin desensitization in patients with severe chronic heart failure. Eur J Heart Fail. 2005;7:865–868. doi: 10.1016/j.ejheart.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Deswal A, Petersen NJ, Feldman AM, et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST) Circulation. 2001;103:2055–2059. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 20.Vaz Pérez A, Doehner W, von Haehling S, et al. The relationship between tumor necrosis factor-alpha, brain natriuretic peptide and atrial natriuretic peptide in patients with chronic heart failure. Int J Cardiol. 2010;141:39–43. doi: 10.1016/j.ijcard.2008.11.146. [DOI] [PubMed] [Google Scholar]
- 21.Bozkurt B, Kribbs SB, Clubb FJ, Jr, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 22.Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;104:826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 23.von Haehling S, Schefold JC, Lainscak M, et al. Inflammatory biomarkers in heart failure revisited: much more than innocent bystanders. Heart Fail Clin. 2009;5:549–560. doi: 10.1016/j.hfc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Lundvig DM, Immenschuh S, Wagener FA. Heme oxygenase, inflammation, and fibrosis: the good, the bad, and the ugly? Front Pharmacol. 2012;3:81. doi: 10.3389/fphar.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alcaraz MJ, Fernández P, Guillén MI. Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharm Des. 2003;9:2541–2551. doi: 10.2174/1381612033453749. [DOI] [PubMed] [Google Scholar]
- 26.Guillén M, Megías J, Gomar F, et al. Haem oxygenase-1 regulates catabolic and anabolic processes in osteoarthritic chondrocytes. J Pathol. 2008;214:515–522. doi: 10.1002/path.2313. [DOI] [PubMed] [Google Scholar]
- 27.Megías J, Guillén MI, Clérigues V, et al. Heme oxygenase-1 induction modulates microsomal prostaglandin E synthase-1 expression and prostaglandin E(2) production in osteoarthritic chondrocytes. Biochem Pharmacol. 2009;77:1806–1813. doi: 10.1016/j.bcp.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Madias JE. Standard electrocardiographic and signal-averaged electrocardiographic changes in congestive heart failure. Congest Heart Failure. 2005;11:266–267. doi: 10.1111/j.1527-5299.2005.04484.x. [DOI] [PubMed] [Google Scholar]
- 29.Cao J, Vecoli C, Neglia D, et al. Cobalt-Protoporphyrin improves heart function by blunting oxidative stress and restoring NO synthase equilibrium in an animal model of experimental diabetes. Front Physiol. 2012;3:160. doi: 10.3389/fphys.2012.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen TM, Li J, Liu L, et al. Effects of heme oxygenase-1 upregulation on blood pressure and cardiac function in an animal model of hypertensive myocardial infarction. Int J Mol Sci. 2013;14:2684–2706. doi: 10.3390/ijms14022684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Botros FT, Schwartzman ML, Stier CT, Jr, et al. Increase in heme oxygenase-1 levels ameliorates renovascular hypertension. Kidney Int. 2005;68:2745–2755. doi: 10.1111/j.1523-1755.2005.00745.x. [DOI] [PubMed] [Google Scholar]
- 32.Ozkan KU, Ozokutan BH, Inanç F, et al. Does maternal nicotine exposure during gestation increase the injury severity of small intestine in the newborn rats subjected to experimental necrotizing enterocolitis. J Pediatr Surg. 2005;40:484–488. doi: 10.1016/j.jpedsurg.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Lu S, Ge J, et al. Increased heme oxygenase-1 expression in infarcted rat hearts following human bone marrow mesenchymal cell transplantation. Microvasc Res. 2005;69:64–70. doi: 10.1016/j.mvr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Pachori AS, Melo LG, Zhang L, et al. Chronic recurrent myocardial ischemic injury is significantly attenuated by pre-emptive adeno-associated virus heme oxygenase-1 gene delivery. J Am Coll Cardiol. 2006;47:635–643. doi: 10.1016/j.jacc.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 35.Yang JJ, Yang X, Liu ZQ, et al. Transplantation of adipose tissue-derived stem cells overexpressing heme oxygenase-1 improves functions and remodeling of infarcted myocardium in rabbits. Tohoku J Exp Med. 2012;226:231–241. doi: 10.1620/tjem.226.231. [DOI] [PubMed] [Google Scholar]
- 36.Mallick IH, Winslet MC, Seifalian AM. Ischemic preconditioning of small bowel mitigates the late phase of reperfusion injury: heme oxygenase mediates cytoprotection. Am J Surg. 2010;199:223–231. doi: 10.1016/j.amjsurg.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Morisaki H, Katayama T, Kotake Y, et al. Carbon monoxide modulates endotoxin-induced microvascular leukocyte adhesion through platelet-dependent mechanisms. Anesthesiology. 2002;97:701–709. doi: 10.1097/00000542-200209000-00025. [DOI] [PubMed] [Google Scholar]
- 38.Gende OA. Carbon monoxide inhibits capacitative calcium entry in human platelets. Thromb Res. 2004;114:113–119. doi: 10.1016/j.thromres.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Onyiah JC, Sheikh SZ, Maharshak N, et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology. 2013;144:789–798. doi: 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clérigues V, Murphy CL, Guillén MI, et al. Haem oxygenase-1 induction reverses the actions of interleukin-1beta on hypoxia-inducible transcription factors and human chondrocyte metabolism in hypoxia. Clin Sci (Lond) 2013;125:99–108. doi: 10.1042/CS20120491. [DOI] [PubMed] [Google Scholar]
- 41.Flierl MA, Rittirsch D, Nadeau BA, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449:721–725. doi: 10.1038/nature06185. [DOI] [PubMed] [Google Scholar]
- 42.Pautz A, Art J, Hahn S, et al. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Beck PL, Xavier R, Wong J, et al. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am J Physiol Gastrointest Liver Physiol. 2004;286:G137–147. doi: 10.1152/ajpgi.00309.2003. [DOI] [PubMed] [Google Scholar]
- 44.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 45.Chatterjee A, Black SM, Catravas JD. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul Pharmacol. 2008;49:134–140. doi: 10.1016/j.vph.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moilanen E, Vapaatalo H. Nitric oxide in inflammation and immune response. Ann Med. 1995;27:359–367. doi: 10.3109/07853899509002589. [DOI] [PubMed] [Google Scholar]
- 47.Lowry JL, Brovkovych V, Zhang Y, et al. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J Biol Chem. 2013;288:4174–4193. doi: 10.1074/jbc.M112.436022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.López A, Lorente JA, Steingrub J, et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Mao K, Zeng Y, et al. Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J Immunol. 2010;185:7699–7705. doi: 10.4049/jimmunol.1001099. [DOI] [PubMed] [Google Scholar]
- 50.Mao K, Chen S, Chen M, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23:201–212. doi: 10.1038/cr.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomes RN, Teixeira-Cunha MG, Figueiredo RT, et al. Bacterial clearance in septic mice is modulated by MCP-1/CCL2 and nitric oxide. Shock. 2013;39:63–69. doi: 10.1097/SHK.0b013e31827802b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linares E, Giorgio S, Mortara RA, et al. Role of peroxynitrite in macrophage microbicidal mechanisms in vivo revealed by protein nitration and hydroxylation. Free Radic Biol Med. 2001;30:1234–1242. doi: 10.1016/s0891-5849(01)00516-0. [DOI] [PubMed] [Google Scholar]
- 53.Brown JD, Lin CY, Duan Q, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]