

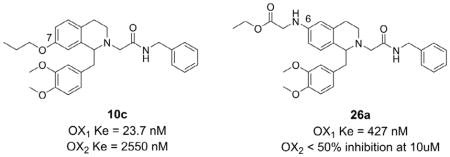
Abstract
Selective antagonism of the orexin 1 (OX1) receptor has been proposed as a potential mechanism for treatment of drug addiction. We have previously reported studies on the structure-activity relationships of tetrahydroisoquinoline-based antagonists. In this report, we elucidated the respective role of the 6- and 7-substitutions by preparation of a series of either 6-substituted tetrahydroisoquinolines (with no 7-substituents) or vice versa. We found that 7-substituted tetrahydroisoquinolines showed potent antagonism of OX1, indicating that the 7-position is important for OX1 antagonism (10c, Ke = 23.7 nM). While the 6-substituted analogs were generally inactive, several 6-amino compounds bearing ester groups showed reasonable potency (26a, Ke = 427 nM). Further, we show evidence that suggests several compounds initially displaying insurmountable antagonism at the OX1 receptor are competitive antagonists with slow dissociation rates.
Keywords: Orexin, antagonist, selective, tetrahydroisoquinoline
Graphical abstract
1. Introduction
Addiction is a chronic, often relapsing brain disease that causes compulsive drug seeking and use, despite harmful consequences. Drug abuse and addiction represent a major public health problem and a significant unmet medical need (www.drugabuse.gov). Unfortunately, there are few effective pharmacotherapies for addiction, and currently approved medications predominantly cover alcohol, nicotine and opiates.1 The orexin system consists of two neuropeptides, orexin-A and B (also known as the hypocretins), and two G protein-coupled receptors, orexin 1 and 2 (OX1 and OX2).2,3 While the orexin system has been extensively implicated in the regulation of arousal and sleep-wake cycles, OX1 in particular has more recently been considered a candidate target for addiction treatment.4–6 The orexins are produced in the hypothalamus and the orexin neurons project to brain regions implicated in incentive motivation, such as the dopamine producing neurons in the ventral tegmental area (VTA) and their afferents in the nucleus accumbens.7,8 Knockout mice lacking the prepro-orexin gene demonstrate reduced morphine-induced conditioned place preference, as well as reduced morphine- and cocaine-induced dopamine release.9–12 Moreover, selective blockade of the OX1 receptor has been shown to attenuate addictive behaviors such as cocaine-, alcohol- and morphine-seeking.13–16 Thus a selective OX1 antagonist would be a potentially useful therapeutic agent for drug addiction.
It is generally accepted that arousal is most closely associated with activation of the OX2 receptor and reward with OX1 receptor activation, although modulation of both receptors simultaneously may prove to be more effective than OX2 alone for sleep disorders.13,17 A number of orexin receptor antagonists that inhibit both receptors have been developed, including almorexant (1, Fig. 1) and suvorexant, the latter of which was approved by the FDA for the treatment of insomnia;18 however, much less has been done on developing OX1 selective antagonists. While the availability of the partially selective OX1 antagonist SB-334867 (~50 fold for OX1 over OX2) allowed the initial elucidation of the function of the OX1 receptor in drug abuse and addiction,19 recent studies suggest that OX2 may also play a role in certain aspects of reward and addiction. For instance, OX2 selective antagonists attenuated alcohol self-administration and conditioned place preference,20 as well as cue-induced nicotine reinstatement.21 On the contrary, OX2 antagonist TCS-OX2-29 (2) showed no effect on cocaine self-administration or cue-induced reinstatement.22 Together, these findings underscore the importance of the development of OX1 antagonists with improved selectivity in order to further probe the individual roles of the OX1 and OX2 receptors. Since the discovery of SB334867, several other OX1 receptor selective antagonists have been reported, but some OX2 activity still remains for most of these compounds (e.g. 3, ACT-335827).23,24
Figure 1.

Tetrahydroisoquinoline-based orexin antagonists
We previously reported our efforts in developing selective OX1 antagonists,25–27 in particular those based on the tetrahydroisoquinoline (THIQ) scaffold found in other orexin antagonists such as dual antagonist almorexant (1) and the OX2 selective antagonist TCS-OX2-29 (2). We have synthesized and pharmacologically evaluated a series of 6,7-disubsituted THIQs and identified several highly potent and selective OX1 antagonists such as RTIOX-276 (4).26 However, the highly electron rich aromatic rings with poly-methoxy groups present in these compounds may be susceptible to oxidative metabolism as these methoxy groups could be metabolically cleaved.28,29 While the dimethoxybenzyl group at the 1-position can be replaced with other aromatic groups that retain potency and OX1 selectivity,25 most orexin antagonists based on this scaffold share the 6,7-dimethoxy substitution pattern on the THIQ core.30 Moreover, the 6- and 7-positions seem to be highly sensitive to substitutions;26,31 therefore, the SARs around these positions need to be further probed. In this study, we examined the importance of substitution at the 6- and 7-positions of the THIQ core, respectively, and investigated the impact of removing one of the methoxy groups on orexin receptor activity.
2. Methods
2.1 Chemistry
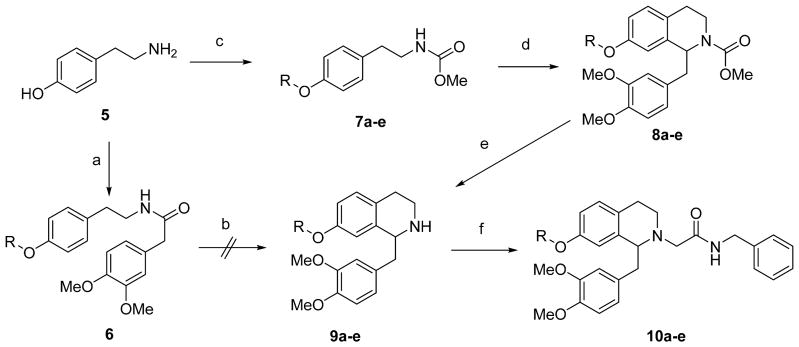
In the 6,7-dialkoxy series, analogs with electron deficient groups at the 7-position demonstrated excellent OX1 potency and selectivity.26 Therefore, we attempted to prepare 7-substituted analogs with substituents of different nature. However, the synthetic route previously used for 6,7-disubstituted analogs did not afford any desired products in the Bischler-Napieralski cyclization reaction on the appropriately 7-substituted amide 6 (R = H or alkyl), even with increased heat and time (Scheme 1). After protection of the tetrahydroisoquinoline nitrogen as the methyl carbamate followed by alkylation of the phenol with the desired substituents (7a–e), the tetrahydroisoquinoline 8a–e (R = alkyl) was obtained in 20–27% yield under modified Pictet-Spengler conditions in neat trifluoroacetic acid at 70 °C. The aldehyde used in this step was obtained via Dess-Martin oxidation from 2-(3,4-dimethoxyphenyl)ethanol. However, neither conditions gave any cyclization product with other electron-withdrawing substituents such as a trifluoroethyl group or the free phenol (R = H). Several protecting groups were investigated but none afforded any of the desired compounds. Removal of the methyl carbamate protecting group on 7a–e was also problematic. Several conditions were examined and the best conditions found were potassium hydroxide and hydrazine monohydrate in ethylene glycol at 120 °C (yields 32–68%). Finally, N-alkylation of 9a–e with N-benzylbromoacetamide gave the final compounds 10a–e.
Scheme 1.
Synthesis of 7-alkoxytetrahydroisoquinoline derivatives
Reagents and Conditions: (a) (i) 3,4-Dimethoxyphenylacetic acid, BOP, iPr2EtN, DMF; (ii) R-I or R-Br, K2CO3, DMF; (b) (i) POCl3, toluene; (ii) NaBH4, MeOH; (c) (i) MeOCOCl, iPr2EtN, CH2Cl2; (ii) R-I or R-Br, K2CO3, DMF; (d) 3,4-dimethoxyphenylacetaldehyde, TFA; (e) KOH, NH2NH2.H2O, (CH2OH)2; (f) BrCH2CONHBn, iPr2EtN, Bu4NI, DMF.
6-Substituted tetrahydroisoquinoline derivatives were synthesized by methods adapted from our earlier work,25,26 summarized in Schemes 2 and 3. The 6-alkoxy series first required synthesis of the phenylethylamines (Scheme 2). 3-Hydroxybenzaldehyde 11 was protected as the benzyl ether with benzyl bromide in the presence of potassium carbonate followed by a Henry reaction with nitromethane to give the nitrostyrene 12, which upon reduction with lithium aluminum hydride gave phenylethylamine 13.32 Coupling with 3,4-dimethoxyphenylacetic acid gave the amide 14, then Bischler-Napieralski cyclization followed by sodium borohydride reduction gave the tetrahydroisoquinoline 15. N-alkylation with N-benzylbromoacetamide followed by benzyl ether deprotection via transfer hydrogenation using palladium on carbon in the presence of ammonium formate gave 16. The phenol 16 was then elaborated via alkylation with alkyl halides in the presence of potassium carbonate (17a–f), sulfonylation with sulfonyl chlorides (18a–b) or acylation with acid chlorides (19).
Scheme 2.

Synthesis of 6-alkoxytetrahydroisoquinoline derivatives
Reagents and Conditions: (a) (i) BnBr, K2CO3, DMF; (ii) MeNO2, NH4OAc, AcOH; (b) LiAlH4, Et2O, CH2Cl2; (c) 3,4-dimethoxyphenylacetic acid, BOP, iPr2EtN, DMF; (d) (i) POCl3, toluene; (ii) NaBH4, MeOH; (e) (i) BrCH2CONHBn, iPr2EtN, Bu4NI, DMF; (ii) NH4HCOO, Pd/C, EtOH; (f) R-Br, K2CO3, Bu4NI, DMF or R-I, K2CO3, DMF; (g) RSO2Cl, Et3N, CH2Cl2; (h) RCOCl, iPr2EtN, CH2Cl2.
Scheme 3.

Synthesis of 6-aminotetrahydroisoquinoline derivatives.
Reactions and Conditions: (a) 3,4-dimethoxyphenylacetic acid, BOP, iPr2EtN, DMF; (b) (i) Raney Ni, NH2NH2.H2O, EtOH; (ii) MeOCOCl, iPr2EtN, CH2Cl2; (c) (i) POCl3, toluene; (ii) NaBH4, MeOH; (iii) BrCH2CONHBn, iPr2EtN, Bu4NI, DMF; (d) 2N NaOH (aq), MeOH; (e) aldehyde, Na(OAc)3BH, 1–2-DCE or alkyl halide, iPr2EtN, DMF; (f) (i) MsCl, Et3N, CH2Cl2; (ii) 2N NaOH (aq); (g) RCO2H, BOP, iPr2EtN, CH2Cl2 or RCOCl/(RCO)2O, iPr2EtN, CH2Cl2 or isocyanate, toluene.
For the 6-amino series (Scheme 3), 3-nitrophenylethylamine 20 was coupled with 3,4-dimethoxyphenylacetic acid to give amide 21. The nitro group was then reduced with Raney nickel and the aniline was protected as the methyl carbamate (22) using methyl chloroformate. Bischler-Napieralski reaction gave the dihydroisoquinoline which was readily reduced to the tetrahydroisoquinoline using sodium borohydride. The amine was alkylated with the bromoacetamide in the presence of DIPEA to give 23a and then the protecting group was removed by basic hydrolysis. Hydrolysis required extended heating time and an attempt to speed up this process in ethanol resulted in an improved reaction rate but also produced some transesterification product in the form of the ethyl carbamate 23b. This aniline 24 could then be elaborated further via reductive amination (25a–d, 25g–i, m, n, r–u) or base-catalyzed alkylation (25e, f, j–l, o–q, 26a–h), sulfonylation (27), acylation (28a–c) or urea formation (28d–e).
2.2. Biological Assays
Activity of the target compounds at the OX1 and OX2 receptors was evaluated in calcium mobilization based functional curve shift assays as previously described.25–27,33 In these assays, EC50 curves of the agonist orexin-A were obtained alone and together with the test compound (15 minute pre-incubation), respectively, and the right-shift of the agonist curve was measured. The apparent dissociation constant Ke was then calculated from compound-mediated inhibition of orexin A activity as previously described.25,26 The EC50 values for orexin A at OX1 and OX2 were 0.13 ± 0.02 nM and 4.2 ± 0.2 nM, respectively. All the compounds that had OX1 Ke values < 1 μM were also tested alone at 10 μM for agonist activity; none of the compounds showed any agonist activity at either receptor.
3. Results and Discussion
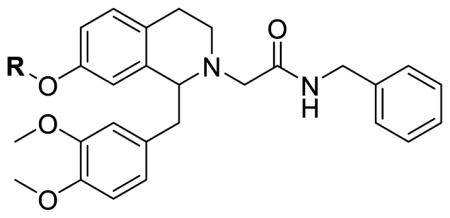
Given the importance of the 7-position substitutions for activity in the 6,7-disubstituted THIQs reported earlier, we first examined a series of compounds that are only substituted at the 7-position (Table 1). To this end, several 7-substituted alkoxy analogs were synthesized and tested. Because of the synthetic difficulties described above, other substitutions (e.g. electron withdrawing groups) were not examined. Compared to the 6,7-disubstituted analogs, the -7-substituted analogs showed similar activity at the OX1 receptor and three (10b, 10c, 10d) showed excellent selectivity against the OX2 receptor (>100-fold). The n-propyl derivative 10c was the most potent compound of the series, with a Ke value of 23.7 nM and was 108-fold more selective for OX1 over OX2. The ethyl analog 10b and isopropyl analog 10d were slightly less potent than 10c, with Ke values of 37.3 nM and 49.7 nM, respectively, but had higher OX1 selectivities (268- and 201-fold, respectively). The 7-methoxy analog 10a had a Ke of 512 nM, and was only two-fold less potent than compound 4. The previously described trend of potency first increasing then decreasing as the alkyl groups increase in size is also present in the 6,7-disubstituted analogs.26 Both ethyl 10b and isopropyl 10d were almost as potent as 10c, but potency decreased for the butyl analog 10e. These results clearly indicate that a substituent with appropriate size is preferred for activity at the 7-position.
Table 1.
7-Alkoxy tetrahydroisoquinoline orexin antagonists.
| |||
|---|---|---|---|
| No | R | OX1 Ke (nM)a | OX2 Ke (nM)a or % inhibition at 10 μM |
| 10a | Methyl | 512 ± 36 | 1680 ± 330 |
| 10b | Ethyl | 37.3 ± 7.8 | b |
| 10c | n-Propyl | 23.7 ± 7.9 | 2550 ± 260 |
| 10d | iso-Propyl | 49.7 ± 11 | b |
| 10e | n-Butyl | 230 ± 70 | b |
Ke values are the mean ± SEM of three independent experiments performed in duplicate. Results where Ke > 10 μM are N=2.
Less than 50% inhibition at 10 μM.
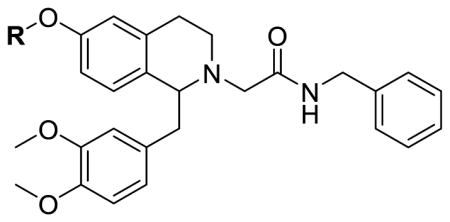
The importance of the 6-position had not been previously examined. Therefore, we prepared a series of analogs with different substituents to replace the methyl group at the 6-methoxy position, including both electron rich and electron deficient alkyl and aryl groups. As shown in table 2, these compounds were mostly inactive at the OX1 receptor, showing Ke values greater than 10 μM. Interestingly, the butanoate analog 17e showed a Ke of 1000 nM, and was only slightly less potent than the 6,7-dimethoxy analog 4, suggesting that the added ester group may have additional interaction with the receptor, or potentially occupy the same space of the 7-methoxy as in compound 4. When screened at the OX2 receptor for antagonist activity at 10 μM, these compounds showed little inhibition.
Table 2.
6-Alkoxy Tetrahydroisoquinoline Orexin Antagonists.
| |||
|---|---|---|---|
| No. | R | OX1 Ke (nM)a | OX2 Ke (nM)a or % inhibition at 10 μM |
| 17a | n-Propyl | >10000 | b |
| 17b | 2,2,2-Trifluoroethyl | >10000 | b |
| 17c | iso-Propyl | >10000 | b |
| 17d | Benzyl | >10000 | b |
| 17e |
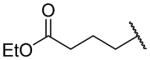
|
1000 ± 190c | b |
| 17f |
|
>10000 | b |
| 18a | Methylsulfonyl | >10000 | 1330 ± 260 |
| 18b | Benzenesulfonyl | >10000 | b |
| 19 | Propionoyl | >10000 | 1290 ± 130 |
Ke values are the mean ± SEM of three independent experiments performed in duplicate. Results where Ke > 10 μM are N=2.
Less than 50% inhibition at 10 μM.
Pre-incubation of antagonist and test compound was 1 hr.
We next prepared a series of 6-amino derivatives as shown in Table 3. Compared to the alkoxy series, the 6-position nitrogen can be di-substituted; one of those substituents may be able to take up similar space as the 7-position substituents and therefore make up for the potency lost by removal of the 7-substituent. Given the difficulties faced in the synthesis of the 7-substituted analogs, these 6-amino analogs may provide an alternative way to improve the potency. The 6-alkylamino analogs showed greater potency than the 6-alkoxy series, although mostly in the low micromolar range. The potencies in general were improved by capping the nitrogen with a methyl group (25b vs. 25c; 25f vs. 25g), though dialkylation gave similar potency to monoalkylation (25b vs. 25d). Larger alkyl groups (25r–u) gave no detectable potency, as was the case with the cyclic piperidinyl group 25q, though the slightly smaller pyrrolidinyl 25p showed some potency. The benzyl substituent showed the opposite effect; the monobenzyl derivative 25h had a Ke of 1100 nM but this was lost with the methylated analog 25i. Extending the linker to the phenethyl 25j and the phenylpropyl 25k showed little change. Pyridylmethyl substituents 25m and 25n showed a decline in potency, indicating that a basic nitrogen was not favorable but was tolerated for the 3-pyridyl, though making ethylpiperidinyl 25o caused a loss of detectable potency. The alkyl ester series (26a–e) showed some activity, with Ke values ranging from 427 to 949 nM, with the exception of the branched analog 26b, suggesting that there is limited space. Reinforcing that idea is the drop in potency for the longer alkyl esters 26f and 26g. Replacing the alkyl ester with an alkyl amide 26h caused a large decline in potency, and all the amide derivatives (28a–c) showed no detectable activity. The methyl carbamate 23a showed slight potency, though the ethyl carbamate 23b was not active, while the sulfonamide 27 and ureas 28d and 28e showed no significant potency. This may be due to a lack of flexibility in the side chain, giving unfavorable interactions with the binding site.
Table 3.
6-Amino tetrahydroisoquinoline orexin antagonists.
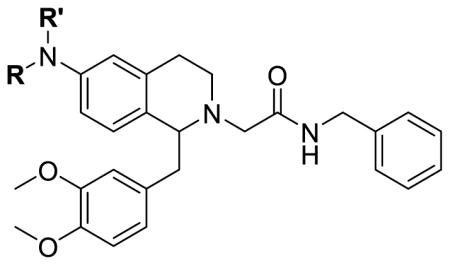
| ||||
|---|---|---|---|---|
| No. | R | R′ | OX1 Ke (nM)a | OX2 Ke (nM)a |
| 25a | Methyl | Methyl | 1370 ± 100 | b |
| 25b | n-Propyl | H | >10000 | b |
| 25c | n-Propyl | Methyl | 619 ± 96 | b |
| 25d | n-Propyl | n-Propyl | 2350 ± 490 | b |
| 25e | iso-Propyl | H | >10000 | b |
| 25f | Cyclopropylmethyl | H | >10000 | b |
| 25g | Cyclopropylmethyl | Methyl | 1400 ± 240 | b |
| 25h | Benzyl | H | 1100 ± 290 | b |
| 25i | Benzyl | Methyl | >10000 | b |
| 25j |
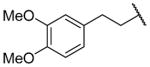
|
H | 2180 ± 360c | b |
| 25k | 3-Phenylpropyl | H | 1180 ± 80 | b |
| 25l | 4-Phenoxybutyl | H | 2080 ± 20 | b |
| 25m | 4-Pyridylmethyl | H | >10,000 | b |
| 25n | 3-Pyridylmethyl | H | 1270 ± 110 | b |
| 25o |
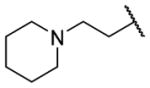
|
H | >10000 | b |
| 25p | Pyrrolidine | 2480 ± 6 | b | |
| 25q | Piperidine | >10000 | b | |
| 25r | n-Pentyl | H | >10000 | b |
| 25s | n-Pentyl | Methyl | >10000 | b |
| 25t | n-Hexyl | H | >10000 | b |
| 25u | n-Hexyl | Methyl | >10000 | b |
| 26a |
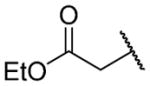
|
H | 427 ± 69 | b |
| 26b |
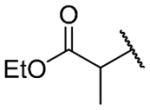
|
H | >10000 | b |
| 26c |
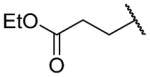
|
H | 809 ± 200 | b |
| 26d |
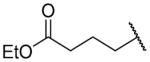
|
H | 591 ± 67 | >10000 |
| 26e |
|
H | 1010 ± 200d | b |
| 26f |
|
H | 1600 ± 190 | b |
| 26g |
|
H | >10000 | b |
| 26h |
|
H | >10000 | b |
| 27 | Methylsulfanyl | H | >10000 | b |
| 28a | Acetyl | H | >10000 | b |
| 28b | Hexanoyl | H | >10000 | >10000 |
| 28c | Benzoyl | H | >10000 | b |
| 23a | Methyl carbamoyl | H | 2400 ± 320 | >10000 |
| 23b | Ethyl carbamoyl | H | >10000 | >10000 |
| 28d | PhNHCO | H | >10000 | b |
| 28e | n-Hexyl-NHCO | H | 5230 ± 680 | b |
Ke values are the mean ± SEM of three independent experiments performed in duplicate. Results where Ke > 10 μM are N=2.
Less than 50% inhibition at 10 μM.
Pre-incubation of antagonist and receptor was 45 min.
Pre-incubation of antagonist and receptor was 1 hr.
Interestingly, while most of the compounds tested in the calcium mobilization curve shift assays displayed competitive antagonism, including the most potent compound 10c, several of the 6-substituted analogs demonstrated insurmountable antagonism under the testing conditions, such as 17e, 25j and 26e. They not only shifted the orexin agonist curve to the right as expected with antagonists, but also suppressed the maximal orexin response. Almorexant has been known to behave as an insurmountable antagonist at the OX2 receptor.34 While it is commonly observed in allosteric modulation due to the nature of the indirect interaction between allosteric ligands and orthosteric agonists, insurmountable antagonism can also be the result of competitive orthosteric antagonists with slow dissociations rates. One way to help distinguish between these two mechanisms involves performing curve shift assays as described above with longer antagonist-receptor incubation periods, which allows for the system to reach equilibrium.35 Steiner and co-workers used another method to distinguish between allosteric and competitive orthosteric antagonists by performing calcium mobilization curve shift experiments with simultaneous addition of antagonist and Orexin A as described in a recent publication.36 To help elucidate the mechanism of antagonism for the insurmountable antagonists in our study, we tested both methods with compounds 17e, 25j and 26e (Figure 2). The 15 minute pre-incubation clearly depicts insurmountable antagonism for all three compounds. As the pre-incubation times were increased to 45 min and 60 min, the compounds appeared increasingly to be competitive antagonists. Interestingly, all three compounds also appeared to be competitive antagonists under the simultaneous addition. Thus, with our compounds, both methods appear to distinguish between allosteric and competitive orthosteric antagonists.
Figure 2.

Effect of incubation time on calcium mobilization of compounds 17e, 25j and 26e in OX1 cells. Each data point represents the composite of at least three individual experiments performed in duplicate.
4. Conclusions
In conclusion, we prepared a series of 6- and 7-mono-substituted THIQ derivatives to investigate the contribution of the 6- and 7-positions to OX1 potency. The 7-substituted analogs with alkyl groups indeed showed potency at the OX1 receptor comparable to the corresponding 6,7-disubstituted analogs, though synthetic challenges made further SAR exploration difficult. The 6-substituted analogs were also examined and the observed Ke values were only modest, in contrast to the much more active 6,7-disubstituted derivatives. However, several 6-position analogs with terminal ester groups (26a, 26c, 26d) showed some activity at the OX1 receptor, potentially introducing additional interactions between the ester group and the receptor or alternatively, occupying the same space of the 7-methoxy as in compound 4. It is clear that the 7-substituent is the dominant position for determining potency in this series, though some contribution remains from the 6-substituent, as a slight drop in potency is observed as compared with the 6-methoxy analogs. Several 6-substituted compounds displayed insurmountable antagonism at the OX1 receptor under the assay conditions (15 min pre-incubation). Under increased pre-incubation time (45 min and 1 hour) or simultaneous addition of antagonist and Orexin A, these compounds appeared to be competitive antagonists, indicating that the insurmountable antagonism observed with a 15 min pre-incubation is most likely the result of slow dissociation rates.
5. Experimental Procedures
General
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all were purchased from commercial vendors and used as received. Flash column chromatography was done on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used were hexane, ethyl acetate (EtOAc), dichloromethane (DCM), methanol and chloroform:methanol:ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds was established by a combination of high pressure liquid chromatography (HPLC), thin layer chromatography (TLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in Hz. TLC was done on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Low resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). High resolution mass spectra were obtained using an Agilent 6230 time-of-flight mass spectrometer. Melting points were determined using a Mel Temp II melting point apparatus and are uncorrected. All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN, with a flow rate of 1.0 mL/min.
Methyl N-[2-(4-hydroxyphenyl)ethyl]carbamate
To a suspension of tyramine (1.0 g, 7.29 mmol) in DCM (35 mL) cooled in an ice bath under N2 was added DIPEA (1.41 g, 1.9 mL, 10.94 mmol); then methyl chloroformate (0.86 g, 0.7 mL, 9.11 mmol) was slowly added. A homogeneous solution formed and the reaction was allowed to warm slowly to RT overnight. The reaction was washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0–40% EtOAc/hexane) to give the desire carbamate as a clear oil (0.74 g, 52%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.04 (d, J = 8.48 Hz, 2H), 6.75 – 6.82 (m, 2H), 5.49 (br. s., 1H), 4.71 (br. s., 1H), 3.66 (s, 3H), 3.35 – 3.45 (m, 2H), 2.73 (t, J = 6.97 Hz, 2H).
Methyl N-[2-(4-methoxyphenyl)ethyl]carbamate (7a)
This was prepared as per methyl N-[2-(4-hydroxyphenyl)ethyl]carbamate but using 4-methoxyphenylethylamine. Yield quantitative. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.11 (d, J = 8.48 Hz, 2H), 6.85 (d, J = 8.67 Hz, 2H), 4.68 (br. s., 1H), 3.79 (d, J = 0.57 Hz, 3H), 3.66 (s, 3H), 3.35 – 3.45 (m, 2H), 2.75 (t, J = 6.88 Hz, 2H).
Methyl N-[2-(4-ethoxyphenyl)ethyl]carbamate (7b)
Methyl N-[2-(4-hydroxyphenyl)ethyl]carbamate (0.4 g, 2.05 mmol) and potassium carbonate (0.85 g, 6.15 mmol) were combined in dry DMF (10 mL) and iodoethane (0.48 g, 0.25 mL, 3.07 mmol) was added and the reaction was stirred at RT overnight. The reaction was diluted with EtOAc, then washed with NaHCO3 solution, water and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0–30% EtOAc/hexane) to give the ether as a white solid (0.36 g, 78%).1H NMR (300 MHz, CHLOROFORM-d) δ 7.09 (d, J = 8.67 Hz, 2H), 6.80 – 6.87 (m, 2H), 4.67 (br. s., 1H), 4.01 (q, J = 7.03 Hz, 2H), 3.66 (s, 3H), 3.35 – 3.46 (m, 2H), 2.74 (t, J = 6.97 Hz, 2H), 1.41 (t, J = 7.06 Hz, 3H)
Methyl N-[2-(4-propoxyphenyl)ethyl]carbamate (7c)
This was prepared as per 7b but using 1-iodopropane to get the desired ether as a white solid (0.56 g, 76%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.05 – 7.17 (m, 2H), 6.80 – 6.91 (m, 2H), 4.68 (br. s., 1H), 3.86 – 3.98 (m, 2H), 3.67 (s, 3H), 3.34 – 3.49 (m, 2H), 2.70 – 2.82 (m, J = 3.60 Hz, 2H), 1.73 – 1.88 (m, 2H), 0.99 – 1.11 (m, 3H).
Methyl N-{2-[4-(propan-2-yloxy)phenyl]ethyl}carbamate (7d)
Methyl N-[2-(4-hydroxyphenyl)ethyl]carbamate (0.4 g, 2.05 mmol), potassium carbonate (0.85 g, 6.15 mmol) and tetrabutylammonium iodide (0.15 g, 0.41 mmol) were combined in dry DMF (10 mL) and 2-bromopropane (0.38 g, 0.29 μL, 3.07 mmol) was added and the reaction was stirred at 50 °C overnight. The reaction was diluted with EtOAc, then washed with NaHCO3 solution, water and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0–30% EtOAc/hexane) to give the ether as a clear oil (0.31 g, 63%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.08 (d, J = 8.48 Hz, 2H), 6.79 – 6.86 (m, 2H), 4.68 (br. s., 1H), 4.51 (td, J = 6.03, 12.06 Hz, 1H), 3.66 (s, 3H), 3.34 – 3.45 (m, 2H), 2.74 (t, J = 6.97 Hz, 2H), 1.33 (d, J = 6.03 Hz, 6H).
Methyl N-[2-(4-butoxyphenyl)ethyl]carbamate (7e)
This was prepared as per 7b but using 1-bromobutane to get the desired butyl ether as a white solid. Yield 81%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.09 (d, J = 8.48 Hz, 2H), 6.80 – 6.88 (m, 2H), 4.67 (br. s., 1H), 3.94 (t, J = 6.50 Hz, 2H), 3.66 (s, 3H), 3.34 – 3.45 (m, 2H), 2.74 (t, J = 6.97 Hz, 2H), 1.70 – 1.82 (m, 2H), 1.42 – 1.56 (m, 2H), 0.97 (t, J = 7.35 Hz, 3H).
Methyl 1-[(3,4-dimethoxyphenyl)methyl]-7-methoxy-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (8a)
Carbamate 7a (209 mg, 1.0 mmol) and 3,4-dimethoxyphenylacetaldehyde (216 mg, 1.2 mmol) were combined in trifluoroacetic acid (3 mL) and heated to 70 °C overnight. The reaction was carefully quenched by pouring into saturated NaHCO3 solution then extracted with EtOAc, The organic fraction was washed with brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–20% EtOAc/hexane) to give the tetrahydroisoquinoline 8a (89 mg, 24%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.02 (dd, J = 8.48, 11.68 Hz, 1H), 6.69 – 6.81 (m, 2H), 6.61 (dd, J = 8.19, 14.03 Hz, 1H), 6.32 – 6.56 (m, 2H), 5.14 – 5.33 (m, 1H), 3.48 – 3.89 (m, 13H), 3.20 – 3.38 (m, 1H), 2.93 – 3.17 (m, 2H), 2.46 – 2.90 (m, 2H).
Methyl 1-[(3,4-dimethoxyphenyl)methyl]-7-ethoxy-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (8b)
This was prepared as per 8a using 7b. Yield 21%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 (dd, J = 8.38, 11.77 Hz, 1H), 6.69 – 6.80 (m, 2H), 6.61 (dd, J = 8.10, 15.07 Hz, 1H), 6.35 – 6.56 (m, 2H), 5.13 – 5.33 (m, 1H), 3.87 – 4.00 (m, 2H), 3.46 – 3.87 (m, 11H), 3.18 – 3.36 (m, 1H), 2.93 – 3.14 (m, 2H), 2.44 – 2.89 (m, 2H), 1.30 – 1.43 (m, 2H).
Methyl 1-[(3,4-dimethoxyphenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (8c)
This was prepared as per 8a using 7c. Yield 27%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 (dd, J = 8.48, 11.49 Hz, 1H), 6.69 – 6.80 (m, 2H), 6.61 (dd, J = 8.95, 13.09 Hz, 1H), 6.33 – 6.56 (m, 2H), 5.14 – 5.32 (m, 1H), 3.93 – 4.05 (m, 2H), 3.46 – 3.89 (m, 10H), 3.19 – 3.36 (m, 1H), 2.92 – 3.15 (m, 2H), 2.66 – 2.89 (m, 1H), 2.45 – 2.64 (m, 1H), 1.75 (dt, J = 6.97, 13.56 Hz, 2H), 0.94 – 1.08 (m, 3H).
Methyl 1-[(3,4-dimethoxyphenyl)methyl]-7-(propan-2-yloxy)-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (8d)
This was prepared as per 8a using 7d. Yield 20%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 (dd, J = 8.48, 11.30 Hz, 1H), 6.68 – 6.79 (m, 2H), 6.61 (dd, J = 9.04, 14.13 Hz, 1H), 6.35 – 6.55 (m, 2H), 5.13 – 5.32 (m, 1H), 4.27 – 4.47 (m, 1H), 3.47 – 3.88 (m, 10H), 3.18 – 3.36 (m, 1H), 2.93 – 3.15 (m, 2H), 2.45 – 2.88 (m, 2H), 1.20 – 1.33 (m, 6H).
Methyl 7-butoxy-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (8e)
This was prepared as per 8a using 7e. Yield 24%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 (dd, J = 8.38, 11.96 Hz, 1H), 6.69 – 6.80 (m, 2H), 6.34 – 6.67 (m, 3H), 5.13 – 5.33 (m, 1H), 3.46 – 3.92 (m, 12H), 3.19 – 3.36 (m, 1H), 2.93 – 3.15 (m, 2H), 2.45 – 2.88 (m, 2H), 1.63 – 1.79 (m, 2H), 1.38 – 1.55 (m, 2H), 0.92 – 1.01 (m, 3H).
1-[(3,4-Dimethoxyphenyl)methyl]-7-methoxy-1,2,3,4-tetrahydroisoquinoline (9a)
Carbamate 8a (84 mg, 0.226 mmol), potassium hydroxide (89 mg, 1.583 mmol) and hydrazine monohydrate (79 mg, 77 μL, 1.583 mmol) were combined in ethylene glycol (1 mL) and heated to 120 °C for 24 hr. the reaction was cooled, diluted with water and extracted 3 times with DCM. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–5% MeOH/DCM) to give the amine 9a (37 mg, 52%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.03 (d, J = 8.29 Hz, 1H), 6.71 – 6.86 (m, 5H), 4.17 (dd, J = 3.86, 9.14 Hz, 1H), 3.88 (s, 3H), 3.85 (s, 3H), 3.79 (s, 3H), 3.15 – 3.25 (m, 2H), 2.64 – 2.94 (m, 4H).
1-[(3,4-Dimethoxyphenyl)methyl]-7-ethoxy-1,2,3,4-tetrahydroisoquinoline (9b)
This was prepared as per 9a using 8b. Yield 67%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.02 (d, J = 8.48 Hz, 1H), 6.76 – 6.85 (m, 3H), 6.71 – 6.76 (m, 2H), 4.16 (dd, J = 3.77, 9.42 Hz, 1H), 4.01 (q, J = 6.97 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.15 – 3.24 (m, 2H), 2.64 – 2.95 (m, 4H), 1.41 (t, J = 6.97 Hz, 3H).
1-[(3,4-Dimethoxyphenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinoline (9c)
This was prepared as per 9a using 8c. Yield 32%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.02 (d, J = 8.29 Hz, 1H), 6.71 – 6.86 (m, 5H), 4.17 (dd, J = 3.39, 9.23 Hz, 1H), 3.88 – 3.93 (m, 2H), 3.86 – 3.88 (m, 3H), 3.85 (s, 3H), 3.16 – 3.26 (m, 2H), 2.64 – 2.95 (m, 4H), 1.74 – 1.87 (m, 2H), 1.04 (t, J = 7.44 Hz, 3H).
1-[(3,4-Dimethoxyphenyl)methyl]-7-(propan-2-yloxy)-1,2,3,4-tetrahydroisoquinoline (9d)
This was prepared as per 9a using 8d. Yield 65%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.01 (d, J = 8.29 Hz, 1H), 6.77 – 6.85 (m, 3H), 6.70 – 6.76 (m, 2H), 4.44 – 4.57 (m, 1H), 4.14 (dd, J = 3.77, 9.42 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.15 – 3.24 (m, 2H), 2.63 – 2.94 (m, 4H), 1.33 (dd, J = 2.17, 6.12 Hz, 6H).
7-Butoxy-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinoline (9e)
This was prepared as per 9a using 8e. Yield 68%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.01 (d, J = 8.29 Hz, 1H), 6.77 – 6.86 (m, 3H), 6.70 – 6.77 (m, 2H), 4.15 (dd, J = 3.67, 9.51 Hz, 1H), 3.94 (t, J = 6.50 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.15 – 3.25 (m, 2H), 2.63 – 2.94 (m, 4H), 1.70 – 1.81 (m, 2H), 1.43 – 1.56 (m, 2H), 0.98 (t, J = 7.35 Hz, 3H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (10a)
9a (37 mg, 0.118 mmol), N-benzyl-2-bromoacetamide (40 mg, 0.177 mmol) and tetrabutylammonium iodide (9 mg, 0.024 mmol) were combined in dry DMF (1 mL) and DIPEA (38 mg, 51 μL, 0.295 mmol) was added. The reaction was stirred at RT overnight under N2. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, and then dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–60% EtOAc in hexane) to give the desired product (54 mg, quantitative). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.34 (m, 3H), 7.01 – 7.13 (m, 3H), 6.89 – 6.97 (m, 1H), 6.57 – 6.79 (m, 5H), 4.49 (dd, J = 8.01, 14.98 Hz, 1H), 3.80 (s, 3H), 3.77 (s, 3H), 3.74 (s, 3H), 3.58 – 3.72 (m, 2H), 3.37 – 3.48 (m, 1H), 3.11 – 3.34 (m, 2H), 2.82 – 2.99 (m, 4H), 2.46 – 2.56 (m, 1H). m/z 461 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-ethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (10b)
This was prepared as per 10a from 9b. Yield quantitative. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.33 (m, 3H), 7.06 – 7.12 (m, 2H), 7.02 (d, J = 8.48 Hz, 1H), 6.93 (br. s., 1H), 6.59 – 6.78 (m, 5H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 4.00 (q, J = 7.10 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.56 – 3.72 (m, 2H), 3.36 – 3.48 (m, 1H), 3.11 – 3.33 (m, 2H), 2.82 – 2.99 (m, 4H), 2.45 – 2.55 (m, 1H), 1.41 (t, J = 6.97 Hz, 3H). m/z 475 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (10c)
This was prepared as per 10a from 9c. Yield 90%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.38 (m, 3H), 7.09 (d, J = 6.78 Hz, 2H), 7.02 (d, J = 8.48 Hz, 1H), 6.89 – 6.97 (m, 1H), 6.66 – 6.79 (m, 3H), 6.56 – 6.66 (m, 2H), 4.48 (dd, J = 8.29, 15.07 Hz, 1H), 3.88 (t, J = 6.50 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.55 – 3.71 (m, 2H), 3.35 – 3.51 (m, 1H), 3.09 – 3.34 (m, 2H), 2.81 – 3.00 (m, 4H), 2.44 – 2.57 (m, 1H), 1.73 – 1.87 (m, 2H), 1.04 (t, J = 7.44 Hz, 3H). m/z 489 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-(propan-2-yloxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (10d)
This was prepared as per 10a from 9d. Yield 98%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.32 (m, 4H), 7.06 – 7.12 (m, 1H), 7.02 (d, J = 8.48 Hz, 1H), 6.90 – 6.97 (m, 1H), 6.66 – 6.77 (m, 3H), 6.63 (s, 1H), 6.61 (s, 1H), 4.43 – 4.54 (m, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.56 – 3.72 (m, 2H), 3.36 – 3.48 (m, 1H), 3.12 – 3.33 (m, 2H), 2.82 – 2.98 (m, 4H), 2.45 – 2.54 (m, 1H), 1.33 (d, J = 4.71 Hz, 3H), 1.31 (d, J = 4.52 Hz, 3H). m/z 489 (M+H).
N-Benzyl-2-{7-butoxy-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (10e)
This was prepared as per 10a from 9e. Yield quantitative. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.33 (m, 3H), 7.06 – 7.11 (m, 2H), 7.02 (d, J = 8.48 Hz, 1H), 6.89 – 6.96 (m, 1H), 6.67 – 6.78 (m, 3H), 6.64 (s, 1H), 6.59 – 6.61 (m, 1H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.92 (t, J = 6.50 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.55 – 3.72 (m, 2H), 3.36 – 3.48 (m, 1H), 3.11 – 3.33 (m, 2H), 2.82 – 2.99 (m, 4H), 2.45 – 2.55 (m, 1H), 1.71 – 1.81 (m, 2H), 1.43 – 1.56 (m, 2H), 0.98 (t, J = 7.44 Hz, 3H). m/z 503 (M+H).
2-[3-(Benzyloxy)phenyl]ethan-1-amine (13) was prepared in 3 steps from 3-hydroxybenzaldehyde according to literature procedure.32
N-{2-[3-(Benzyloxy)phenyl]ethyl}-2-(3,4-dimethoxyphenyl)acetamide (14)
Phenylethylamine 13 (1.0 g, 4.40 mmol), 3,4-dimethoxyphenylacetic acid (0.95 g, 4.84 mmol) and BOP (2.53 g, 5.72 mmol) were combined in dry DMF (40 mL). DIPEA (2.27 g, 3.1 mL, 17.6 mmol) was added and the reaction was stirred under N2 at RT overnight. The reaction was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure to give the amide (1.78 g, quantitative). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.29 – 7.46 (m, 5H), 7.14 (t, J = 7.86 Hz, 1H), 6.69 – 6.88 (m, 4H), 6.67 (s, 1H), 6.62 (d, J = 7.54 Hz, 1H), 5.41 (br. s., 1H), 5.02 (s, 2H), 3.84 (s, 3H), 3.81 (s, 3H), 3.41 – 3.51 (m, 4H), 2.71 (t, J = 6.83 Hz, 2H).
6-(Benzyloxy)-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinoline (15)
Amide 14 (1.78 g, 4.40 mmol) was dissolved with warming in toluene (25 mL) then phosphorus oxychloride (3.55 g, 2.2 mL, 23.18 mmol) added. The solution was heated to 90 °C for 2 hr. The reaction was cooled, poured into water and stirred for 10 min. Layers were separated, and the aqueous portion was treated with 2N NaOH solution until a pH of 8–9 was reached; then it was extracted 3 times with DCM. Combined DCM fractions were dried over MgSO4, and the solvent was removed under reduced pressure. The crude dihydroisoquinoline was redissolved in methanol (20 mL) and cooled in ice. Sodium borohydride (0.75 g, 19.87 mmol) was added portionwise. After initial reaction had subsided, the ice bath was removed and the reaction was stirred at RT for 30 min. The reaction was quenched with water and then the methanol was removed under reduce pressure. The aqueous solution was extracted 3 times with DCM, the combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give the tetrahydroisoquinoline 15 (1.42 g, 92%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.28 – 7.47 (m, 5H), 7.16 (d, J = 8.57 Hz, 1H), 6.67 – 6.89 (m, 5H), 5.05 (s, 2H), 4.15 (dd, J = 3.53, 9.28 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.80 – 3.86 (m, 1H), 3.16 – 3.25 (m, 1H), 2.66 – 3.00 (m, 4H).
N-Benzyl-2-[6-(benzyloxy)-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (17d)
This was prepared from 14 using the method for 10a. Purified by chromatography on silica (0–60% EtOAc/hexane). Yield 35%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.47 (m, 8H), 7.05 – 7.13 (m, 2H), 6.99 (d, J = 8.57 Hz, 1H), 6.89 – 6.97 (m, 1H), 6.82 (dd, J = 2.59, 8.43 Hz, 1H), 6.68 – 6.75 (m, 2H), 6.66 (d, J = 1.79 Hz, 1H), 6.59 – 6.64 (m, 1H), 5.05 (s, 2H), 4.48 (dd, J = 8.10, 14.98 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.57 – 3.72 (m, 2H), 3.35 – 3.47 (m, 1H), 3.12 – 3.33 (m, 2H), 2.79 – 3.03 (m, 4H), 2.53 (dd, J = 4.29, 16.62 Hz, 1H). m/z 537 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-hydroxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (16)
Benzyl ether 15 (90 mg, 0.168 mmol), ammonium formate (32 mg, 0.503 mmol) and palladium on carbon (10%, 45 mg, 50% w/w) were combined in ethanol (2 mL) and heated to reflux for 2 hr. The reaction was cooled, filtered through Celite, rinsed thoroughly with ethanol, and then the solvent was removed under reduced pressure to give the desired phenol (75 mg, quantitative). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.37 (m, 3H), 7.08 (d, J = 7.44 Hz, 2H), 6.95 – 7.02 (m, 1H), 6.89 (d, J = 8.19 Hz, 1H), 6.60 – 6.73 (m, 4H), 6.55 (s, 1H), 4.48 (dd, J = 8.19, 14.79 Hz, 1H), 3.80 (s, 3H), 3.72 – 3.75 (m, 3H), 3.54 – 3.65 (m, 2H), 3.32 – 3.47 (m, 1H), 3.05 – 3.30 (m, 2H), 2.71 – 2.94 (m, 4H), 2.37 – 2.49 (m, 1H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (17a)
Phenol 16 (25 mg, 0.056 mmol) was combined with potassium carbonate (23 mg, 0.168 mmol) and tetrabutylammonium iodide (4 mg, 0.011 mmol) in DMF (0.5 mL) and 1-bromopropane (10 mg, 8 μL, 0.084 mmol) was added. The reaction was stirred at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–50% EtOAc/hexane) to give the ether (12 mg, 44%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.36 (m, 3H), 7.09 (d, J = 7.91 Hz, 2H), 6.90 – 7.02 (m, 2H), 6.58 – 6.79 (m, 5H), 4.49 (dd, J = 8.19, 14.98 Hz, 1H), 3.90 (t, J = 6.55 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.56 – 3.72 (m, 2H), 3.35 – 3.47 (m, 1H), 3.12 – 3.32 (m, 2H), 2.79 – 3.03 (m, 4H), 2.48 – 2.58 (m, 1H), 1.82 (s, 2H), 1.04 (t, J = 7.39 Hz, 3H). m/z 489 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (17b)
Phenol 16 (25 mg, 0.056 mmol) was combined with cesium carbonate (55 mg, 0.168 mmol) in DMF (0.5 mL) and 2,2,2-trifluoroiodoethane (24 mg, 11 μL, 0.112 mmol) was added. The reaction was stirred at 50 °C overnight. The reaction was cooled, diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–50% EtOAc/hexane) to give the ether (10 mg, 33%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 – 7.34 (m, 3H), 7.09 (d, J = 7.91 Hz, 2H), 7.01 (d, J = 8.48 Hz, 1H), 6.87 – 6.96 (m, 1H), 6.78 (dd, J = 2.26, 8.48 Hz, 1H), 6.60 – 6.73 (m, 4H), 4.49 (dd, J = 8.19, 14.98 Hz, 1H), 4.34 (q, J = 8.23 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.57 – 3.73 (m, 2H), 3.36 – 3.48 (m, 1H), 3.11 – 3.33 (m, 2H), 2.79 – 3.03 (m, 4H), 2.51 – 2.60 (m, 1H). 19F NMR (282 MHz, CHLOROFORM-d) δ–73.96. m/z 529 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(propan-2-yloxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (17c)
This was prepared as for 17a but using 2-bromopropane at 50 °C. Yield 48%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.33 (m, 3H), 7.06 – 7.13 (m, 2H), 6.90 – 7.00 (m, 2H), 6.68 – 6.76 (m, 2H), 6.59 – 6.67 (m, 3H), 4.43 – 4.59 (m, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.57 – 3.72 (m, 2H), 3.34 – 3.47 (m, 1H), 3.12 – 3.32 (m, 2H), 2.79 – 3.01 (m, 4H), 2.51 (dd, J = 4.33, 16.58 Hz, 1H), 1.33 (d, J = 6.03 Hz, 6H). m/z 489 (M+H).
Ethyl 4-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}oxy)butanoate (17e)
This was prepared as for 17a but using ethyl 4-bromobutyrate. Yield 23%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 – 7.36 (m, 3H), 7.05 – 7.12 (m, 2H), 6.89 – 7.01 (m, 2H), 6.59 – 6.78 (m, 5H), 4.48 (dd, J = 8.10, 14.88 Hz, 1H), 4.15 (q, J = 7.16 Hz, 2H), 3.99 (t, J = 6.03 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.54 – 3.72 (m, 2H), 3.34 – 3.49 (m, 1H), 3.10 – 3.33 (m, 2H), 2.78 – 3.03 (m, 4H), 2.44 – 2.59 (m, 3H), 2.10 (quin, J = 6.59 Hz, 2H), 1.27 (t, J = 7.16 Hz, 3H). m/z 561 (M+H).
Ethyl 7-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}oxy)heptanoate (17f)
This was prepared as for 17a but using ethyl 7-bromoheptanoate. Yield 65%.1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.06 – 7.13 (m, 2H), 6.97 (d, J = 8.48 Hz, 2H), 6.58 – 6.78 (m, 5H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 4.13 (q, J = 7.16 Hz, 2H), 3.93 (t, J = 6.40 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.55 – 3.72 (m, 2H), 3.35 – 3.49 (m, 1H), 3.11 – 3.33 (m, 2H), 2.78 – 3.03 (m, 4H), 2.53 (dd, J = 4.14, 16.58 Hz, 1H), 2.31 (t, J = 7.44 Hz, 2H), 1.72 – 1.84 (m, 2H), 1.59 – 1.72 (m, 2H), 1.34 – 1.55 (m, 4H), 1.26 (t, J = 7.16 Hz, 3H). m/z 603 (M+H).
2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl methanesulfonate (18a)
To phenol 16 (25 mg, 0.056 mmol) and TEA (17 mg, 23 μL, 0.168 mmol) in DCM (0.5 mL) was added methanesulfonyl chloride (13 mg, 9 μL, 0.112 mmol) and the reaction was stirred at RT overnight. The reaction was taken directly into purification by chromatography on silica (0–80% EtOAc/hexane) to give the sulfonate (17 mg, 59%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.34 (m, 3H), 7.09 (t, J = 4.71 Hz, 5H), 6.83 – 6.93 (m, 1H), 6.60 – 6.75 (m, 3H), 4.49 (dd, J = 8.10, 14.88 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.58 – 3.78 (m, 2H), 3.36 – 3.49 (m, 1H), 3.16 (s, 3H), 3.10 – 3.35 (m, 2H), 2.80 – 3.06 (m, 4H), 2.61 (dd, J = 4.33, 16.77 Hz, 1H). m/z 525 (M+H).
2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl benzenesulfonate (18b)
This was prepared as per 18a using benzenesulfonyl chloride. Yield 67%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.83 – 7.91 (m, 2H), 7.64 – 7.73 (m, 1H), 7.50 – 7.60 (m, 2H), 7.19 – 7.34 (m, 3H), 7.04 – 7.13 (m, 2H), 6.94 (d, J = 8.48 Hz, 1H), 6.80 – 6.89 (m, 2H), 6.59 – 6.74 (m, 4H), 4.48 (dd, J = 8.10, 14.88 Hz, 1H), 3.78 (s, 3H), 3.74 (s, 3H), 3.57 – 3.72 (m, 2H), 3.34 – 3.46 (m, 1H), 3.07 – 3.31 (m, 2H), 2.75 – 2.97 (m, 4H), 2.45 – 2.58 (m, 1H). m/z 587 (M+H).
2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl propanoate (19)
This was prepared as per 18a using propionyl chloride. Yield 89%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.03 – 7.13 (m, 3H), 6.83 – 6.96 (m, 3H), 6.67 – 6.74 (m, 1H), 6.59 – 6.67 (m, 2H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.58 – 3.73 (m, 2H), 3.35 – 3.47 (m, 1H), 3.11 – 3.34 (m, 2H), 2.81 – 3.04 (m, 4H), 2.52 – 2.65 (m, 3H), 1.27 (t, J = 7.54 Hz, 3H). m/z 503 (M+H).
2-(3,4-Dimethoxyphenyl)-N-[2-(3-nitrophenyl)ethyl]acetamide (21)
3-Nitrophenylethylamine hydrochloride (1.0 g, 4.94 mmol), 3,4-dimethoxyphenylacetic acid (1.07 g, 5.43 mmol) and BOP (2.84 g, 6.42 mmol) were combined in dry DMF (50 mL). DIPEA (2.55 g, 3.4 mL, 19.74 mmol) was added and the reaction was stirred under N2 at RT overnight. The reaction was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure to give the amide (1.7 g, quantitative). 1H NMR (300 MHz, METHANOL-d4) δ 7.93 – 8.08 (m, 2H), 7.37 – 7.56 (m, 2H), 6.78 – 6.87 (m, 2H), 6.67 – 6.74 (m, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.48 (t, J = 6.78 Hz, 2H), 3.35 (s, 2H), 2.86 – 2.94 (m, 2H).
N-[2-(3-Aminophenyl)ethyl]-2-(3,4-dimethoxyphenyl)acetamide
To the amide 21 (1.70 g, 4.94 mmol) in ethanol (65 mL) was added hydrazine monohydrate (3.26 g, 3.2 mL, 65.1 mmol) and the reaction was heated to 50 °C. Raney nickel (2880 type as slurry in water, 0.45 g) was added and the reaction was stirred at 50 °C for 1 hr. The reaction was cooled, filtered through Celite, rinsed thoroughly with ethanol. The solvent was removed under reduced pressure to give the desired aniline (1.55 g, 98%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 (t, J = 7.68 Hz, 1H), 6.79 – 6.86 (m, 2H), 6.66 – 6.76 (m, 2H), 6.51 (dd, J = 2.26, 8.01 Hz, 1H), 6.40 (d, J = 7.54 Hz, 1H), 6.33 (s, 1H), 5.43 (br. s., 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.46 – 3.49 (m, 2H), 3.39 – 3.46 (m, 2H), 2.58 – 2.65 (m, 2H).
Methyl N-(3-{2-[2-(3,4-dimethoxyphenyl)acetamido]ethyl}phenyl)carbamate (22)
To the aniline (1.55 g, 4.84 mmol) in DCM (30 mL) cooled in an ice bath under N2 was added DIPEA (1.23 g, 1.65 mL, 9.50 mmol) and then dropwise addition of methyl chloroformate (0.75 g, 0.61 mL, 7.91 mmol). The reaction was stirred in ice for 15 min then at RT overnight. The reaction completion was checked by TLC (5% MeOH/DCM). The reaction was washed with water and the aqueous fraction was extracted once with DCM. The combined organic fractions were washed with 2N HCl solution, then NaHCO3 solution, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude mixture was purified by chromatography on silica (0–5% MeOH in DCM) to give the carbamate as a yellow viscous oil (1.53 g, 85%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.13 – 7.24 (m, 2H), 7.10 (s, 1H), 6.77 – 6.82 (m, 1H), 6.68 – 6.75 (m, 3H), 6.65 (br. s., 1H), 5.41 (br. s., 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.75 – 3.79 (m, 3H), 3.41 – 3.50 (m, 4H), 2.71 (t, J = 6.73 Hz, 2H).
Methyl N-{1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}carbamate
Amide 22 (1.53 g, 4.11 mmol) was dissolved with warming in toluene (20 mL) and then phosphorus oxychloride (3.15 g, 1.9 mL, 20.54 mmol) was added. The reaction was heated to 90 °C for 2 hr. The reaction was cooled, poured into water and stirred for 10 min. Layers were separated, and the aqueous portion was treated with 2N NaOH solution until a pH of 8–9 was reached and then it was extracted 3 times with DCM. Combined DCM fractions were dried over MgSO4 and the solvent was removed under reduced pressure. The crude dihydroisoquinoline was redissolved in methanol (40 mL) and cooled in ice. Sodium borohydride (0.77 g, 20.5 mmol) was added portionwise. After initial reaction had subsided, the ice bath was removed and the reaction was stirred at RT for 30 min. The reaction was quenched with water then the methanol was removed under reduce pressure. The aqueous solution was extracted 3 times with DCM, the combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give the tetrahydroisoquinoline (1.17 g, 80%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.11 – 7.21 (m, 3H), 6.73 – 6.86 (m, 3H), 6.55 (s, 1H), 4.14 (dd, J = 3.81, 9.65 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.77 (s, 3H), 3.16 – 3.25 (m, 2H), 2.68 – 2.95 (m, 4H).
Methyl N-{2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}carbamate (23a)
Tetrahydroisoquinoline (1.17 g, 3.28 mmol), N-benzyl-2-bromoacetamide (1.12 g, 4.92 mmol) and tetrabutylammonium iodide (0.24 g, 0.66 mmol) were combined in dry DMF (30 mL) and DIPEA (1.06 g, 1.4 mL, 8.21 mmol) was added. The reaction was stirred at RT overnight under N2. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, and then dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0–70% EtOAc in hexane) to give the desired product (0.85 g, 52%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.32 (m, 4H), 7.06 – 7.15 (m, 2H), 7.00 (d, J = 8.29 Hz, 1H), 6.92 (dd, J = 4.76, 7.49 Hz, 1H), 6.59 – 6.74 (m, 4H), 6.57 (s, 1H), 4.48 (dd, J = 8.10, 14.98 Hz, 1H), 3.80 (s, 3H), 3.78 (s, 3H), 3.74 (s, 3H), 3.57 – 3.73 (m, 2H), 3.36 – 3.47 (m, 1H), 3.11 – 3.32 (m, 2H), 2.80 – 3.02 (m, 4H), 2.56 (dd, J = 4.57, 16.44 Hz, 1H). m/z 504 (M+H).
2-{6-Amino-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (24)
To a solution of the carbamate 23a (0.85 g, 1.69 mmol) in methanol (10 mL) was added 2N sodium hydroxide solution (1.69 mL, 3.38 mmol) and the reaction was heated to 50 °C overnight. Two additional aliquots of 2N NaOH solution (1.7 mL) was added after 24 hr and 48 hr, respectively, and heating continued at 50 °C until all starting material was gone by TLC (4:1 EtOAc:hexane). Methanol was removed under reduced pressure and the aqueous solution diluted with water. The solution was extracted 3 times with EtOAc and the combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give the free amine as a white solid (0.63 g, 84%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.34 (m, 3H), 7.09 (d, J = 7.82 Hz, 2H), 6.95 (br. s., 1H), 6.86 (d, J = 8.19 Hz, 1H), 6.58 – 6.73 (m, 3H), 6.53 (dd, J = 1.84, 8.15 Hz, 1H), 6.44 (s, 1H), 4.48 (dd, J = 8.15, 15.02 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.56 – 3.68 (m, 2H), 3.33 – 3.47 (m, 1H), 3.22 (q, J = 17.02 Hz, 2H), 2.77 – 2.96 (m, 4H), 2.39 – 2.51 (m, 1H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(dimethylamino)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25a)
To a solution of aniline 24 (25 mg, 0.056 mmol) and formaldehyde (37% aqueous, 23 mg, 21 μL, 0.281 mmol) in 1,2-dichloroethane (0.5 mL) was added portionwise sodium triacetoxyborohydride (59 mg, 0.281 mmol) and the reaction was stirred at RT overnight. The solvent was removed under reduced pressure, and the crude was redissolved in EtOAc, washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude sample was purified by chromatography on silica (0–100% EtOAc/hexane) to give the desired dimethylamine (23 mg, 88%). 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 2.50 (dd, J=16.58, 4.24 Hz, 1 H) 2.77 – 3.01 (m, 4 H) 2.92 (s, 6 H) 3.11 – 3.32 (m, 2 H) 3.33 – 3.46 (m, 1 H) 3.55 – 3.69 (m, 2 H) 3.73 (s, 3 H) 3.78 (s, 3 H) 4.47 (dd, J=14.98, 8.19 Hz, 1 H) 6.44 (d, J=2.54 Hz, 1 H) 6.57 – 6.73 (m, 4 H) 6.94 (d, J=8.48 Hz, 2 H) 7.05 – 7.11 (m, 1 H) 7.17 – 7.32 (m, 4 H). m/z 474 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(propylamino)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25b)
To a solution of aniline 24 (50 mg, 0.112 mmol) and propionaldehyde (7 mg, 8 μL, 0.112 mmol) in 1,2-dichloroethane (1 mL) was added portionwise sodium triacetoxyborohydride (36 mg, 0.168 mmol) and the reaction was stirred at RT overnight. The solvent was removed under reduced pressure, and the crude was redissolved in EtOAc, washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude sample was purified by chromatography on silica (0–60% EtOAc/hexane) to give the desired amine (37 mg, 67%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 – 7.34 (m, 3H), 7.05 – 7.12 (m, 2H), 6.92 – 7.02 (m, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.68 – 6.74 (m, 1H), 6.67 (s, 1H), 6.58 – 6.64 (m, 1H), 6.43 – 6.51 (m, 1H), 6.31 – 6.36 (m, J = 2.00 Hz, 1H), 4.48 (dd, J = 8.01, 14.98 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.53 – 3.69 (m, 3H), 3.33 – 3.45 (m, 1H), 3.13 – 3.32 (m, 2H), 3.07 (t, J = 7.11 Hz, 2H), 2.77 – 2.98 (m, 4H), 2.41 – 2.52 (m, 1H), 1.60 – 1.71 (m, 2H), 0.96 – 1.05 (m, 3H). m/z 488 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[methyl(propyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25c)
This was prepared as per 25a from 25b. Yield 100%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.06 – 7.12 (m, 2H), 6.96 – 7.02 (m, 1H), 6.92 (d, J = 8.57 Hz, 1H), 6.68 – 6.75 (m, 1H), 6.65 (d, J = 1.79 Hz, 1H), 6.61 (d, J = 8.10 Hz, 1H), 6.54 – 6.59 (m, 1H), 6.38 (d, J = 2.54 Hz, 1H), 4.48 (dd, J = 8.15, 15.02 Hz, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 3.56 – 3.69 (m, 2H), 3.33 – 3.45 (m, 1H), 3.13 – 3.32 (m, 4H), 2.91 (s, 3H), 2.76 – 3.01 (m, 4H), 2.49 (dd, J = 3.96, 16.48 Hz, 1H), 1.59 – 1.67 (m, 2H), 0.93 (t, J = 7.39 Hz, 3H). m/z 502 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(dipropylamino)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25d)
This was prepared as per 25b but with 2 eq. of propionaldehyde and 2.5 eq. of sodium triacetoxyborohydride. Yield 94%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 – 7.32 (m, 3H), 7.05 – 7.12 (m, 2H), 6.98 (dd, J = 5.04, 7.58 Hz, 1H), 6.90 (d, J = 8.48 Hz, 1H), 6.67 – 6.73 (m, 1H), 6.63 (d, J = 1.79 Hz, 1H), 6.56 – 6.61 (m, 1H), 6.50 (dd, J = 2.64, 8.57 Hz, 1H), 6.31 (d, J = 2.45 Hz, 1H), 4.47 (dd, J = 8.05, 15.02 Hz, 1H), 3.76 (s, 3H), 3.72 (s, 3H), 3.56 – 3.68 (m, 2H), 3.36 (d, J = 3.11 Hz, 1H), 3.13 – 3.27 (m, 6H), 2.75 – 2.98 (m, 4H), 2.45 (dd, J = 3.67, 16.39 Hz, 1H), 1.51 – 1.66 (m, 4H), 0.88 – 0.97 (m, 6H). m/z 530 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(propan-2-yl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25e)
Aniline 24 (25 mg, 0.056 mmol), 2-bromopropane (7 mg, 5 μL, 0.056 mmol) and tetrabutylammonium iodide (4 mg, 0.011 mmol) were combined in anhydrous DMF (0.5 mL). DIPEA (18 mg, 24 μL, 0.140 mmol) was added and the reaction was heated at 50 °C overnight. The reaction was cooled, diluted with EtOAc and washed with NaHCO3 solution and brine, and then dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0–60% EtOAc/hexane) to give the desired amine (5 mg, 19%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.09 (d, J = 7.91 Hz, 2H), 6.97 (br. s., 1H), 6.87 (d, J = 8.29 Hz, 1H), 6.58 – 6.74 (m, 3H), 6.44 (dd, J = 2.17, 8.19 Hz, 1H), 6.31 (s, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.53 – 3.68 (m, 3H), 3.33 – 3.46 (m, 1H), 3.13 – 3.32 (m, 2H), 2.76 – 2.97 (m, 4H), 2.41 – 2.52 (m, 1H), 1.21 (d, J = 6.22 Hz, 6H). m/z 488 (M+H).
N-Benzyl-2-{6-[(cyclopropylmethyl)amino]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25f)
This was prepared as per 25e using bromomethylcyclopropane. After stirring overnight at 50 °C, the reaction was stirred an additional 6 hr at 70 °C. Yield 43%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 4H), 7.05 – 7.12 (m, 2H), 6.96 (dd, J = 4.90, 7.72 Hz, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.58 – 6.63 (m, 1H), 6.48 (dd, J = 2.45, 8.29 Hz, 1H), 6.34 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.54 – 3.68 (m, 2H), 3.33 – 3.45 (m, 1H), 3.22 (q, J = 16.95 Hz, 2H), 2.94 (d, J = 6.97 Hz, 2H), 2.77 – 2.92 (m, 4H), 2.42 – 2.51 (m, 1H), 1.02 – 1.17 (m, 1H), 0.51 – 0.60 (m, 2H), 0.20 – 0.27 (m, 2H). m/z 500 (M+H).
N-Benzyl-2-{6-[(cyclopropylmethyl)(methyl)amino]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25g)
This was prepared as per 25a from 25f. Yield 94%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.33 (m, 3H), 7.09 (d, J = 6.97 Hz, 2H), 6.89 – 7.04 (m, 2H), 6.58 – 6.76 (m, 4H), 6.47 (s, 1H), 4.48 (dd, J = 8.01, 14.98 Hz, 1H), 3.78 (s, 3H), 3.74 (s, 3H), 3.57 – 3.70 (m, 2H), 3.34 – 3.46 (m, 1H), 3.12 – 3.33 (m, 4H), 2.96 (s, 3H), 2.78 – 3.01 (m, 4H), 2.50 (dd, J = 3.67, 16.67 Hz, 1H), 0.97 – 1.09 (m, 1H), 0.47 – 0.56 (m, 2H), 0.17 – 0.25 (m, 2H). m/z 514 (M+H).
N-Benzyl-2-[6-(benzylamino)-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (25h)
Aniline 23 (30 mg, 0.067 mmol), sodium bicarbonate (2 mg, 0.027 mmol) and benzaldehyde (8 mg, 8 μL, 0.074 mmol) were combined in anhydrous methanol (0.5 mL) and heated to 40 °C for 1 hr. The reaction was then cooled in an ice bath and sodium borohydride was added. The reaction was allowed to warm slowly to RT overnight. The solvent was removed under reduced pressure and the crude purified by chromatography on silica (0–100% EtOAc/hexane) to obtain the desired amine (19 mg, 53%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.36 – 7.42 (m, 3H), 7.20 – 7.35 (m, 4H), 7.10 (d, J = 7.63 Hz, 2H), 6.97 (br. s., 1H), 6.90 (d, J = 8.19 Hz, 1H), 6.57 – 6.76 (m, 4H), 6.51 (dd, J = 2.17, 8.19 Hz, 1H), 6.39 (d, J = 2.07 Hz, 1H), 4.49 (dd, J = 8.15, 15.02 Hz, 1H), 4.32 (s, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.54 – 3.70 (m, 2H), 3.34 – 3.49 (m, 1H), 3.13 – 3.33 (m, 2H), 2.77 – 3.00 (m, 4H), 2.41 – 2.54 (m, 1H). m/z 536 (M+H).
N-Benzyl-2-{6-[benzyl(methyl)amino]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25i)
This was prepared as per 25a from 25h. Yield 67%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.39 (m, 8H), 7.05 – 7.12 (m, 2H), 6.88 – 7.01 (m, 2H), 6.67 – 6.75 (m, 1H), 6.57 – 6.67 (m, 3H), 6.47 (d, J = 2.07 Hz, 1H), 4.51 (s, 2H), 4.42 – 4.49 (m, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 3.55 – 3.69 (m, 2H), 3.33 – 3.46 (m, 1H), 3.13 – 3.32 (m, 2H), 2.99 (s, 3H), 2.76 – 2.97 (m, 4H), 2.42 – 2.54 (m, 1H). m/z 550 (M+H).
N-Benzyl-2-(6-{[2-(3,4-dimethoxyphenyl)ethyl]amino}-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (25j)
This was prepared as per 25e using 3,4-dimethoxyphenethyl bromide, stirring at RT overnight. Yield 12%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.08 (d, J = 6.78 Hz, 2H), 6.96 (br. s., 1H), 6.65 – 6.91 (m, 6H), 6.58 – 6.64 (m, 1H), 6.47 (dd, J = 2.26, 8.29 Hz, 1H), 6.34 (d, J = 1.70 Hz, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.88 (s, 6H), 3.79 (s, 3H), 3.73 (s, 3H), 3.50 – 3.68 (m, 2H), 3.32 – 3.46 (m, 3H), 3.22 (q, J = 16.95 Hz, 2H), 2.77 – 2.98 (m, 6H), 2.41 – 2.51 (m, 1H). m/z 611 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(3-phenylpropyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25k)
This was prepared as per 25e using 3-phenyl-1-bromopropane, stirring at RT overnight. Yield 41%.1H NMR (300 MHz, CHLOROFORM-d) δ 7.16 – 7.36 (m, 8H), 7.04 – 7.13 (m, 2H), 6.92 – 7.01 (m, 1H), 6.87 (d, J = 8.29 Hz, 1H), 6.64 – 6.74 (m, 2H), 6.58 – 6.63 (m, 1H), 6.44 (dd, J = 2.26, 8.29 Hz, 1H), 6.29 (d, J = 2.26 Hz, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.52 – 3.69 (m, 2H), 3.32 – 3.47 (m, 1H), 3.11 (s, 2H), 3.05 – 3.32 (m, 2H), 2.78 – 2.98 (m, 4H), 2.74 (t, J = 7.63 Hz, 2H), 2.38 – 2.50 (m, 1H), 1.95 (quin, J = 7.30 Hz, 2H). m/z 564 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(4-phenoxybutyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25l)
This was prepared as per 25e using 3-phenyl-1-bromopropane, stirring at RT overnight. Yield 41%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.36 (m, 6H), 7.04 – 7.12 (m, 2H), 6.84 – 7.01 (m, 5H), 6.64 – 6.74 (m, 2H), 6.60 (s, 1H), 6.47 (dd, J = 2.26, 8.29 Hz, 1H), 6.33 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 7.91, 15.07 Hz, 1H), 4.02 (t, J = 5.93 Hz, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.53 – 3.69 (m, 2H), 3.33 – 3.46 (m, 1H), 3.10 – 3.32 (m, 4H), 2.76 – 2.99 (m, 4H), 2.41 – 2.53 (m, 1H), 1.75 – 1.98 (m, 4H). m/z 594 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(pyridin-4-ylmethyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25m)
This was prepared as per 25b from 24 using 4-pyridine carboxaldehyde. Yield 33%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.57 (d, J = 5.75 Hz, 2H), 7.19 – 7.37 (m, 6H), 7.04 – 7.12 (m, 2H), 6.94 (s, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.57 – 6.75 (m, 3H), 6.45 (dd, J = 2.45, 8.29 Hz, 1H), 6.30 (d, J = 2.26 Hz, 1H), 4.47 (dd, J = 8.15, 15.02 Hz, 1H), 4.37 (s, 2H), 3.79 (s, 3H), 3.75 (s, 3H), 3.53 – 3.67 (m, 2H), 3.33 – 3.46 (m, 1H), 3.21 (q, J = 16.95 Hz, 2H), 2.76 – 2.95 (m, 4H), 2.36 – 2.49 (m, 1H). m/z 537 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(pyridin-3-ylmethyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25n)
This was prepared as per 25b from 24 using 3-pyridine carboxaldehyde. Yield 53%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.64 (s, 1H), 8.54 (d, J = 4.71 Hz, 1H), 7.71 (d, J = 7.72 Hz, 1H), 7.19 – 7.35 (m, 4H), 7.09 (d, J = 7.72 Hz, 2H), 6.91 – 6.99 (m, 1H), 6.89 (d, J = 8.29 Hz, 1H), 6.58 – 6.75 (m, 3H), 6.50 (d, J = 8.29 Hz, 1H), 6.36 (s, 1H), 4.48 (dd, J = 7.96, 15.02 Hz, 1H), 4.36 (s, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.54 – 3.68 (m, 2H), 3.33 – 3.47 (m, 1H), 3.21 (q, J = 16.99 Hz, 2H), 2.77 – 2.97 (m, 4H), 2.38 – 2.53 (m, 1H). m/z 537 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-{[2-(piperidin-1-yl)ethyl]amino}-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25o)
This was prepared as per 25e using 1-(2-chloroethyl)piperidine hydrochloride. Yield 16%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 – 7.34 (m, 3H), 7.08 (d, J = 6.59 Hz, 2H), 6.92 – 7.01 (m, 1H), 6.89 (d, J = 8.29 Hz, 1H), 6.64 – 6.75 (m, 2H), 6.57 – 6.63 (m, 1H), 6.51 (dd, J = 2.26, 8.29 Hz, 1H), 6.36 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 8.19, 15.16 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.53 – 3.68 (m, 2H), 3.33 – 3.48 (m, 1H), 3.09 – 3.32 (m, 4H), 2.75 – 2.99 (m, 4H), 2.58 (t, J = 6.03 Hz, 2H), 2.47 – 2.54 (m, 1H), 2.35 – 2.45 (m, 3H), 1.59 (td, J = 5.30, 10.69 Hz, 4H), 1.46 (d, J = 5.09 Hz, 2H). m/z 558 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(pyrrolidin-1-yl)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25p)
This was prepared as per 25e using 1-bromo-4-chlorobutane. Yield 16%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.33 (m, 5H), 7.06 – 7.12 (m, 1H), 6.97 – 7.02 (m, 1H), 6.94 (d, J = 8.48 Hz, 1H), 6.66 – 6.76 (m, 1H), 6.58 – 6.65 (m, 1H), 6.45 (dd, J = 2.45, 8.48 Hz, 1H), 6.28 (d, J = 2.26 Hz, 1H), 4.48 (dd, J = 8.19, 14.79 Hz, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.53 – 3.69 (m, 2H), 3.36 – 3.48 (m, 1H), 3.13 – 3.33 (m, 6H), 2.79 – 3.02 (m, 4H), 2.51 (dd, J = 4.05, 16.67 Hz, 1H), 1.99 (td, J = 3.34, 6.50 Hz, 4H). m/z 500 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(piperidin-1-yl)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25q)
Aniline 24 (25 mg, 0.056 mmol), potassium carbonate (9 mg, 0.062 mmol) and 1,5- dibromopentane (14 mg, 8 μL, 0.062 mmol) were combined in DI water and heated to 120 °C for 20 min in the microwave at 50W power. The aqueous was removed via pipette and the residue was dissolved in EtOAc, then washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–50% EtOAc/hexane) to give the piperidine (17 mg, 59%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.09 (d, J = 7.91 Hz, 2H), 6.95 (d, J = 8.29 Hz, 2H), 6.80 (dd, J = 2.26, 8.48 Hz, 1H), 6.68 – 6.74 (m, 1H), 6.57 – 6.67 (m, 3H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.55 – 3.71 (m, 2H), 3.33 – 3.45 (m, 1H), 3.08 – 3.32 (m, 6H), 2.78 – 3.01 (m, 4H), 2.45 – 2.55 (m, 1H), 1.66 – 1.76 (m, 4H), 1.58 (d, J = 5.09 Hz, 2H). m/z 514 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(pentylamino)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25r)
This was prepared as per 25b from 24 using valeraldehyde. Yield 46%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.05 – 7.13 (m, 2H), 6.93 – 7.01 (m, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.58 – 6.64 (m, 1H), 6.46 (dd, J = 2.26, 8.29 Hz, 1H), 6.33 (s, 1H), 4.48 (dd, J = 8.29, 15.07 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.51 – 3.68 (m, 3H), 3.33 – 3.46 (m, 1H), 3.13 – 3.32 (m, 2H), 3.08 (t, J = 7.06 Hz, 2H), 2.77 – 2.99 (m, 4H), 2.42 – 2.52 (m, 1H), 1.58 – 1.68 (m, 2H), 1.32 – 1.44 (m, 4H), 0.88 – 0.97 (m, 3H). m/z 516 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[methyl(pentyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25s)
This was prepared as per 25a from 25r. Yield 82%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.06 – 7.13 (m, 2H), 6.96 – 7.02 (m, 1H), 6.93 (d, J = 8.48 Hz, 1H), 6.69 – 6.75 (m, 1H), 6.54 – 6.68 (m, 3H), 6.39 (s, 1H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 3.56 – 3.70 (m, 2H), 3.39 (dt, J = 4.71, 12.15 Hz, 1H), 3.14 – 3.33 (m, 4H), 2.91 (s, 3H), 2.77 – 3.01 (m, 4H), 2.49 (dd, J = 3.77, 16.58 Hz, 1H), 1.57 (td, J = 7.23, 14.36 Hz, 2H), 1.23 – 1.42 (m, 4H), 0.91 (t, J = 6.88 Hz, 3H). m/z 530 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-(hexylamino)-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25t)
This was prepared as per 25b from 24 using 1-bromohexane. Yield 33%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.09 (d, J = 6.59 Hz, 2H), 6.96 (dd, J = 4.80, 7.82 Hz, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.57 – 6.64 (m, 1H), 6.46 (dd, J = 2.26, 8.29 Hz, 1H), 6.33 (d, J = 1.88 Hz, 1H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.52 – 3.69 (m, 3H), 3.33 – 3.46 (m, 1H), 3.13 – 3.32 (m, 2H), 3.08 (t, J = 7.06 Hz, 2H), 2.77 – 2.98 (m, 4H), 2.42 – 2.51 (m, 1H), 1.53 – 1.67 (m, 2H), 1.27 – 1.47 (m, 6H), 0.86 – 0.94 (m, 3H). m/z 530 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[hexyl(methyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (25u)
This was prepared as per 25a from 25t. Yield 90%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.09 (d, J = 7.16 Hz, 2H), 6.99 (dd, J = 5.27, 7.72 Hz, 1H), 6.93 (d, J = 8.48 Hz, 1H), 6.68 – 6.75 (m, 1H), 6.53 – 6.67 (m, 3H), 6.38 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.78 (s, 3H), 3.74 (s, 3H), 3.55 – 3.70 (m, 2H), 3.39 (dt, J = 4.62, 12.20 Hz, 1H), 3.13 – 3.32 (m, 4H), 2.90 (s, 3H), 2.77 – 3.01 (m, 4H), 2.49 (dd, J = 3.96, 16.58 Hz, 1H), 1.50 – 1.62 (m, 2H), 1.31 (br. s., 6H), 0.86 – 0.94 (m, 3H). m/z 544 (M+H).
Ethyl 2-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)acetate (26a)
This was prepared as per 25e from 24 using ethyl bromoacetate. Yield 90%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.09 (d, J = 7.91 Hz, 2H), 6.92 – 7.00 (m, 1H), 6.90 (d, J = 8.29 Hz, 1H), 6.58 – 6.74 (m, 3H), 6.48 (dd, J = 2.12, 8.24 Hz, 1H), 6.33 (s, 1H), 4.47 (dd, J = 8.10, 15.07 Hz, 1H), 4.25 (q, J = 7.16 Hz, 2H), 3.89 (s, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.57 – 3.69 (m, 2H), 3.33 – 3.47 (m, 1H), 3.11 – 3.32 (m, 2H), 2.76 – 2.98 (m, 4H), 2.42 – 2.53 (m, 1H), 1.31 (t, J = 7.11 Hz, 3H). m/z 532 (M+H).
Ethyl 2-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)propanoate (26b)
This was prepared as per 25e from 24 using ethyl 2-bromopropionate. Yield 65%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.32 (m, 3H), 7.08 (d, J = 6.78 Hz, 2H), 6.91 – 6.99 (m, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.58 – 6.73 (m, 3H), 6.47 (dd, J = 2.07, 8.10 Hz, 1H), 6.31 – 6.35 (m, 1H), 4.47 (dd, J = 8.01, 14.98 Hz, 1H), 4.20 (q, J = 7.10 Hz, 2H), 4.06 – 4.14 (m, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.55 – 3.67 (m, 2H), 3.32 – 3.45 (m, 1H), 3.21 (q, J = 17.08 Hz, 2H), 2.76 – 2.97 (m, 4H), 2.38 – 2.51 (m, 1H), 1.47 (d, J = 5.46 Hz, 3H), 1.27 (dt, J = 2.26, 7.16 Hz, 3H). m/z 546 (M+H).
Ethyl 3-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)propanoate (26c)
This was prepared as per 25e from 24 using ethyl 3-bromopropionoate. Yield 12%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.08 (d, J = 6.97 Hz, 2H), 6.96 (t, J = 6.31 Hz, 1H), 6.89 (d, J = 8.29 Hz, 1H), 6.58 – 6.74 (m, 3H), 6.48 (dd, J = 2.26, 8.29 Hz, 1H), 6.36 (d, J = 1.88 Hz, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 4.16 (q, J = 7.16 Hz, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.54 – 3.67 (m, 2H), 3.44 (t, J = 6.30 Hz, 2H), 3.36 – 3.49 (m, 1H), 3.22 (q, J = 17.08 Hz, 2H), 2.77 – 2.97 (m, 4H), 2.61 (t, J = 6.22 Hz, 2H), 2.42 – 2.53 (m, 1H), 1.22 – 1.31 (m, 3H). m/z 546 (M+H).
Ethyl 4-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)butanoate (26d)
This was prepared as per 25e from 24 using ethyl 4-bromobutanoate. Yield 35%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.34 (m, 3H), 7.05 – 7.12 (m, 2H), 6.96 (dd, J = 4.71, 7.72 Hz, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.75 (m, 2H), 6.59 – 6.64 (m, 1H), 6.47 (dd, J = 2.26, 8.29 Hz, 1H), 6.33 (d, J = 2.26 Hz, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 4.15 (q, J = 7.16 Hz, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.54 – 3.67 (m, 2H), 3.34 – 3.45 (m, 1H), 3.11 – 3.32 (m, 4H), 2.77 – 2.99 (m, 4H), 2.49 (d, J = 5.27 Hz, 1H), 2.43 (t, J = 7.16 Hz, 2H), 1.95 (quin, J = 6.97 Hz, 2H), 1.26 (t, J = 7.16 Hz, 3H). m/z 560 (M+H).
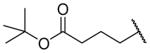
Tert-butyl 4-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)butanoate (26e)
This was prepared as per 25e from 24 using t-butyl 4-bromobutanoate. Yield 21%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.33 (m, 3H), 7.09 (d, J = 6.78 Hz, 2H), 6.98 (br. s., 1H), 6.87 (d, J = 8.29 Hz, 1H), 6.65 – 6.73 (m, 2H), 6.58 – 6.64 (m, 1H), 6.46 (dd, J = 2.17, 8.19 Hz, 1H), 6.33 (d, J = 2.07 Hz, 1H), 4.47 (dd, J = 8.01, 14.98 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.54 – 3.69 (m, 2H), 3.32 – 3.47 (m, 1H), 3.14 (t, J = 6.88 Hz, 2H), 3.07 – 3.32 (m, 2H), 2.76 – 2.99 (m, 4H), 2.41 – 2.54 (m, 1H), 2.34 (t, J = 7.16 Hz, 2H), 1.90 (quin, J = 7.02 Hz, 2H), 1.46 (s, 9H). m/z 588 (M+H).
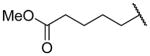
Methyl 5-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)pentanoate (26f)
This was prepared as per 25e from 24 using methyl 5-bromopentanoate, stirring overnight at RT. Yield 19%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.35 (m, 3H), 7.05 – 7.12 (m, 2H), 6.93 – 7.01 (m, J = 4.30 Hz, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.57 – 6.64 (m, 1H), 6.46 (dd, J = 2.26, 8.29 Hz, 1H), 6.32 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 8.10, 15.07 Hz, 1H), 3.79 (s, 3H), 3.73 (s, 3H), 3.68 (s, 3H), 3.53 – 3.66 (m, 2H), 3.33 – 3.45 (m, 1H), 3.06 – 3.32 (m, 4H), 2.76 – 2.98 (m, 4H), 2.42 – 2.53 (m, 1H), 2.33 – 2.41 (m, 2H), 1.56 – 1.83 (m, 4H). m/z 560 (M+H).
Ethyl 7-({2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)heptanoate (26g)
This was prepared as per 25e from 24 using ethyl 5-bromoheptanoate, stirring overnight at RT. Yield 18%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 – 7.36 (m, 3H), 7.03 – 7.15 (m, 2H), 6.97 (dd, J = 4.62, 7.63 Hz, 1H), 6.88 (d, J = 8.29 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.58 – 6.64 (m, 1H), 6.46 (dd, J = 2.35, 8.19 Hz, 1H), 6.32 (d, J = 2.07 Hz, 1H), 4.48 (dd, J = 8.01, 14.98 Hz, 1H), 4.13 (q, J = 7.16 Hz, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.52 – 3.70 (m, 2H), 3.34 – 3.46 (m, 1H), 3.13 – 3.32 (m, 2H), 3.09 (t, J = 7.06 Hz, 2H), 2.76 – 2.99 (m, 4H), 2.41 – 2.52 (m, 1H), 2.31 (t, J = 7.44 Hz, 2H), 1.55 – 1.71 (m, 4H), 1.33 – 1.49 (m, 4H), 1.22 – 1.30 (m, 3H). m/z 602 (M+H).
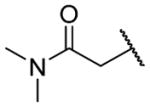
2-({2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}amino)-N,N-dimethylacetamide (26h)
Ester 25q (18 mg, 0.034 mmol) was dissolved in ethanol (0.5 mL) and 2N sodium hydroxide solution (0.1 mL, 0.2 mmol) was added. The reaction was stirred at RT overnight. The ethanol was removed under reduced pressure, the reaction was diluted with water and the pH was adjusted to 7 with 2N HCl. All solvents were removed under reduced pressure, and the crude was dissolved as far as possible in methanol. The solution was filtered to remove solids and the solvent removed under reduced pressure to give the acid.
The acid was combined with dimethylamine hydrochloride (7 mg, 0.079 mmol) and HATU (23 mg, 0.060 mmol) in DMF (0.5 mL) and DIPEA (26 mg, 35 μL, 0.199 mmol) was added. The reaction was stirred at RT overnight, diluted with EtOAc, washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–10% CMA-80/EtOAc) to give the amide (9 mg, 43%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.35 (m, 3H), 7.09 (d, J = 7.72 Hz, 2H), 6.94 – 7.02 (m, 1H), 6.81 – 6.93 (m, 2H), 6.59 – 6.75 (m, 3H), 6.52 (d, J = 8.29 Hz, 1H), 6.34 (s, 1H), 4.48 (dd, J = 8.19, 14.98 Hz, 1H), 3.85 (s, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.55 – 3.69 (m, 2H), 3.33 – 3.47 (m, 1H), 3.13 – 3.32 (m, 2H), 3.01 – 3.08 (m, 4H), 2.83 – 2.99 (m, 4H), 2.81 (s, 2H), 2.43 – 2.55 (m, 1H). m/z 531 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-methanesulfonamido-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (27)
To aniline 24 (40 mg, 0.090 mmol) and TEA (36 mg, 50 μL, 0.359 mmol) in DCM (0.5 mL) was added methanesulfonyl chloride (31 mg, 21 μL, 0.269 mmol) and the reaction was stirred at RT overnight. The solution was taken directly into purification by chromatography on silica (0–70% EtOAc/hexane) to give the bis-sulfonamide (30 mg, 56%).
The bis-sulfonamide was suspended in 2N NaOH solution (2 mL) and heated to 80 °C overnight. The reaction was cooled and acidified with 2N HCl solution. The precipitate formed was collected by filtration to give the sulfonamide as a white solid (19 mg, 73%). 1H NMR (300 MHz, DMSO-d6) δ 7.18 – 7.33 (m, 3H), 6.97 – 7.13 (m, 4H), 6.81 – 6.92 (m, 3H), 6.75 (s, 2H), 4.29 (dd, J = 7.72, 15.07 Hz, 1H), 3.70 – 3.74 (m, 1H), 3.68 (s, 3H), 3.61 (s, 3H), 3.53 – 3.60 (m, 1H), 3.21 – 3.31 (m, 2H), 2.85 – 2.98 (m, 3H), 2.82 (s, 3H), 2.70 – 2.80 (m, 3H), 2.39 – 2.46 (m, 1H). m/z 524 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-acetamido-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (28a)
To a solution of aniline 24 (25 mg, 0.056 mmol) in DCM (0.5 mL) was added DIPEA (22 mg, 29 μL, 0.168 mmol) and then acetic anhydride (11 mg, 11 μL, 0.112 mmol) and the resulting solution was stirred at RT overnight. The solution was applied directly to column for chromatography on silica (0–100% EtOAc/hexane) to give the amide (27 mg, quantitative). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.37 (s, 1H), 7.17 – 7.33 (m, 4H), 7.14 (s, 1H), 7.09 (d, J = 8.01 Hz, 2H), 7.01 (d, J = 8.29 Hz, 1H), 6.92 (s, 1H), 6.66 – 6.74 (m, 2H), 6.60 – 6.66 (m, 1H), 4.49 (dd, J = 8.15, 14.93 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.56 – 3.72 (m, 2H), 3.36 – 3.49 (m, 1H), 3.10 – 3.33 (m, 2H), 2.79 – 3.03 (m, 4H), 2.52 – 2.63 (m, 1H), 2.17 (s, 3H). m/z 488 (M+H).
N-{2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}hexanamide (28b)
Aniline 24 (30 mg, 0.067 mmol) and BOP (30 mg, 0.067 mmol) were combined in DCM (1 mL) and hexanoic acid (8 mg, 8.5 μL, 0.067 mmol) was added, followed by DIPEA (17 mg, 24 μL, 0.135 mmol) and the reaction was stirred at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, and dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–75% EtOAc/hexane) to give the desired amide (34 mg, 97%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.42 (s, 1H), 7.17 – 7.35 (m, 4H), 7.05 – 7.14 (m, 3H), 7.01 (d, J = 8.29 Hz, 1H), 6.87 – 6.96 (m, 1H), 6.59 – 6.74 (m, 3H), 4.48 (dd, J = 8.15, 15.02 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.60 (dd, J = 4.71, 14.98 Hz, 2H), 3.35 – 3.51 (m, 1H), 3.08 – 3.33 (m, 2H), 2.78 – 3.03 (m, 4H), 2.51 – 2.63 (m, 1H), 2.34 (t, J = 7.49 Hz, 2H), 1.66 – 1.79 (m, 2H), 1.31 – 1.41 (m, 4H), 0.87 – 0.95 (m, 3H). m/z 544 (M+H).
N-{2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}benzamide (28c)
This was prepared as per 27a except using benzoyl chloride. Yield 81%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.84 – 7.90 (m, 2H), 7.82 (s, 1H), 7.45 – 7.60 (m, 4H), 7.36 (d, J = 8.48 Hz, 1H), 7.19 – 7.33 (m, 3H), 7.03 – 7.12 (m, 3H), 6.88 – 6.96 (m, 1H), 6.67 – 6.75 (m, 2H), 6.59 – 6.66 (m, 1H), 4.49 (dd, J = 8.15, 15.02 Hz, 1H), 3.81 (d, J = 0.94 Hz, 3H), 3.73 – 3.76 (m, 3H), 3.55 – 3.73 (m, 2H), 3.38 – 3.51 (m, 1H), 3.11 – 3.34 (m, 2H), 2.81 – 3.08 (m, 4H), 2.61 (dd, J = 4.57, 16.81 Hz, 1H). m/z 550 (M+H).
Ethyl N-{2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-6-yl}carbamate (23b)
This was isolated from the hydrolysis of carbamate 23a in ethanol. Yield 15%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 – 7.37 (m, 4H), 7.05 – 7.16 (m, 3H), 7.00 (d, J = 8.38 Hz, 1H), 6.92 (dd, J = 4.85, 7.77 Hz, 1H), 6.65 – 6.74 (m, 2H), 6.64 (s, 1H), 6.61 (s, 1H), 4.49 (dd, J = 8.10, 14.98 Hz, 1H), 4.22 (q, J = 7.16 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.56 – 3.72 (m, 2H), 3.35 – 3.50 (m, 1H), 3.09 – 3.34 (m, 2H), 2.78 – 3.03 (m, 4H), 2.48 – 2.61 (m, 1H), 1.27 – 1.35 (m, 3H). m/z 518 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(phenylcarbamoyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (28d)
Aniline 24 (25 mg, 0.056 mmol) and phenyl isocyanate (7 mg, 7 μL, 0.062 mmol) were combined in toluene (0.5 mL) and heated to 75 °C for 4 hr. The reaction was cooled and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0–80% EtOAc/hexane) to give the urea (32 mg, quantitative). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.82 (s, 1H), 7.46 (s, 1H), 7.17 – 7.35 (m, 8H), 6.95 – 7.12 (m, 6H), 6.70 – 6.78 (m, 2H), 6.60 – 6.68 (m, 1H), 4.48 (dd, J = 8.19, 15.07 Hz, 1H), 3.81 (s, 3H), 3.72 (s, 3H), 3.56 – 3.68 (m, 2H), 3.35 – 3.49 (m, 1H), 3.05 – 3.34 (m, 2H), 2.70 – 2.91 (m, 4H), 2.40 – 2.53 (m, 1H). m/z 565 (M+H).
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-6-[(hexylcarbamoyl)amino]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (28e)
This was prepared as per 27d using hexyl isocyanate. Yield 89%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 – 7.34 (m, 3H), 7.02 – 7.16 (m, 4H), 6.92 – 7.01 (m, 3H), 6.67 – 6.75 (m, 2H), 6.60 – 6.66 (m, 1H), 5.03 (t, J = 5.56 Hz, 1H), 4.46 (dd, J = 8.19, 15.07 Hz, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.55 – 3.67 (m, 2H), 3.33 – 3.50 (m, 1H), 3.05 – 3.32 (m, 4H), 2.75 – 2.94 (m, 4H), 2.48 (dd, J = 4.66, 15.87 Hz, 1H), 1.38 – 1.52 (m, 2H), 1.22 – 1.35 (m, 6H), 0.82 – 0.91 (m, 3H). m/z 573 (M+H).
Calcium Mobilization Ke Assay for OX1 and OX2
Two individual stable cell lines were created by over-expressing human OX1 and OX2 receptors in CHO-RD-HGA16 (Molecular Devices) cells. The day before the assay, cells were plated into 96-well black-walled assay plates at 25,000 cells/well in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, and 100 μg/mL normocin™. The cells were incubated overnight at 37°C, 5% CO2. Prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to the manufacturer instructions. The reconstituted dye was diluted 1:40 in pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). Growth medium was removed and the cells were gently washed with 100 μL of pre-warmed (37°C) assay buffer. The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 μL of the diluted Calcium 5 dye. A single concentration of each test compound was prepared at 10× the desired final concentration in 2.5% BSA/8% DMSO/assay buffer. Serial dilutions of orexin A were prepared at 10× the desired final concentration in 0.25% BSA/1% DMSO/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37°C. After the dye-loading incubation period, the cells were pre-treated with 25 μL of the test compounds and incubated for 15 min at 37°C. After the pre-treatment incubation period, the plate was read with a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 μL of the orexin A serial dilutions at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against the log of compound concentration. Data were fit to a three-parameter logistic curve to generate EC50 values (Prism, version 6.0, GraphPad Software, Inc., San Diego, CA). Apparent Ke values were calculated using the equation Ke = [L]/((EC50 +/EC50−) – 1) where [L] is the concentration of test compound, EC50 + is the EC50 of orexin A with test compound, and EC50− is the EC50 of orexin A. Ke values were considered valid when the EC50 +/EC50− ratio was at least 4.
Acknowledgments
We are grateful to National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA032837 and DA026582 to Y.Z.) for the financial support of this research. We thank Tiffany Langston, Keith Warner and Dr. Angela Giddings for their valuable technical assistance. We also thank Dr. Gerald Pollard for critical reading of the manuscript.
ABBREVIATIONS
- BOP
(Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HPLC
high performance liquid chromatography
- OX1
orexin 1 receptor
- OX2
orexin 2 receptor
- SAR
structure-activity relationship
- TLC
thin layer chromatography
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierce RC, O’Brien CP, Kenny PJ, Vanderschuren LJ. Cold Spring Harbor perspectives in medicine. 2012;2:a012880. doi: 10.1101/cshperspect.a012880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakurai T. Nippon Rinsho. 2005;63(Suppl 8):600. [PubMed] [Google Scholar]
- 3.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:322. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Journal of Neuroscience. 1998;18:9996. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Brain Research. 1999;827:243. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Hu Z, de Lecea L. Br J Pharmacol. 2014;171:332. doi: 10.1111/bph.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldo BA, Daniel RA, Berridge CW, Kelley AE. J Comp Neurol. 2003;464:220. doi: 10.1002/cne.10783. [DOI] [PubMed] [Google Scholar]
- 8.Fadel J, Deutch AY. Neuroscience. 2002;111:379. doi: 10.1016/s0306-4522(02)00017-9. [DOI] [PubMed] [Google Scholar]
- 9.Espana RA, Melchior JR, Roberts DC, Jones SR. Psychopharmacology (Berl) 2011;214:415. doi: 10.1007/s00213-010-2048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espana RA, Oleson EB, Locke JL, Brookshire BR, Roberts DC, Jones SR. Eur J Neurosci. 2010;31:336. doi: 10.1111/j.1460-9568.2009.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharf R, Guarnieri DJ, Taylor JR, DiLeone RJ. Brain Res. 2010;1317:24. doi: 10.1016/j.brainres.2009.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharf R, Sarhan M, Dileone RJ. Brain Res. 2010;1314:130. doi: 10.1016/j.brainres.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G. Prog Brain Res. 2012;198:79. doi: 10.1016/B978-0-444-59489-1.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris GC, Wimmer M, Aston-Jones G. Nature. 2005;437:556. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 15.Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19168. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. British journal of pharmacology. 2006;148:752. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winrow CJ, Tanis KQ, Reiss DR, Rigby AM, Uslaner JM, Uebele VN, Doran SM, Fox SV, Garson SL, Gotter AL, Levine DM, Roecker AJ, Coleman PJ, Koblan KS, Renger JJ. Neuropharmacology. 2010;58:185. doi: 10.1016/j.neuropharm.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Yang LP. Drugs. 2014;74:1817. doi: 10.1007/s40265-014-0294-5. [DOI] [PubMed] [Google Scholar]
- 19.Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. British journal of pharmacology. 2001;132:1179. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoblock JR, Welty N, Aluisio L, Fraser I, Motley ST, Morton K, Palmer J, Bonaventure P, Carruthers NI, Lovenberg TW, Boggs J, Galici R. Psychopharmacology (Berl) 2011;215:191. doi: 10.1007/s00213-010-2127-x. [DOI] [PubMed] [Google Scholar]
- 21.Uslaner JM, Winrow CJ, Gotter AL, Roecker AJ, Coleman PJ, Hutson PH, Le AD, Renger JJ. Behav Brain Res. 2014;269:61. doi: 10.1016/j.bbr.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Smith RJ, See RE, Aston-Jones G. Eur J Neurosci. 2009;30:493. doi: 10.1111/j.1460-9568.2009.06844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stasi LP, Artusi R, Bovino C, Buzzi B, Canciani L, Caselli G, Colace F, Garofalo P, Giambuzzi S, Larger P, Letari O, Mandelli S, Perugini L, Pucci S, Salvi M, Toro P. Bioorg Med Chem Lett. 2013;23:2653. doi: 10.1016/j.bmcl.2013.02.093. [DOI] [PubMed] [Google Scholar]
- 24.Lebold TP, Bonaventure P, Shireman BT. Bioorg Med Chem Lett. 2013;23:4761. doi: 10.1016/j.bmcl.2013.06.057. [DOI] [PubMed] [Google Scholar]
- 25.Perrey DA, German NA, Decker AM, Thorn D, Li JX, Gilmour BP, Thomas BF, Harris DL, Runyon SP, Zhang Y. ACS chemical neuroscience. 2015;6:599. doi: 10.1021/cn500330v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrey DA, German NA, Gilmour BP, Li JX, Harris DL, Thomas BF, Zhang Y. J Med Chem. 2013;56:6901. doi: 10.1021/jm400720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrey DA, Gilmour BP, Runyon SP, Thomas BF, Zhang Y. Bioorg Med Chem Lett. 2011;21:2980. doi: 10.1016/j.bmcl.2011.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silverman RB. The Organic Chemistry of Drug Design and Drug Action. 2. Elsevier Academic Press; 2012. [Google Scholar]
- 29.Smith DA. Metabolism, Pharmacokinetics, and Toxicity of Functional Groups: Impact of the Building Blocks of Medicinal Chemistry in ADMET. Royal Society of Chemistry; 2010. [Google Scholar]
- 30.Gatfield J, Brisbare-Roch C, Jenck F, Boss C. ChemMedChem. 2010;5:1197. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 31.Koberstein R, Aissaoui H, Bur D, Clozel M, Fischli W, Jenck F, Mueller C, Nayler O, Sifferlen T, Treiber A, Weller T. Chimia. 2003;57:270. [Google Scholar]
- 32.Anderson WK, McPherson HL, Jr, New JS, Rick AC. J Med Chem. 1984;27:1321. doi: 10.1021/jm00376a017. [DOI] [PubMed] [Google Scholar]
- 33.Perrey DA, Gilmour BP, Thomas BF, Zhang Y. ACS medicinal chemistry letters. 2014;5:634. doi: 10.1021/ml4004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malherbe P, Borroni E, Pinard E, Wettstein JG, Knoflach F. Mol Pharmacol. 2009;76:618. doi: 10.1124/mol.109.055152. [DOI] [PubMed] [Google Scholar]
- 35.Kenakin T, Jenkinson S, Watson C. J Pharmacol Exp Ther. 2006;319:710. doi: 10.1124/jpet.106.107375. [DOI] [PubMed] [Google Scholar]
- 36.Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F, Boss C. ChemMedChem. 2013;8:898. doi: 10.1002/cmdc.201300003. [DOI] [PubMed] [Google Scholar]