

Abstract
We developed a novel poly(lactic-co-glycolic acid)-based, microparticle (MP) system providing concurrent delivery of multiple encapsulated immuno-suppressive factors and antigen, for in vivo conditioning of dendritic cells (DCs) toward a tolerance promoting pathway. Subcutaneous administration prevents onset of type 1 diabetes (T1D) in NOD mice. Two MP sizes were made: phagocytosable MPs were fabricated encapsulating vitamin D3 or insulin B(9-23) peptide, while unphagocytosable MPs were fabricated encapsulating TGF-β1 or GM-CSF. The combination of Vit D3/ TGF-β1 MPs confers an immature and LPS activation-resistant phenotype to DCs, and MP-delivered antigen is efficiently and functionally presented. Notably, two subcutaneous injections into 4 week old NOD mice using the combination of MPs encapsulating Vit D3, Ins B, TGF-β1 and GM-CSF protected 40% of mice from T1D development, significant in comparison to the control. This work represents one of the first applications of a biomaterial-based, MP vaccine system to successfully prevent autoimmune diabetes.
Keywords: microparticle, vaccine, PLGA, dendritic cells, type 1 diabetes, NOD, tolerance, biomaterial, microsphere
1. Introduction
Type 1 diabetes (T1D) in both humans and non-obese diabetic (NOD) mice results from a breakdown of self-tolerance that is characterized by T cell-mediated β-cell destruction [1]. Ultimately, glucose metabolism is interrupted and even when exogenous insulin is provided, serious complications can develop such as heart disease, renal failure and ketoacidosis [2]. Recently, this failure in homeostatic regulation of autoimmune responses has been attributed to a deficiency in the number and/ or function of dendritic cells (DCs) and regulatory T cells (Tregs). Boudaly et al. demonstrated that DCs derived from the NOD strain of mice are deficient not only in quantity, but also in phenotype and functional capacity compared to control strains [3]. Additionally, Ohnmacht and associates established that constitutive ablation of DCs in mice result in lack of protection from spontaneous fatal autoimmunity [4]. These findings are consistent with a central role for DCs in the maintenance of peripheral self-tolerance. Dendritic cells circulate throughout the body, sampling the extracellular environments and relaying these local conditions back to effector and regulatory cells in the secondary lymphoid organs [5]. Moreover, these cells are professional antigen presenting cells (APCs) with the capacity to instigate either inflammatory or anti-inflammatory adaptive immunity. The direction and magnitude of the response is influenced by DC phenotype– either an activated phenotype providing an inflammatory reaction, or conversely, a tolerogenic phenotype for regulatory measures [5]. Dendritic cells influence peripheral immune tolerance via a number of modalities including effector T cell anergy and deletion, immune deviation, and the expansion and induction of regulatory T cells (Tregs) [6–8]. Other reports have detailed that Tregs control diabetogenic autoimmunity by restraining the actions of pathogenic T cells at the site of inflammation (i.e., β-islet cells) subsequent to initial infiltration [9]. Notably, DCs with an ‘immature’ phenotype and other tolerogenic features have be identified as inducers of the CD25+ FoxP3+ T cells that are capable of protection from the onset of T1D in NOD mice [10].
These features of DCs have motivated dendritic cell-based immunotherapies for the treatment of autoimmune diseases, including T1D, where patient-derived DCs are manipulated ex vivo and then reintroduced to the patient as a cellular vaccine [11]. Ex vivo manipulation can involve genetic modification of DCs, or exposure to immunosuppressive agents, as well as antigen [12–15]. This personalized immunotherapy is currently in clinical trials for treatment of T1D in humans [16, 17]. However, issues such as consistency and stability of DC phenotype, and exceedingly high manufacturing costs have restricted the widespread application of this therapeutic approach [18].
An emerging strategy focuses on in vivo targeting and conditioning of DCs with injectable, synthetic particulate systems that can deliver vaccine components including immunomodulatory agents. This approach greatly simplifies issues related to manufacturing, storage, and shipping as biomaterial encapsulation provides vaccine stability and good shelf-life [11]. Additionally, microparticle (MP) systems can be engineered to simultaneous deliver both prime & boost doses using time-release materials (e.g., poly lactide-co-glycolide; PLGA). Microparticle systems can be modular and multifunctional, can specifically target DCs, and provide both intracellular and extracellular delivery of immunomodulatory agents [19–21]. Factors of particular interest are the potent immunosuppressive agents – 1α, 25-dihydroxycholecalciferol (Vit D3), transforming growth factor beta-1 (TGF-β1) and granulocyte macrophage colony-stimulating factor (GM-CSF). 1α, 25-dihydroxycholecalciferol is the active metabolite of vitamin D3. It is a steroid hormone that plays an integral role in bone formation as it regulates calcium/ phosphate metabolism [22]. Vitamin D3 also has immunomodulatory effects, particularly on APCs and T cells that highly express the vitamin D3 receptor [22]. In DCs, multiple studies have revealed that Vit D3 impairs DC maturation, with reduced expression of major histocompatibility complex (MHC) II, co-stimulatory molecules (CD40, CD80, CD86) and other maturation markers. Additionally, expression of inflammatory cytokines such as IL-12, are significantly suppressed in DCs when treated with Vit D3 [23, 24].
Transforming growth factor beta-1 is produced and secreted in a latent form by an array of lymphoid cells, particularly DCs and T cells [25]. Interestingly, these cells are not only sources for TGF-β1, but also targets of action for this immunosuppressive cytokine. The scope of impact of TGF-β1 on DCs is still being discovered, but it has been demonstrated TGF-β1 immunomodulatory effects are inhibitory in nature and lead to a tolerogenic DC phenotype that is capable of inducing CD25+ FoxP3+ T cells (induced Tregs) from CD+4 naïve T cell populations [26, 27]. Notably, TGF-β1 treated DCs produce indoleamine 2,3 deoxgenase (IDO), an enzyme involved in tryptophan catabolism and responsible for the generation of kynurenines – thought to be key factors in the spread of ‘infectious tolerance’ [28, 29]. Additionally, granulocyte macrophage colony stimulating factor (GM-CSF) is another pleiotropic cytokine impacting a number of immune cells, particularly APCs. Granulocyte macrophage colony stimulating factor is secreted by peripheral tissues under pathologic conditions and influences DC recruitment, phagocytic activity, antigen presentation capacity and proliferation [45]. Further, Gaudreau et al demonstrated that GM-CSF treated semi-mature DCs play an integral role in prevention of T1D in NOD mice and further, suggest that they induce IL-10 secreting CD4+ CD25+ Tregs that suppress diabetogenic T cells that promote diabetes development [30, 31].
Herein, we exploit the versatility of PLGA to develop a dual MP system which delivers a combinatorial therapy to locally recruit DCs, supply particulate autoantigen, and provide tolerance-promoting immune modulation. This dual MP system is comprised of the combination of two phagocytosable MPs – Vit D3-loaded MPs and antigen-loaded MPs (insulin B[9-23] peptide), as well as two unphagocytosable MPs – TGF-β1-loaded MPs and GM-CSF-loaded MPs.
2. Materials and Methods
2.1 Experimental animals
Female NOD/Ltj, NOD-BDC2.5, C57Bl/6 and Balb/c mice, ages 4 – 8 weeks, were purchased from either The Jackson Laboratories (Bar Harbour, ME) or The University of Florida Animal Care Services (ACS) (Gainesville, FL). All animals were housed in specific free-environment conditions in University of Florida ACS facilities and used in accordance with detailed experimental protocols approved by The University of Florida Institutional Animal Care and Use Committee (IACUC).
2.2 Microparticle preparation
A 50:50 polymer composition of poly(d lactide-co-glycolide) (PLGA) (MW ~ 65,000 g/mol) in methylene chloride (Purac) was used to generate microparticles (MPs). Poly-vinyl alcohol (PVA) (MW ~ 100,000 g/mol) was purchased from Fisher Scientific (NJ, USA) and was used as an emulsion stabilizer. Distilled water (DiH2O) was used as the aqueous phase to form the emulsions while methylene chloride (Fisher Scientific, NJ, USA) was used as the organic solvent to dissolve PLGA polymer. Microparticles were formed using a standard oil-water emulsion solvent evaporation techniques.
Phagocytosable MPs were fabricated using either single- or double-emulsion solvent evaporation techniques based on the solubility of the desired drug in organic solvent. Briefly, 100 mg of PLGA polymer was dissolved in methylene chloride at 5% w/v ratio. Vitamin D3 (MP Biomedical, CA, USA) in methanol (Fisher Scientific, NJ, USA) was loaded into 2 ml of 5% PLGA solution. This solution was added to 2 ml of 5% PVA solution in DiH2O and homogenized at 35,000 rpm for 180 s using a tissue-miser homogenizer (Fisher Scientific, NJ, USA) to form a primary emulsion. This was added to 30 ml of 1% PVA solution. The particles thus formed were agitated using a magnetic stirrer (Fisher Scientific, NJ, USA) for 24 h to evaporate residual methylene chloride. The remaining solution was centrifuged at 10,000 × g for 10 minutes to collect MPs which were subsequently washed three times with DiH2O. The water was aspirated from the centrifuged MPs, which were then flash-frozen in liquid nitrogen and kept under vacuum in dry ice overnight. The MPs were stored at −20 °C until their use.
For insulin B peptide-encapsulated (Anaspec Inc., CA, USA) MPs, 100 mg of PLGA polymer was dissolved in methylene chloride at 5% w/v ratio. 0.1 ml of Insulin B peptide solution (1mg/ ml) was then added the 5% PLGA solution and homogenized at 35,000 rpm for 120 s to form a primary emulsion. This emulsion was added to 2 ml of a 5% PVA solution and homogenized again at 19,500 rpm for 60 s to form the secondary emulsion which was transferred to a beaker containing 30 ml of 1% PVA. The particles thus formed were agitated using a magnetic stirrer (Fisher Scientific, NJ, USA) for 24 h to evaporate residual methylene chloride. The remaining solution was centrifuged at 10,000 x g for 10 minutes to collect MPs which were subsequently washed three times with DiH2O. The water was aspirated from the centrifuged MPs, which were then flash-frozen in liquid nitrogen and kept under vacuum in dry ice overnight. The MPs were stored at −20 °C until their use.
Unphagocytosable MPs encapsulating TGF-β1- and GM-CSF were fabricated using by a double emulsion solvent evaporation technique similar to that described above but using a vortexter (Fisher Scientific) instead of the tissue-miser homogenizer. The TGF-β1 (EMD Millipore, MA, USA) solution was reconstituted in 10 mM citric acid and 2 mg/ml bovine serum albumin in PBS to a final concentration of 50μg/ml. For GM-CSF (Thermo Scientific, MA, USA), the lyophilized powder was reconstituted in DiH2O to a concentration of 100 μg/ ml. Either DMSO or distilled water was used to generate unloaded MPs, depending on the control group being fabricated.
2.3 Microparticle characterization – sizing, loading
The size distributions of MPs were measured by the Beckman Coulter LS13320 (Beckman Coulter Inc., Brea, CA) and the Microtrac Nanotrac Dynamic Light Scattering Particle Analyser (Microtrac, Montgomery, PA). The MP diameter is reported as mean ± standard deviation (SD). The loading efficiency of Vit D3 MPs was measured by dissolving 100 mg of MPs into 2 ml MC and re-precipitating the PLGA with a known volume of methanol (Acros Organics). The suspension was centrifuged and the supernatant removed to a new tube. Following evaporation, residue remaining in the tube is concentrated in a known, small quantity of DMSO and the solution concentration measured by spectrophotometer. For the other MPs (Ins B, TGF-β1, GM-CSF), loading efficiency was measured using a solvent evaporation technique followed by spectrophotometric analysis.
2.4 Bone marrow-derived DC culture and MP incubation
Dendritic cells were obtained from 8 – 12 week old, female, C57BL6/j mice in accordance with guidelines approved by University of Florida using a modified 10 day protocol. For DC culture, mice were euthanized by CO2 asphyxiation followed by cervical dislocation and tibias and femurs harvested for isolating marrow cells. The marrow cells were obtained by flushing the shaft of the bones with a 25g needle using RPMI medium (MP Biomedicals, OH, USA) containing 1% fetal bovine serum (Lonza, Walkersville, MD) and 1% penicillin-streptomycin (Hyclone) and mixed to make a homogenous suspension. The suspension was then strained using 70 μm cell strainers (Becton Dickinson, NJ, USA) and cells collected by centrifugation at 1300 rpm for 7 min. The red blood cells (RBCs) were removed by lysing with ACK lysis buffer (Lonza, Walkersville, MD) followed by centrifugation at 1500 rpm for 5 min to recover leucocytes. Leucocytes were then re-suspended in DMEM/F-12 with L-glutamine (Cellgro, Herndon, VA), 10% fetal bovine serum, 1% sodium pyruvate (Lonza, Walkersville, MD), 1% non-essential amino acids (Lonza, Walkersville, MD), 1% penicillin-streptomycin (Hyclone) and 20 ng/ml GM-CSF (R&D systems, MN, USA) (DC media) and plated on tissue culture flasks for 2 days in order to remove adherent cells. At day 2 the floating cells were transferred to low attachment plates and cultured in fresh DC media for expansion of DC precursor cells. At day 7, cells were transferred to tissue culture plates to allow for DC adhesion and proliferation. At day 10, they were lifted from tissue culture plates and used for various studies [32]. For these studies, MPs were incubated at 37 °C for a period of 48 h prior to analysis or addition of T cells. Phagocytosable MPs (Vit D3 MP) were added at a 10:1 MP to cell ratio, while unphagocytosable MPs (TGF-β1 MP) were incubated at a mass that encapsulated the effective concentration of that respective drug for the incubation media volume. Unloaded MPs and the soluble equivalent of released drugs doses were included as controls.
2.5 Bone marrow-derived DC phenotype - maturation markers
Dendritic cell maturation was quantified by measuring cell surface marker levels by flow cytometry. Following MP incubation, DCs were lifted by incubating with a 5 mM Na2EDTA in PBS solution at 37 °C for 20 min. Dendritic cells were then washed with 1% fetal bovine serum in PBS and incubated with antibodies against CD16/CD32 (Fcγ III/II Receptor; clone 2.4G2, IgG2b, κ; BD Pharmingen, CA, USA) for 15 min at 4 °C to block Fcγ receptors on DCs. Cells were washed and then stained with antibodies against CD80 (clone 16-10A1, IgG2, κ), CD86 (clone GL1, IgG2a, κ), I-A/I-E (clone M5/114.15.2, IgG2b, κ), and CD11c (clone HL3, IgG1, λ2) for 30 min at 4 °C. Appropriate isotypes were used for each antibody species as negative controls. Data acquisition was performed using (Guava Easycyte Flowcytometer, EMD Millipore, Darmstadt, Germany) flow cytometry and the geometric fluorescent intensities as well as percent of positively stained cells determined. More than 10,000 events were acquired for each sample and data analysis was performed using FCS Express version 3 (De Novo Software, Los Angeles, CA).
2.6 CD4+ T cell Isolation
Mouse CD4+ T cells were purified from splenocyte suspensions by negative selection using Miltenyi CD4+ T cell isolation kit II following the manufacturer’s instructions. The purity of CD4+ T cells as determined by flow cytometry was in the 90 – 92% range.
2.7 Mixed lymphocyte coupling – allogeneic T cell suppression
For suppression studies, bone marrow-derived C57BL6 DCs (2.5 × 104/ well) were co-incubated with the VitD3 MP/ TGF-β1 MP combination, as well as the relevant soluble and MP control treatments, in a 96 well tissue culture plate for 48 h at 37 °C in culture media. After thoroughly washing away all unphagoctyosed and unbound MPs, BALB/c CD4+ T cells (1.25 × 105/ well) were added to each well and incubated at 37 °C for 3 d. Bromodeoxyuridine (BrdU) (Beckton Dickinson) was pulsed into the culture media for the last 4 h. T cells were then immunofluorescently stained for BrdU according to manufacturer’s specifications. Flow cytometry was then used to quantify T cell proliferation for the different treatments.
2.8 Antigen presentation by DC after microparticle uptake
Non-obese diabetic mouse-derived DCs (2.5 × 104/ well) were co-incubated with 1040-55 mimotope peptide (Peptides International, Louisville, KY) -loaded MPs, as well as the relevant control treatments, in a 96 well tissue culture plate for 4 h at 37 °C. MPs outnumbered NOD DCs by 10:1. After thoroughly washing away all un-phagocytosed and unbound MPs, NOD-BDC2.5 CD4+ T-cells (1.25 × 105/ well) were added to each well and incubated at 37 °C for 3 d. Bromodeoxyuridine (BrdU) (kit from Beckton Dickinson) was added to the culture for the last 4 h. T-cells were then immunofluorescently stained for BrdU according to manufacturer’s specifications. Flow cytometry was then used to quantify T cell proliferation for the different treatments.
2.9 Diabetes prevention study
A cohort of 4 week old, female NOD mice was divided in 5 groups (n = 13). The groups were given MP formulation injections as follows: Group A - Unloaded MPs only; Group B - GM-CSF MPs + Ins B MPs; Group C – Vit D3 MPs + TGF-β1 MPs + Ins B MPs; Group D – Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs; Group E – Ins B MPs. All injections consisted 10 mg (1:1:1:1 MP mass ratio) of MPs in 0.2 ml PBS (Unloaded MPs were added to control formulations where there was an omitted factor, in order to deliver equivalent PLGA MP mass across groups). Mice were injected twice at a subcutaneous site anatomically proximal to the pancreatic lymph nodes, once at 4 weeks old, then again at five weeks of age. The blood glucose levels of 10 randomly selected mice from each group were then monitored once weekly for the next 27 weeks. Mice were diagnosed as diabetic, once the blood glucose level was over 240 mg/dl for two consecutive days.
2.10 Midpoint mechanistic studies – histopathology, antigen presenting cell and Treg analysis
At 12 weeks of age, 3 mice for each treatment group were randomly removed from the study, sacrificed and organs (spleen, pancreas) excised for analyses at this time point. Histopathology: Pancreata were fixed with formalin, embedded using paraffin, sectioned, mounted and stained with Hematoxylin and Eosin (H&E). Stained sections were blind scored for Islet infiltration using the following grading system: 0 = healthy islet, 1 = peri-insulitis, 2 = >25% leukocytic infiltration of islet area, and 3 = >75% leukocytic infiltration of islet area. At least 50 islets were examined for each group. APC Maturation and Differentiation: Using the cell surface staining protocol described above, splenocytes (1 × 106 cells from each mouse) were stained with antibodies against mouse CD11c (clone N418, IgG), CD11b (M1/70, IgG2b, κ) (eBiosciences) and Gr-1 (clone RB6-8C5, IgG2b, κ). Cells were analyzed using the FCS express V3 software (De Novo Software, Los Angeles, CA). Treg Analysis: Splenocytes (1 × 106 cells from each mouse) were immunofluorescently stained using antibodies against CD4 (clone RM4-5, IgG2a, κ) (BD Pharmingen), and FoxP3(clone FJK-16s, IgG2a, κ) (eBioscience). Cells were analyzed using the FCS express V3 software (De Novo Software, Los Angeles, CA).
2.11 Statistical Analysis
Statistical analyses were performed using general linear nested model ANOVA, followed by posthoc assessment using Tukey test to make pair-wise comparisons. Differences were considered significant when p ≤ 0.05 using Systat (Version 12, Systat Software, Inc., San Jose, CA), except for comparisons shown in Figure 6B,C for which p ≤ 0.1 was used. For the diabetes prevention studies, statistical analysis of survival curves was done in SYSTAT using Kaplan-Meier Non-parametric Survival analysis model. P values were determined by comparisons with the ‘Unloaded MP’ treatment group.
Figure 6. Regulatory cell frequency in microparticle (MP) -vaccinated NOD mice.

A cohort of NOD mice (n = 3/ group) were given two subcutaneous injections of MP formulations, at 4 and 5 weeks of age. The MP vaccine system consisted of both unphagocytosable (30 μm) and phagocytosable (1 μm) MPs as described. At 12 weeks, mice were sacrificed, spleens excised and processed into single cell suspensions. Single cell suspensions were stained for different regulatory cell subsets – including (A) Tregs, (B) Gr1+ dendritic cells (DCs) and (C) Gr1+ macrophages (Mϕs) and analyzed using flow cytometry. Data shown represent the mean percent positive cells ± standard error (n = 3). Pair-wise significant difference from the Unloaded MP group (by ANOVA and Tukey significance test) is denoted by the * symbol for p value ≤ 0.05 and the ** symbol for p value ≤ 0.1.
3. Results
3.1 Fabrication and characterization of two size class MP system
Microparticles loaded with the desired factors were successfully fabricated using either single or double emulsion (depending on solubility of the factor), solvent evaporation techniques for phagocytosable MPs (Vit D3, Ins B). A double emulsion, solvent extraction method was employed for the unphagocytosable MPs (TGF-β1, GM-CSF). These MPs were characterized to determine their size distribution and agent encapsulation efficiency. The size range of phagocytosable MP was determined by dynamic light scattering, calculated by volume, to be approximately 0.5 μm – 2.5 μm, and an average diameter of approximately 1 μm, as shown on the size distribution graph (Figure 1A). On the other hand, the average diameter for the TGF-β1 MPs and GM-CSF MPs, as determined by light diffraction measurements using a Coulter Counter, was approximately 30 μm, ranging from 2 μm to 140 μm (Figure 1A). Shown in Figure 1B are encapsulation efficiencies of each of the MP formulations. The encapsulation efficiencies for the phagocytosable MPs were determined to be 80 ± 7.0% and 50 ± 0.1% for the Vit D3 MPs and Ins B MPs, respectively. Similar encapsulation efficiencies were observed for the unphagocytosable MPs at approximately 62% for TGF-β1 MPs and 72% for GM-CSF MPs.
Figure 1. Physicochemical traits of microparticles (MPs).
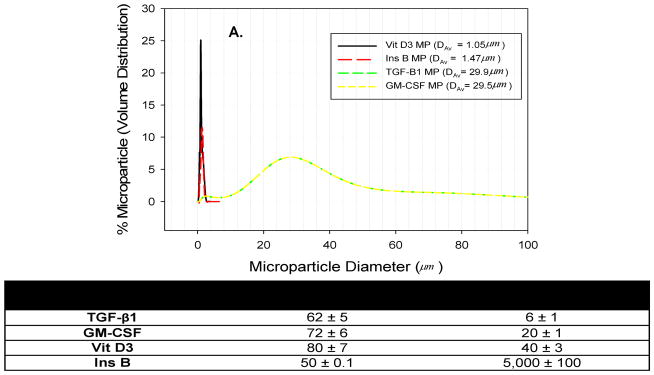
(A) Microparticle size determined using dynamic light scattering techniques for phagocytosable MPs (average diameter: Vit D3 MP ~1 μm; insulin B peptide MP ~ 1μm) and unphagocytosable MPs (average diameter: TGF-β1 MPs ~ 30 μm). (B) Encapsulation efficiencies and loading amounts for MPs loaded with pharmacological and biological agents are shown.
3.2 Vit D3/TGF-β1 MPs modulate DC phenotype and suppress allogeneic T cell proliferation
The immunosuppressive ability of the Vit D3/TGF-β1 combinatorial MP system was evaluated in vitro by determining their effects on DC maturation and downstream suppressive power. It should be noted that GM-CSF MPs were not included in these series of in vitro experiments as GM-CSF was already contained in the media as a growth factor required for maintaining DCs in culture. Following incubation with MPs, the expression was quantified for activation markers of stimulatory (MHC II) and costimulatory molecules (CD80, CD86), which are integral for the formation of the DC-T cell immune synapse, DC production of cytokines, and clonal expansion of specific T cell populations. Vit D3 MPs in combination with TGF-β1 MPs were co-cultured with DCs for 48 h, and control groups were included: untreated immature DCs (iDCs), unloaded MPs, single factor MP controls and soluble lipopolysaccharide (LPS) treatment used a positive control. Lipopolysaccharide is recognized by the toll-like receptor 4 and typically leads to a mature DC phenotype characterized by increased production of stimulatory molecules (e.g., CD80, 86) and inflammatory cytokines (e.g., IL-12) [33, 34]. To compare between treatments, the percent of DCs expressing a given marker was normalized by the expression of the control group, iDC (Figure 2A). The combined Vit D3 MPs and TGF-β1 MPs (and soluble GM-CSF) on DC phenotype was found to exhibit MHC II, CD80 and CD86 expression at levels equivalent to the iDC control group, and in stark contrast to the significantly higher expression levels observed for the LPS control. Maintenance of low levels of surface activation markers for the Vit D3/ TGF-β1 MP-treated DCs indicates an immature DC phenotype. The single factor MP controls – the Vit D3 MP only group, and the TGF-β1 MP only group, also maintained low expression of activation markers. Interestingly, the Vit D3 MP only treatment significantly lowered the expression of MHC II, whilst exposure of DCs to the TGF-β1 MPs only treatment resulted in a considerable decrease in CD80 expression. Importantly, exposure to the PLGA vehicle (unloaded MPs) had no effect on DC phenotype, indicating that the biomaterial did not affect DC activation.
Figure 2. Dual-sized microparticle (MP) system maintains immature dendritic cells which suppress allogeneic T cell activation.

(A) Dual-sized MP system maintains immaturity of DCs. DCs were incubated 48 hr with MP formulations: 1 μm MPs loaded with (Vit D3), 30 μm MPs loaded with TGF-β1, the dual MP system (Vit D3 MPs and/ or TGF MPs), or unloaded MPs. Immature DCs (iDC) and soluble LPS groups were included for comparison. Cells were immunostained for activation markers MHC-II, CD86, and CD80. The percent positively-stained cells were quantified by flow cytometry. (B) Suppression of allogeneic T cells and resistance to maturation stimuli by DCs treated with dual-sized MP system. After pre-incubating C57Bl/6 DCs 48 hr with MP formulations, allogeneic, Balb/cyj CD4+ T-cells were then co-cultured (ratio of 1:5, DCs:T-cells) and this mixed lymphocyte reaction was cultured for 5 d. Cultures were pulsed with BrdU for 4 hr and then fixed, permeablized, immuno-stained for BrdU incorporation (for proliferation) and CD4 (for T-cells), and analyzed by flow cytometry. Pair-wise significant difference from the immature DC (iDC) group (by ANOVA and Tukey significance test) is denoted by the * symbol (p value ≤ 0.05)
To further investigate the effects of treatment on DC function, we evaluated the ability of the combination Vit D3/ TGF-β1 MP-treated DCs to suppress the proliferative responses of allogeneic T cells (Figure 2B). Dendritic cells (from C57BL/6 mice) were cultured under experimental and control conditions for 2 d, and cells were washed to remove free MPs. Dendritic cells were then co-cultured with purified, allogeneic splenic CD4+ T cells (from BALB/c mice) for 5 d. At which point, the percent of proliferating T cells was measured. We found that following incubation with the combined Vit D3 and TGF-β1 MPs, DCs potently inhibited proliferation of CD4+ T cells to levels significantly below that of the iDC control. Impressively, Vit D3/ TGF--β1 MP-treated DCs in the presence of soluble LPS, also similarly suppressed the proliferation of T cells in response to allogeneic stimulus. These results indicate the combination of Vit D3 and TGF-β1 MPs modulate DCs toward an immature phenotype that limits allogeneic T cell activation and is robustly resistant to LPS maturation.
3.3 Dendritic cell presentation of MP-encapsulated antigen
An antigen-specific T cell proliferation assay was employed to demonstrate the functional presentation of MP-encapsulated antigen by NOD DCs. NOD DCs were cultured with either MPs loaded with 1040-55 mimotope peptide or with soluble 1040-55 mimotope peptide (as a positive control, 3 μM) at a 10:1 MP to DC ratio for 4 h, followed by washing to remove free MPs. Subsequently, freshly-isolated BDC2.5 CD4+ T cells were added to culture wells and co-cultured for 3 d. T-cell proliferation was then measured using a BrdU proliferation assay as a measure of functional antigen presentation. The NOD-BDC2.5 mouse is engineered with T-cell receptors that are specifically engaged by the 1040-55 mimotope. Upon binding the 1040-55 peptide (when bound to MHC II molecules presented on APCs), NOD-BDC2.5 T-cells are stimulated to proliferate. Results shown in Figure 3 demonstrate that DCs exposed to 1040-55-loaded MPs are capable of stimulating antigen-dependent T cell proliferation to levels comparable to that of direct loading of peptide on DC MHC II complexes via soluble administration. This outcome highlights the ability of MPs encapsulating antigen to be effectively taken up by NOD DCs and to be functionally presented to T cells.
Figure 3. Presentation of microparticle (MP)-encapsulated antigen.

Non-obese diabetic mouse dendritic cells (DCs) were cultured with either MPs loaded with mimotope 1040-55 peptide or unloaded MPs at a 10:1 MP to DC ratio for 24 h, or with soluble 1040-55 peptide (as a positive control), followed by washing to remove unbound MPs. Subsequently, freshly-isolated BDC2.5 CD4+ T cells were added to culture wells and co-cultured 3 d. T-cell proliferation was then measured using a BrdU proliferation assay as a measure of functional antigen presentation. (A) Data shown represent the mean percent of BrdU positive T cells ± standard deviation (n = 3). Pair-wise significant difference from the immature DC (iDC) group (by ANOVA and Tukey significance test) is denoted by the * symbol (p value ≤ 0.05). (B) Representative dot plots for the different treatment groups are also shown.
3.4 Early treatment with dual MP system prevents the onset of T1D in NOD mice
The above in vitro observations supported investigating the efficacy of this dual MP system to mitigate the onset of diabetes in NOD mice. In this study, 4 week old, female NOD mice were injected twice subcutaneously in the ventral, abdominal region (anatomically proximal to the pancreatic lymph nodes), once at 4 weeks old followed by a booster at five weeks of age, with MP formulations consisting of either – A) Unloaded MPs (control), B) GM-CSF MPs + Ins B MPs, C) Vit D3 MPs + TGF-β1 MPs + Ins B MPs, D) Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs, and E) Ins B MPs. Injections consisted of a total mass of 10 mg of MPs in a 1:1:1:1 mass ratio of the four MP types. This MP dose (10 mg in 200 μl) was chosen based on the considerations of i.) providing a highly concentrated MP suspension which yields minimal loss of MPs upon injection, while ii.) using a high subcutaneous injection volume of 200 μl for mice, which is retained subcutaneously without leaking. Furthermore, where prior controlled release results were available, consideration was given to amount of factor required to elicit immune cell responses in vivo. In particular, Ali et al., previously demonstrated that PLG scaffolds releasing a total of ~50 ng of GM-CSF resulted in in vivo DC recruitment [35], which we recapitulated by delivering that total amount in two injections of GM-CSF-loaded MPs. While this formulation and dosing was chosen for initial studies, there is potential for improvement. The total mass of PLGA MPs was normalized across all groups, by substituting unloaded MPs to make up the deficit in mass for the control groups with omitted factors. The site of injection was chosen based on a previous report which demonstrated that administration of phagocytosable MPs in this site led to MP trafficking to the pancreatic draining lymph nodes by antigen presenting cells [36]. Mice were monitored for diabetes over a 32 week period. Administration of unloaded MPs (control) failed to protect mice, with only 10% of mice from this group remaining disease-free at the end of the study (Figure 4), a result consistent with other reports on diabetes incidence in female NOD mice [37]. Results statistically equivalent to the control group were obtained for three other groups, the GM-CSF MPs + Ins B MPs formulation, the Vit D3 MPs + TGF-β1 MPs + Ins B MPs formulation, and the Ins B MPs formulation, yielding non-diabetic incidences of 20%, 30% and 20%, respectively, at the completion of the study. However, administration of the full complement of MPs (Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs) prevented onset of diabetes in 40% of mice tested. Kaplan-Meier analysis for this treatment group reveals a p-value of 0.025 in comparison to survival proportions for the unloaded MP group. Additionally, the mean survival time for this treatment group was 24 weeks, substantially longer than the 19 week mean survival time for the control group (Figure 4, table).
Figure 4. Combination dual-sized microparticle (MP) system prevents type 1 diabetes in 4 week old NOD mice.

A cohort of NOD mice (n=10 per group) were given two subcutaneous injections of MP formulations at 4 and 5 weeks of age (indicated by arrows). The MP vaccine system consisted of both unphagocytosable (30 μm) and phagocytosable (1 μm) MPs. GM-CSF or TGF-β1 was encapsulated in unphagocytosable MPs while Vit D3 or insulin peptide B:9-23 (antigen) was encapsulated in phagocytosable MPs. The MP vaccine system consisted of these four MPs mixed in an equal 1:1:1:1 mass ratio. Blood glucose levels were monitored through to 32 weeks of age. A blood glucose level over 240 mg/dL for two consecutive days was diagnosed as diabetic. Survival data is fit using the Kaplan-Meier Non-parametric Survival Analysis Model and statistical analysis performed via log-rank test (Mantel method).
In order to characterize the extent of β-cell destruction following immunization with the dual MP system, pancreata from 12 week old, non-diabetic NOD mice, immunized as previously described, were examined histologically. As indicated in Figure 5A, the majority of mice treated with the Vit D3 MPs + TGF-β1 MPs + Ins B MPs group, or the Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs group, particularly the former, were either minimally inflamed or had mild peri-insulitis. In contrast, over 60% of islets from mice that were injected with only unloaded MPs (control treatment) had a much higher level of immune cell infiltration. Similar levels of islet infiltration were observed for mice immunized with the GM-CSF MPs + Ins B MPs group and the Ins B MPs only group, with approximately 55% being highly inflamed. These results are consistent with the indication that the Vit D3/ InsB/ TGF-β1/ GM-CSF dual MP system is capable of affecting the diabetogenic process in the NOD mouse. However, while the insulitis results are broadly similar with the prevention results, some of the MP formulation groups demonstrate small inconsistencies between the prevention and insulitis results. For instance, the best formulation for diabetes prevention – the Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs group, differs from the formulation which showed the least insulitis – the Vit D3 MPs + TGF-β1 MPs + Ins B MPs group, which lacks GM-CSF MPs. Notably, incongruence between prevention and insulitis has been reported extensively, where mononuclear infiltration of islets has been shown (in both T1D animal models and humans) to be often inconsistent with β-cell destruction, and may not be the best clinical marker for autoimmune diabetes [38–41].
Figure 5. (A) Insulitis scoring of mouse pancreata.

A cohort of NOD mice (n = 3/ group) were given two subcutaneous injections of microparticle (MP) formulations, at 4 and 5 weeks of age. The MP vaccine system consisted of both unphagocytosable (~ 30 μm) and phagocytosable (~ 1 μm) MPs as described. At 12 weeks, mice were sacrificed, pancreata excised, processed, sectioned and stained with hematoxylin and eosin (H&E). Sections were examined and scored for insulitis in a blinded fashion (0 = no insulitis; 1 = peri-insulitis; 2 = 25 – 75% insulitis; 3 >75% insulitis). (B) The number of islets examined, as well as assigned scores for each group is displayed in tabular form.
3.5 Administration of dual MP system in NOD mice stimulates increased frequency of regulatory cells
The frequency of regulatory cells, Tregs and Gr1 suppressor cells, at 8 weeks post-treatment was determined. A number of reports have linked type 1 diabetes progression with decreasing numbers of functional FoxP3+ regulatory T cells [9, 42]. To ascertain whether Treg numbers were altered, we enumerated the percentage of FoxP3-expressing, CD4+ T cells in the splenocyte population from NOD mice in the prevention treatment groups. Flow cytometric analysis revealed that the percent of splenic Tregs were conserved compared to the unloaded MP control when mice were treated with the GM-CSF MPs + Ins B MPs formulation, and the Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs formulation (Figure 6A). On the other hand, a significant increase in the percent of FoxP3+ CD4+ T cells was observed when mice were treated with the Vit D3 MPs + TGF-β1 MPs + Ins B MPs formulation. Additionally, expression of Gr1 was evaluated on splenic APCs (CD11b+ and CD11c+ cells, representing macrophages and DCs respectively). Studies have shown that Gr1+ monocytes, derived from the bone marrow, can differentiate into CD11b+ and CD11c+ cells in the presence of GM-CSF and TGF-β1, with immunosuppressive capabilities to prevent autoimmune diabetes in 4 week old NOD mice [43]. Treatment with the Vit D3 MPs + TGF-β1 MPs + Ins B MPs formulation promoted an increase in levels of splenic CD11c Gr1+ cells (Figure 6B) in comparison to mice treated with the unloaded MPs control group, at a lower level of significance (p ≤ 0.1). Similarly, an increase in splenic Gr1+ CD11b+ cells was also observed for the Vit D3 MPs + TGF-β1 MPs + Ins B MPs group in comparison to the control group (Figure 6C), also at a lower level of significance (p ≤ 0.1).
4. Discussion
Antigen-specific, dendritic cell-based immunotherapy is a growing strategy being developed for treatment of autoimmune diabetes [1],[44]. However, due to a number of major limitations, such as the clinical grade ex vivo manipulation of cells, DC-based immunotherapy faces major hurdles to achieve widespread application [11]. Alternatively, formulation of immuno-active agents in synthetic biodegradable microparticles (MPs) offers an approach to provide in vivo conditioning of dendritic cells for immunomodulatory applications. In this regard, we demonstrate the utility of immuno-active, factor-loaded, PLGA MPs as a vaccine which suppresses type 1 diabetes. In this study, we developed a dual microparticle vaccine system that significantly protects NOD mice from the onset of T1D, upon early immunization. This dual MP system is comprised of two size classes of MPs: 1) phagocytosable MPs for intracellular delivery – containing either of Vit D3 or antigen (insulin B peptide) and 2) unphagocytosable MPs for extracellular release – containing either of TGF-β1 or GM-CSF; the aim being to develop an injectable system that delivers factors combinatorially to modulate DCs. Others, including Phillips et al. and Yeste et al., have reported on the formulation of bioactive agents into polymeric particulate matter as a means of modulating DC phenotype and ultimately, curtailing autoimmunity in mice [36, 45]. However, this dual MP vaccine system represents the first multi-modal platform to deliver multiple pharmaceutical and biological agents, and antigen simultaneously to mitigate auto-antigen reactivity.
The principal outcome of these studies is the capability of a combinatorial formulation of PLGA MPs encapsulating antigen (insulin B peptide), 1α 2,5-dihydroxyvitamin D3, GM-CSF and TGF-β1 to suppress autoimmune diabetes in female NOD mice immunized at 4 weeks of age. The latter three factors have been previously evaluated individually by various groups, including ours, for their effects on DC motility, phenotype and suppressive capabilities. For instance, Ali et al. demonstrated that GM-CSF is a powerful recruiter of CD11c+ cells [35]. The pleiotropic cytokine TGF-β1, has been implicated as an effective chemoattractant of immune cells including DCs and Mϕs, and is considered one of the most potent tolerance-inducing, biological soluble mediators [46, 47]. Similarly, in DCs, 1α 2,5-dihydroxyvitamin D3 binding impairs DC maturation, with reduced expression of MHC II, co-stimulatory molecules (CD40, CD80, CD86) and other maturation markers [24]. Our hypothesis was that combined exposure of these agents, along with auto-antigen, to DCs would generate antigen-specific, immunosuppressive dendritic cells capable of downregulating diabetogenic responses. The dual sized MP system was conceptualized to provide local, subcutaneous controlled release of GM-CSF and TGF-β1 (via unphagocytosable MPs) to cell surface receptors while also simultaneously targeting particulate auto-antigen and vitamin D3 to intracellular compartments of antigen presenting cells (via phagocytosable MPs). Use of phagocytosable MPs can promote delivery of 1α 2,5-dihydroxyvitamin D3, which is membrane permeable, to its receptor, found in the nuclear compartment of DCs. We have previously demonstrated that PLGA MPs (~1 μm in diameter), injected subcutaneously, are readily phagocytosed by DCs and macrophages (Mϕs) and translocated to draining LNs within 48 h [32]. Moreover, others have reported that subcutaneous injection of similarly-sized particles results in trafficking to the spleen and proximal lymph nodes, by DCs and other phagocytic cells in NOD mice [36]. Conversely, large, unphagocytosable MPs (diameter > 10 μm) are too large to leave the subcutaneous injection site, and thus release their contents into the surrounding extracellular milieu, in a manner capable of recruiting and conditioning DCs [48].
Given these prior observations, we fabricated phagocytosable MPs containing either Vit D3 or Ins B, and unphagocytosable MPs containing either TGF-β1 or GM-CSF. Herein, our data first demonstrates that we can reliably fabricate these two size types of PLGA MPs, individually loaded with appreciable quantities of respective immuno-active agents. Further, through in vitro testing, we elucidated the effects of these immuno-modulatory factors encapsulated in MPs, singly and in combination on DCs. Because GM-CSF was always included in vitro in the media as a required supplement to support DC cultures, GM-CSF MPs were not incorporated in the in vitro studies. Predictably, DC exposure to combinations of these bioactive agents in MP form resulted in reduced expression of MHCII, CD80 and CD86. And when all three immune modulating factors (Vit D3, TGF-β1 and GM-CSF) are present, expression levels for these activation molecules were equivalent to untreated, immature DCs. Furthermore, the combination MP-treated DCs robustly limited the extent of proliferation of allogeneic T cells, supporting the possibility for T cell anergy as one plausible mechanism for the dampening of immunity. Moreover, the Vit D3/ TGF-β1 MPs formulation provided resistance to LPS maturation, which is advantageous because sites of autoreactivity, like inflamed pancreatic islets can stimulate activation of immature DCs [49]. Antigen specific T cell proliferation studies demonstrated the ability of DCs to take up MP-encapsulated antigen, incorporate it into MHC II complexes and functionally present to T cells, in vitro. This is an important consideration for antigen-specific DC therapy. Therapies providing exogenous auto-antigen have been effective for autoimmune diseases, and have established safety [45, 50, 51] On the other hand, the possibility exists for heterogeneity in the particles which are taken up, wherein a fraction of DCs may be recruited to the injection site or nearby, become modulated by the released factors, but not simultaneously take up antigen-loaded MPs. In this case, such modulated DCs may also potentially migrate to the pancreatic lymph node and gain antigen-specificity as a consequence of auto-antigen drainage from the islets, as was hypothesized by Phillips et al. [36], in the case where effective treatment for autoimmune diabetes was realized without the exogenous provision of antigen.
The most significant result is the demonstration that delivery of the combination dual-sized MP system encapsulating immunosuppressive agents and antigen can effectively halt autoimmune diabetes in mice predisposed to develop the disease. A cohort of four week old, female NOD mice were randomized into groups, and vaccinated with the MP formulations, followed by a booster shot at 5 weeks of age. At this stage, NOD mice show no clinical symptoms of diabetes onset, and variations can exist in the glucose tolerance of individual mice. Therefore, randomization and large cohorts are used to average out the biological variability associated with the NOD mouse model, which allows significant differences in treatment arms to be detected.
The survival proportion for female, NOD mice treated with the Vit D3 MPs + TGF-β1 MPs + GM-CSF MPs + Ins B MPs formulation is 40%, significantly greater than the 10% observed for the control group. While other treatment groups had a higher proportion of protected mice than the control group, only when all the immuno-active factors and auto-antigen was provided was there a significant prevention of autoimmune diabetes progress and substantially longer survival time, suggesting that all factors are necessary. Although further investigation involving more treatment groups is required to comprehensively evaluate the system, this result is notable, especially given that only two doses were administered. Other studies achieving similar results gave a greater number of doses of the respective treatments [30, 50, 52]. Insulitis scoring results for MP treatments, while broadly consistent with prevention data, discordance in specific MP groups adds to the growing body of literature suggesting insulitis may not be the best clinical marker for autoimmune diabetes [38–41].
In an effort to gain some insight into mechanisms underlying disease prevention in treated NOD mice, we surveyed different regulatory cell populations. While the percent of splenic regulatory cells were conserved compared to the unloaded MP control when mice were treated with most MP formulations, only treatment with the Vit D3 MPs + TGF-β1 MPs + Ins B MPs formulation increased Tregs, CD11c Gr1+, and Gr1+ CD11b+ cells at the single time point examined, and then only modestly. Clearly, a number of other regulatory mechanisms could be at play, and it is possible that tracking regulatory dynamics over multiple time points and physiologic locations will be required to determine specific mechanisms involved. Other considerations include issues such as the biodistribution of the components, whether in particulate or fully degraded and fully released form. For instance, competition by macrophages invading the injection site and taking up MPs, could lessen the impact of DCs on immune modulation, the effect of which is unclear. In moving forward, improving efficacy will be prioritized, followed by elucidation of the cellular compartments involved in conferring protection. Given the demonstrated capabilities of the combination dual-sized MP system, and tunability of the controlled release parameters, it is likely that the treatment could be optimized in terms of the loading of immuno-active agents, antigen selection, dose, injection schedule, and active targeting of DCs for improved diabetes prevention.
5. Conclusion
We encapsulated multiple immunosuppressive factors as well as antigen into a dual-sized polymeric microparticle platform. In vitro, this combination MP formulation promotes a DC phenotype which strongly suppresses allogeneic T cells, even upon LPS stimulation. Most importantly, protection from T1D was observed in NOD mice vaccinated with the full complement of MPs. However, the need for elucidation of the precise in vivo mechanism(s) involved in disease protection remains. Taken altogether, these results represent the first iteration of a biodegradable platform with tremendous potential to mitigate the effects of T1D.
Highlights.
microparticles encapsulating multiple factors are fabricated in two size classes
microparticles encapsulate vitamin D3, insulin B peptide, TGF-β1 or GM-CSF
microparticle formulations maintain dendritic cell immature state
microparticle treated dendritic cells suppress allogeneic T cell proliferation
microparticles prevent T1D in NOD mice when administered at 4 and 5 weeks of age
Acknowledgments
The efforts herein were supported by the NIH NIDDK (R01DK098589, R01 DK091658 – to B.G.K.) and NIAID (AI42288 – to M.A.A.; R21AI094360 – to B.G.K.), and The Leona M. and Harry B. Helmsley Charitable Trust (to M.A.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Santamaria P. The Long and Winding Road to Understanding and Conquering Type 1 Diabetes. Immunity. 2010;32(4):437–445. doi: 10.1016/j.immuni.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: Etiology, presentation, and management. Pediatric Clinics of North America. 2005;52(6):1553. doi: 10.1016/j.pcl.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Boudaly S, Morin J, Berthier R, Marche P, Boitard C. Altered dendritic cells (DC) might be responsible for regulatory T cell imbalance and autoimmunity in nonobese diabetic (NOD) mice. European Cytokine Network. 2002;13(1):29–37. [PubMed] [Google Scholar]
- 4.Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. Journal of Experimental Medicine. 2009;206(3):549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. 2000. [DOI] [PubMed] [Google Scholar]
- 6.Cobbold SP, Nolan KF, Graca L, Castejon R, Le Moine A, Frewin M, Humm S, Adams E, Thompson S, Zelenika D, Paterson A, Yates S, Fairchild PJ, Waldmann H. Regulatory T cells and dendritic cells in transplantation tolerance: molecular markers and mechanisms. Immunological Reviews. 2003;196(1):109–124. doi: 10.1046/j.1600-065x.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 7.Penna G, Giarratana N, Amuchastegui S, Mariani R, Daniel KC, Adorini L. Manipulating dendritic cells to induce regulatory T cells. Microbes and Infection. 2005;7(7–8):1033–1039. doi: 10.1016/j.micinf.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Turnquist HR, Thomson AW. Taming the lions: manipulating dendritic cells for use as negative cellular vaccines in organ transplantation. Current Opinion in Organ Transplantation. 2008;13(4):350–357. doi: 10.1097/MOT.0b013e328306116c. [DOI] [PubMed] [Google Scholar]
- 9.Chen ZB, Herman AE, Matos M, Mathis D, Benoist C. Where CD4(+) CD25(+) T reg cells impinge on autoimmune diabetes. Journal of Experimental Medicine. 2005;202(10):1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specitic, regulatory T cells that suppress autoimmunity. Seminars in Immunology. 2006;18(2):93–102. doi: 10.1016/j.smim.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Keselowsky BG, Xia CQ, Clare-Salzler M. Multifunctional dendritic cell-targeting polymeric microparticles Engineering new vaccines for type 1 diabetes. Human Vaccines. 2011;7(1):37–44. doi: 10.4161/hv.7.1.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita Y, Yang JM, Gupta R, Shimizu K, Shelden EA, Endres J, Mule JJ, McDonagh KT, Fox DA. Dendritic cells genetically engineered to express IL-4 inhibit murine collagen-induced arthritis. Journal of Clinical Investigation. 2001;107(10):1275–1284. doi: 10.1172/JCI11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SH, Kim S, Evans CH, Ghivizzani SC, Oligino T, Robbins PD. Effective treatment of established murine collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express IL-4. Journal of Immunology. 2001;166(5):3499–3505. doi: 10.4049/jimmunol.166.5.3499. [DOI] [PubMed] [Google Scholar]
- 14.Trepiakas R, Berntsen A, Hadrup SR, Bjorn J, Geertsen PF, Straten PT, Andersen MH, Pedersen AE, Soleimani A, Lorentzen T, Johansen JS, Svane IM. Vaccination with autologous dendritic cells pulsed with multiple tumor antigens for treatment of patients with malignant melanoma: results from a phase I/II trial. Cytotherapy. 2010;12(6):721–734. doi: 10.3109/14653241003774045. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen AE, Gad M, Walter MR, Claesson MH. Induction of regulatory dendritic cells by dexamethasone and 1 alpha,25-dihydroxyvitamin D-3. Immunology Letters. 2004;91(1):63–69. doi: 10.1016/j.imlet.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 16.ClinicalTrials.gov Identifier: NCT00445913. 2015. Ref Type: Generic
- 17.Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase 1 (Safety) Study of Autologous Tolerogenic Dendritic Cells in Type 1 Diabetic Patients. Diabetes Care. 2011;34(9):2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tacken PJ, de Vries I, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nature Reviews Immunology. 2007;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 19.Jain S, O’Hagan DT, Singh M. The long-term potential of biodegradable poly(lactide-co-glycolide) microparticles as the next-generation vaccine adjuvant. Expert Review of Vaccines. 2011;10(12):1731–1742. doi: 10.1586/erv.11.126. [DOI] [PubMed] [Google Scholar]
- 20.O’Hagan DT, Singh M, Gupta RK. Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Advanced Drug Delivery Reviews. 1998;32(3):225–246. [PubMed] [Google Scholar]
- 21.Jiang WL, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Advanced Drug Delivery Reviews. 2005;57(3):391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Baeke F, Van Etten E, Overbergh L, Mathieu C. Vitamin D(3) and the immune system: maintaining the balance in health and disease. Nutrition Research Reviews. 2007;20(1):106–118. doi: 10.1017/S0954422407742713. [DOI] [PubMed] [Google Scholar]
- 23.Penna G, Roncari A, Amuchastegui S, Daniel KC, Berti E, Colonna M, Adorini L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4(+)Foxp3(+) regulatory T cells by 1,25-dihydroxyvitamin D-3. Blood. 2005;106(10):3490–3497. doi: 10.1182/blood-2005-05-2044. [DOI] [PubMed] [Google Scholar]
- 24.Penna G, Adorini L. 1 alpha,25-dihydroxyvitamin D-3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. Journal of Immunology. 2000;164(5):2405–2411. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- 25.Mantel PY, Schmidt-Weber CB. Transforming Growth Factor-Beta: Recent Advances on Its Role in Immune Tolerance. 2011. [DOI] [PubMed] [Google Scholar]
- 26.Luo X, Tarbell KV, Yang H, Pothoven K, Bailey SL, Ding R, Steinman RM, Suthanthiran M. Dendritic cells with TGF-beta 1 differentiate naive CD4+CD25(-) T cells into islet-protective Foxp3(+) regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(8):2821–2826. doi: 10.1073/pnas.0611646104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Cheng EY, Sharma VK, Lagman M, Chang C, Song P, Ding R, Muthukumar T, Suthanthiran M. Dendritic Cells With TGF-beta 1 and IL-2 Differentiate Naive CD4+T Cells Into Alloantigen-Specific and Allograft Protective Foxp3+Regulatory T Cells. Transplantation. 2012;93(6):580–588. doi: 10.1097/TP.0b013e318244dd67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pallotta MT, Orabona C, Bianchi R, Vacca C, Fallarino F, Belladonna ML, Volpi C, Mondanelli G, Gargaro M, Allegrucci M, Talesa VN, Puccetti P, Grohmann U. Forced IDO1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. Journal of Cellular and Molecular Medicine. 2014;18(10):2082–2091. doi: 10.1111/jcmm.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belladonna ML, Orabona C, Grohmann U, Puccetti P. TGF-beta and kynurenines as the key to infectious tolerance. Trends in Molecular Medicine. 2009;15(2):41–49. doi: 10.1016/j.molmed.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Cheatem D, Ganesh BB, Gangi E, Vasu C, Prabhakar BS. Modulation of dendritic cells using granulocyte-macrophage colony-stimulating factor (GM-CSF) delays type 1 diabetes by enhancing CD4+CD25+regulatory T cell function. Clinical Immunology. 2009;131(2):260–270. doi: 10.1016/j.clim.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaudreau S, Guindi C, Besin G, Dupuis G, Amrani A. Granulocyte macrophage-colony stimulating factor (GM-CSF) induces precursors of bone marrow-derived dendritic cells in vivo to acquire a tolerogenic phenotype in NOD mice. Diabetologia. 2008;51:S228. [Google Scholar]
- 32.Lewis JS, Zaveri TD, Crooks CP, Keselowsky BG. Microparticle surface modifications targeting dendritic cells for non-activating applications. Biomaterials. 2012;33(29):7221–7232. doi: 10.1016/j.biomaterials.2012.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawai T, Akira S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity. 2011;34(5):637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 34.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunology. 2001;2(8):675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 35.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nature Materials. 2009;8(2):151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes. 2008;57(6):1544–1555. doi: 10.2337/db07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson MS, Bluestone JA. The NOD mouse: A model of immune dysregulation. 2005. [DOI] [PubMed] [Google Scholar]
- 38.Mcculloch DK, Klaff LJ, Schoenfeld SL, Greenbaum CJ, Benson EA, Palmer JP. Subclinical Beta-Cell Dysfunction Is Not Always Progressive Among 1St Degree Relatives of Type-I Diabetics - 5 Year Follow-Up of the Seattle Family Study. Clinical Research. 1989;37(1):A132. [Google Scholar]
- 39.Verge CF, Gianani R, Yu LP, Pietropaolo M, Smith T, Jackson RA, Soeldner JS, Eisenbarth GS. Late Progression to Diabetes and Evidence for Chronic Beta-Cell Autoimmunity in Identical-Twins of Patients with Type-I Diabetes. Diabetes. 1995;44(10):1176–1179. doi: 10.2337/diab.44.10.1176. [DOI] [PubMed] [Google Scholar]
- 40.Kolb H, Freytag G, Kiesel U, Kolbbachofen V. Spontaneous Autoimmune Reactions Against Pancreatic-Islets in Mouse Strains with Generalized Autoimmune-Disease. Diabetologia. 1980;19(3):216–221. doi: 10.1007/BF00275272. [DOI] [PubMed] [Google Scholar]
- 41.Flohr K, Kiesel U, Freytag G, Kolb H. Insulitis As A Consequence of Immune Dysregulation - Further Evidence. Clinical and Experimental Immunology. 1983;53(3):605–613. [PMC free article] [PubMed] [Google Scholar]
- 42.Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4(+)CD25(+) T-Cells in type 1 diabetes. Diabetes. 2005;54(5):1407–1414. doi: 10.2337/diabetes.54.5.1407. [DOI] [PubMed] [Google Scholar]
- 43.Steptoe RJ, Ritchie JM, Jones LK, Harrison LC. Autointmune diabetes is suppressed by transfer of proinsulin-encoding Gr-1(+) myeloid progenitor cells that differentiate in vivo into resting dendritic cells. Diabetes. 2005;54(2):434–442. doi: 10.2337/diabetes.54.2.434. [DOI] [PubMed] [Google Scholar]
- 44.Claresalzler MJ, Brooks J, Chai A, Vanherle K, Anderson C. Prevention of Diabetes in Nonobese Diabetic Mice by Dendritic Cell Transfer. Journal of Clinical Investigation. 1992;90(3):741–748. doi: 10.1172/JCI115946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wahl SM. Transforming growth factor-beta: innately bipolar. Current Opinion in Immunology. 2007;19(1):55–62. doi: 10.1016/j.coi.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Wahl SM, Hunt DA, Wakefield LM, Mccartneyfrancis N, Wahl LM, Roberts AB, Sporn MB. Transforming Growth-Factor Type-Beta Induces Monocyte Chemotaxis and Growth-Factor Production. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(16):5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. Journal of Materials Chemistry B. 2014;2(17):2562–2574. doi: 10.1039/C3TB21460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertera S, Alexander A, Giannoukakis N, Robbins PD, Trucco M. Immunology of type 1 diabetes - Intervention and prevention strategies. Endocrinology and Metabolism Clinics of North America. 1999;28(4):841. doi: 10.1016/s0889-8529(05)70105-5. [DOI] [PubMed] [Google Scholar]
- 50.Lo J, Clare-Salzler MJ. Dendritic cell subsets and type I diabetes: Focus upon DC-based therapy. Autoimmunity Reviews. 2006;5(6):419–423. doi: 10.1016/j.autrev.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, Reijonen H, Wolfsdorf J, Ricker A, Matthews JB, Tchao N, Sayre P, Bianchine P. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. Journal of Autoimmunity. 2010;34(4):408–415. doi: 10.1016/j.jaut.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karounos DG, Bryson JS, Cohen DA. Metabolically inactive insulin analog prevents type I diabetes in prediabetic NOD mice. Journal of Clinical Investigation. 1997;100(6):1344–1348. doi: 10.1172/JCI119654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Seminars in Immunology. 2008;20(2):86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]