

Abstract
Ceramide is a bioactive sphingolipid involved in mitochondrial-mediated apoptosis. Our data suggest that ceramides directly regulate a key initiation step in apoptosis: mitochondrial outer membrane permeabilization (MOMP). MOMP allows release of intermembrane space proteins to the cytosol, inducing the execution of the cell. Ceramides form channels in planar phospholipid membranes and outer membranes of isolated mitochondria, channels large enough to facilitate passage of proteins released during MOMP. Bcl-xL inhibits MOMP in vivo and inhibits the formation of ceramide channels in vitro. However the significance of Bcl-xL’s regulation of ceramide channel formation within cells was untested. We engineered Bcl-xL point mutations that specifically affect the interaction between ceramide and Bcl-xL to probe the mechanism of ceramide channel regulation and the role of ceramide channels in apoptosis. Using these mutants and fluorescently-labeled ceramide, we identified the hydrophobic groove on Bcl-xL as the critical ceramide binding site and regulator of ceramide channel formation. Bcl-xL mutants with weakened interaction with ceramide also have reduced ability to interfere with ceramide channel formation. Some mutants have similar altered ability to inhibit both ceramide and Bax channel formation, whereas others act differentially, suggesting distinct but overlapping binding sites. To probe the relative importance of these channels in apoptosis, Bcl-xL mutant proteins were stably expressed in Bcl-xL deficient cells. Weakening the inhibition of either Bax or ceramide channels decreased the ability of Bcl-xL to protect cells from apoptosis in a stimulus-dependent manner. These studies provide the first in vivo evidence for the role of ceramide channels in MOMP.
Keywords: Bax, apoptosis, mitochondria, sphingolipid, outer membrane, Bcl-2
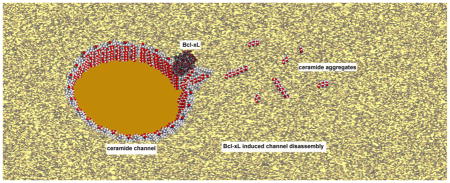
GRAPHICAL ABSTRACT
1. INTRODUCTION
The release of mitochondrial intermembrane space (MIS) proteins into the cytosol is a key decision-making step in the apoptotic process. The released proteins initiate the execution phase of apoptosis via a series of steps that ultimately lead to the activation of caspases, specific proteases that lead to the transformation of the cell into apoptotic bodies that are engulfed by phagocytes. Various mechanisms exist by which proteins can be released from the MIS (1–4). Some of these involve the formation of large channels in the mitochondrial outer membrane (MOM). Two such channels are formed by either the translocation to the MOM and oligomerization of a cytosolic protein called Bax or the self-assembly of a MOM lipid called ceramide (5–9). Bax is a member of the Bcl-2 family of proteins that regulate apoptosis (10–12). Both Bax and Bak are pore-forming proapoptotic members of this family. Ceramide is a pore-forming member of the sphingolipid family of cell lipids, some of which are involved in the apoptotic process (13). The functions of Bax, Bak and ceramide are interdependent in a number of ways. In some systems, the induction of apoptosis by ceramide requires the presence of Bax (14, 15). The Bak protein is required for ceramide synthase-dependent long-chain ceramide synthesis during apoptosis (16). There is evidence for synergy between Bax and ceramide in MOM permeabilization (MOMP) in isolated mitochondria (17, 18).
The role of Bax in MOMP is well established (12) although the structure of the Bax channels is only poorly understood (19). Ceramide’s role in inducing apoptosis is also widely acknowledged (20, 21) although its ability to directly form channels in the MOM is still viewed with skepticism. Published work strongly supports the ability of ceramide to form channels in the MOM at physiologically relevant concentrations (4). Ceramide forms large channels (typically 10 nm in diameter) in phospholipid membranes lacking proteins and these have been visualized by electron microscopy (22). It has also been shown to release a variety of MIS proteins from isolated mitochondria while not releasing matrix proteins (5). Functional studies (23) and molecular dynamic (MD) simulations (24) support a model of the ceramide channels that consists of barrel-like structure whose staves are membrane-spanning strings of ceramide molecules organized in an anti-parallel manner (S1). These thin-walled pores behave like elastic discs that can be deformed but will regain their natural structure after relaxation (25). Yet all evidence indicates (5) that they are in dynamic equilibrium with ceramide monomers or non-conducting ceramide assemblies. Thus, there is a great deal of dynamic motion and structural changes associated with these channels.
The formation of both Bax and ceramide channels is inhibited by the anti-apoptotic protein, Bcl-xL (26, 27). Whereas, it is generally agreed that Bcl-xL inhibits Bax channel formation by forming heterodimers with activated Bax monomers (27, 28), the mechanism by which Bcl-xL inhibits ceramide channels is not well understood. Since ceramide channels are large assemblies of ceramide monomers, there is no precedent to indicate possible mechanisms by which such a structure could be prevented from forming or be destabilized by a protein although a mechanism has been proposed (29). Experiments with ceramide analogs demonstrated that the hydrophobic tails of ceramide are important for the inhibition by Bcl-xL (29). In addition, small molecule inhibitors (2-methoxyantimycin A3, ABT-737 and ABT-263), known as BH3 peptide mimetics because they bind to a hydrophobic grove on Bcl-xL and thus block its ability to bind to Bax, were also found to inhibit the ability of Bcl-xL to inhibit the formation of ceramide channels in the MOM (29). These inhibitors interfere with the anti-apoptotic activity of Bcl-xL (30–32) suggesting that the ability of Bcl-xL to interfere with ceramide channel formation may somehow be linked to its anti-apoptotic function. Here we provide insight into the molecular basis by which Bcl-xL acts to inhibit ceramide channels by providing evidence for direct binding and using point mutations to identify the region on Bcl-xL responsible for the inhibition. These same Bcl-xL mutants were expressed in cells lacking Bcl-xL to assess the importance of ceramide channels in the in vivo apoptotic process.
2. MATERIALS AND METHODS
2.1 Isolation of rat liver mitochondria
Mitochondria were isolated from the liver of male Sprague Dawley rats as described originally (33) and as modified (29). The animal use protocols were approved by the Institutional Animal Care and Use Committee. The animals were euthanized by a procedure consistent with the Panel on Euthanasia of the AVMA (American Veterinary Medical Association). The animal facility used to house the animals is accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care).
2.2 Purification of recombinant proteins
The construct of full-length human Bcl-xL (Bcl-xL wild-type), Bax and t-Bid were gifts from Dr. Marie Hardwick, Dr. Richard Youle and Dr. Donald Newmeyer, respectively. All single mutations of Bcl-xL were introduced using QuickChange Site-Directed Mutagenesis kit (Stratagene) and verified by DNA sequencing. Recombinant Bcl-xL and its mutants were purified as previously described (34) and as modified (29). In brief, GST-tagged Bcl-xL and GST-tagged Bid were produced in BL21(DE3) pLysS. The cells were induced with 10 μM IPTG for 2 hours at 37°C for GST-tagged Bcl-xL and the cells were induced with 0.4 mM IPTG for 3 hours at 37°C for GST-tagged Bid, resuspended in PBS with 1.25 kU/L cells of lysozyme and 35 μM PMSF and lysed using a French press. After removal of cell fragments, the GST-tagged Bcl-xL and GST-tagged t-Bid were purified using glutathione agarose beads and the GST tag was cleaved with 5 U biotinylated thrombin which was removed using streptavidin beads. Recombinant human Bax was purified using a chitin column (New England Biolabs Inc.) and dialyzed with a 12,000 MW cut-off dialysis membrane to remove dithiothreitol (17, 35, 36). All purified proteins were supplemented with glycerol to 10 % and filtered sterilized through a 0.2 μm filter prior to being rapidly shell-frozen in ethanol and dry ice and stored at −80 °C. The concentration of proteins was determined with a MicroBCA Protein Kit (Pierce Chemical).
2.3 Binding of ceramide to Bcl-xL
nine microliters of fluorescently-labeled ceramide (C11 TopFluor ceramide; Avanti Polar Lipids) dissolved in isopropanol at 42 μg/ml was dispersed to 590 μl containing 1.8 μM Bcl-xL wild-type or its mutants in 20 mM Tris, pH 7.4, 10% sucrose. The dispersal was performed by slow delivery of the ceramide solution into the protein solution while it was being vortexed. Entrainment of air was carefully avoided. In order to remove ceramide micelles, 50 μl of 20 mM Tris, pH 7.4, 5% sucrose was applied to the top of the mixture to produce a density gradient. Then the gradient was spun in MLA-130 at 55,000 rpm at 25°C for 30 min. Ceramide micelles float into the upper layer and so a portion of the solution in the 10% sucrose phase was removed, excited by 490 nm incident light and the emission spectrum from 495 to 520 nm was recorded in a FluoroMax-4. The fluorescence was not detected in the non-protein controls, so all fluorescence was due to ceramide associated with protein. The fluorescent quantum yield of the bound ceramide varied depending on the protein used. Thus, in order to assess the amount of ceramide bound, a portion of the fluorescent solution was mixed with the same volume of isopropanol (to denature the protein and extract the ceramide) and spun at 18,000 rcf at 4°C for 40 min. The emission spectrum of the supernatant was recorded.
2.4 Cytochrome c accessibility assay
The rate of oxidation of exogenous reduced cytochrome c by cytochrome c oxidase in the isolated mitochondria indicates the permeability of the MOM, because translocation through the outer membrane is a rate limiting step (37). The procedure used was as previously described (29) except that different concentrations of Bcl-xL and its mutants were mixed with isolated mitochondria in reaction buffer (280 mM mannitol, 0.1 mM EGTA, 5 mM HEPES, pH 7.25, 5 mM, 2,4-Dinitrophenol and 1.3 μM antimycin A) before N-palmitoyl-D-erythro-sphingosine (C16-ceramide) dispersal. The amount of cytochrome c that was oxidized was determined by measuring the change in absorbance 550 nm and quantitated using an extinction coefficient of 18.5 mM−1 · cm−1 (the difference between the reduced and oxidized form). At dose used there is no significant effect of antimycin A of the ability of Bcl-xL to inhibit ceramide (29).
2.5 Adenylate kinase assay
This assay measures the release of adenylate kinase from the MIS. This indicates the portion of mitochondria whose outer membrane was made permeable to proteins. The procedure was performed as previously described (38) except that the final concentration of KCl was kept at 60 mM. This assay was used to assess the permeabilization of the MOM by Bax.
2.6 Molecular dynamic simulations
The structure of full-length Bcl-xL-ceramide assembly was based on the published model from MD studies (29) that were performed using the crystallographic structure of the Bcl-xL/Bim fragment complex (PDB ID: 1PQ1). In the experimental setting, the N-terminal part of Bcl-xL is preceded by a few extra amino acids (GSPRRS) inherited after thrombin cleavage. To better match with the experiments, we attached this fragment to N-terminus of Bcl-xL (however being so remote from the ceramide binding site, this modification should not have any significant effect). Fragments of Bcl-xL, unresolved in the crystal structure (residues 29 to 77) as well as the added N-terminal fragment were modelled using Robetta (39). N- and C-termini of the final Bcl-xL structure were modeled in the dissociated state.
The complete system (96080 atoms) contained 3686 protein atoms (239 residues), 106 atoms of C16-ceramide (one residue), and 2392 atoms of D-mannitol (92 residues – matching 160 mM mannitol in the experimental medium). To maintain the electroneutrality of the system K+ and Cl− ions were added up to an equivalent of 60 mM salt concentration, 43 K+ and 34 Cl−. Assembly of the simulation system, introduction of the single mutations into the Bcl-xL structure (see the main text), analysis of the results and visualization were performed in VMD v1.9 (40) using custom-written Tcl scripts.
MD stimulations were done using the NAMD package (41). All the simulations were performed as an NPT ensemble using the CHARMM36 force field (42) and TIP3P water model (43). Parameters for ceramide were taken from the previously published simulations (24). The acidic and basic residues of Bcl-xL were set in their default protonation state at pH 7.0 (estimated using PROPKA for neutral pH (44)). Langevin Dynamics (45, 46) was used to maintain constant pressure (1 atm) and constant temperature (295.15° K). Periodic boundary conditions were maintained and the particle mesh used the Ewald method (47) with a real space cutoff distance of 1.2 nm and a grid width of 0.1 nm. Energy Minimization steps were performed using the steepest descent in the first 2000 steps and then a conjugate gradient in the subsequent 2000 steps. To attain the equilibrium, the system was subjected to gradual heating until it reached to 295.15°K (22°C), first with the protein backbone harmonically restrained (100 kcal/mol/nm2 per backbone atom) to the initial coordinates for 1 ns, then simulated unrestrained for 30 ns.
2.7 Statistics for all studies except the whole-cell experiments
The results are reported as the mean ± SE of at least 3 independent experiments. Significance was determined by using the Student’s t-test. Single, double, triple and quadruple symbols, indicate significance with P-values < 0.05, < 0.01, <0.001 and <0.0001 respectively.
2.8 Cell culture
Mouse embryonic fibroblast (MEF) Bcl-x-KO cell line was kindly provided by Dr. Chi Li (University of Louisville, Louisville, KY) (48). Cells were maintained in DMEM (Thermo Scientific, SH30243.01) containing 10% FBS (Thermo Scientific, SH30070.03) supplemented with 1X pen-strep (Gibco 15140-122). Cells were routinely tested to verify that they were free of mycoplasma infection.
2.9 Generation of cell lines
Full length human Bcl-xL cDNA and single point mutation Bcl-xL mutants (generated by using QuickChange II XL Site-Directed Mutagenesis kit from Agilent Technologies, # 200521-5 and verified by DNA sequencing) were cloned into the GFP expressing mammalian expression retroviral vector MIGRX EcoRI/XhoI to generate MIG-Bcl-xL or MIG-Bcl-mutant plasmids. MEF Bcl-xL-KO cells were infected with viruses expressing these mutants and infected cell populations were selected by sorting of GFP expressing cells by flow cytometry to generate stable cell lines. Presence of Bcl-xL WT and mutants in MEF-Bcl-xL-KO stable cell lines was confirmed by western blot as described below.
2.10 Western blot
Cell pellets were collected and lysed in NP-40 lysis buffer (Boston BioProducts, BP-119). Protein concentrations were determined using BCA protein assay reagent (Pierce #23223 and #23224). Twenty micrograms of protein samples were loaded on a 4–12% Bolt Bis-Tris Plus gel (Life technologies, BG04120BOX) and transferred to PVDF membrane (Bio-Rad, #162-0177). Membranes were blocked in TBS-T with 5% milk for 1 hour at room temperature. Membranes were incubated overnight at 4°C with primary antibodies; anti-Bcl-xL (54H6) (Cell signaling, # 2764), anti-GFP (Abcam, ab6673) and anti-tubulin (Sigma Aldrich, #T5168) at 1: 20,000 dilutions. Anti-mouse and anti-rabbit peroxidases labelled secondary antibodies were added at a dilution of 1:40,000 for 1 h at room temperature. Chemiluminescent detection was performed using Pierce ECL Western Blot Substrate (Thermo Scientific, #32106).
2.11 Cell Viability Assay
MEF cells were cultured in 96- well plates (2,500 cells per well) overnight and then treated with different apoptotic stimuli at indicated drug concentrations for 24 h. Cells were then incubated with 10% Alamar Blue reagent (Invitrogen, DAL1100) for 4 h and the fluorescence of Alamar Blue reduction was determined using BioTek HT Synergy plate reader (540 nm excitation, 594 nm emission). Relative viability of treated cells was calculated by normalizing to vehicle treated cells. For bar graphs, relative cell viability was presented as normalized with respect to WT at that dose of a drug. Cisplatin (# 4394), Thapsigargin (# T9033), Etoposide (#, E1383) and Gemcitabine (#, G6423) were purchased from Sigma. Hydrogen peroxide was purchased from Fisher scientific (# H325-100). Doxorubicin (#, 15007) was purchased from Cayman chemical company. Bortezomib was purchased from ChemieTek (#, CT-BZ001). All treatments were done in triplicate and each graph shown is a representative experiment of at least three biological replicates. Statistical analysis was performed using one-way ANOVA with Tukey’s test. p < 0.05 was considered significant.
3. RESULTS
Previous studies demonstrated that Bcl-xL inhibits ceramide channel formation in isolated mammalian mitochondria (26, 29). In addition, the use of ceramide analogs indicated that the hydrophobic regions of ceramide are important for this action of Bcl-xL (29). To distinguish between direct and indirect action, it would be desirable to determine whether Bcl-xL binds a ceramide channel. However, direct binding studies are not possible because ceramide channels are in dynamic equilibrium with ceramide monomers (5) and thus cannot be isolated and purified. The possibility of examining the binding of Bcl-xL to ceramide channels formed in liposomes is also not feasible because Bcl-xL binds to membranes regardless of the presence of ceramide channels and current methods of generating ceramide channels in liposomes cause only partial release of contents indicating that only one channel is formed per liposome (49, 50). Thus any binding studies would be plagued by very low signal to noise ratios. An alternative method is to determine whether a ceramide molecule is capable of binding to Bcl-xL. The binding of a ceramide molecule to Bcl-xL may mimic the binding between Bcl-xL and ceramide channels.
3.1 Binding of ceramide to Bcl-xL
Binding studies between Bcl-xL and ceramide are complicated by the physical properties of ceramide. Ceramide is inherently insoluble in water and Bcl-xL cannot be expected to function normally in an organic solvent. Therefore, an equilibrium binding experiment is not possible. However, when ceramide dissolved in isopropanol is dispersed in an aqueous environment, ceramide molecules exist transiently in aqueous solution and these can either bind to Bcl-xL or combine with each other to form micelles. If the Bcl-xL/ceramide complex is formed it is unlikely to dissociate because of ceramide’s insolubility in water. Thus the complex should be stable and can be detected. However, a binding constant cannot be determined. Thus fluorescently-labeled ceramide (C11 TopFluor ceramide) was dispersed into a solution containing Bcl-xL (Fig. 1A). Any fluorescent ceramide that does not bind must form micelles. The fluorescent ceramide micelles were removed by centrifugation taking advantage of the fact that the density of the micelles is less than that of the aqueous solution and this difference was increased by using 10 % sucrose. The fluorescence of the subphase was recorded. In the absence of Bcl-xL, the centrifugation cleared the solution of ceramide micelles resulting in no detectable fluorescence. However, in the presence of Bcl-xL substantial fluorescence was recorded (Fig. 1B). The results indicate that the ceramide molecule binds to Bcl-xL directly.
Fig 1.

Fluorescently-labeled ceramide binds to Bcl-xL. A. Separation of fluorescently-labeled ceramide bound Bcl-xL from ceramide micelles. F1 is fluorescently-labeled ceramide: C11 TopFluor ceramide. B. Fluorescence spectrum of final sample from the separation shown in “A” (see methods for details). “WT” indicates the medium contained wild-type Bcl-xL and fluorescence was due to bound fluorescently-labeled ceramide. “buffer” indicates that there was no Bcl-xL in that separation procedure. The lack of fluorescence indicates that the procedure was effective at eliminating ceramide micelles. The inset is the structure of fluorescently-labeled ceramide.
3.2 Determination of the location of the binding region on Bcl-xL
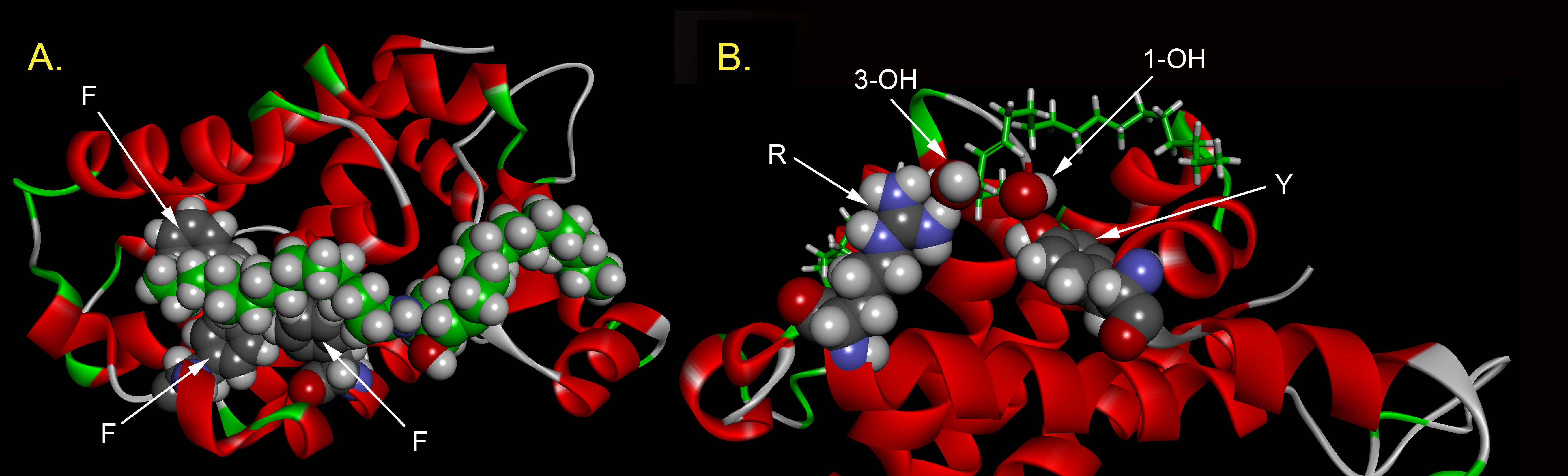
Previous studies showed that drug molecules that bind to hydrophobic groove on Bcl-xL (ABT-263 and ABT-737) interfered with the protein’s ability to inhibit ceramide channel formation (29). Thus this region of Bcl-xL is a good candidate for the putative binding site by which Bcl-xL inhibits ceramide channels. To identify residues in the hydrophobic groove that may be important in regulation of ceramide channels, molecular docking of a ceramide molecule to Bcl-xL was conducted and two low-energy modes were used (pose1 and 2) (29) (Fig 2A). Single amino acid substitutions were made so as to change the strength of the interaction at the putative docking sites. Changes in charge (E96L and R100L), polarity (F97Q, F105Q, V126Q), and physical volume (V126W, F146A, Y195A) were engineered into full-length Bcl-xL (Fig. 2A). These were tested in isolated mitochondria for their ability to interfere with ceramide channel formation.
Fig. 2.

A. One docking pose of ceramide on the crystal structure of Bcl-xL. The amino acids to be mutated are illustrated (yellow). Ceramide is in green with the two chains specified. The ceramide amide linkage is in the middle with the amide nitrogen in blue and the carbonyl oxygen in red. The pose was obtained from the publication of Perera et al., 2012 (29). B. Permeabilization of the MOM to cytochrome c estimated by measuring the initial rate of oxidation of added cytochrome c (24.7 μM final). To 730 μL of 0.25 mg/mL rat liver mitochondrial suspension was added 15 μL of either isopropanol (iso) or C16 ceramide (cer). The results were normalized by the absorbance of cytochrome c at zero time. WT, F105Q, R100L indicated that mitochondria had been preincubated for 5 min with 47 nM wild-type Bcl-xL or the respective mutants.
As illustrated in Fig. 2B, the amount of reduced cytochrome c in the medium decreased as the added mitochondria oxidized the protein. The cytochrome c could only be oxidized if it could cross the outer membrane and bind to cytochrome c oxidase on the outer surface of the inner membrane. Thus the rate of oxidation of the cytochrome c is related to the degree of MOMP by ceramide channels. The baseline rate of oxidation occurred when the vehicle (isopropanol) was added to the mitochondria. When ceramide dissolved in isopropanol was dispersed into the mitochondrial suspension, the rate of oxidation increased. This rate was diminished by the presence of Bcl-xL in the medium. Many of the altered proteins had lost some of their ability to inhibit ceramide-induced MOMP, but one mutant was actually a more potent inhibitor than the wild-type. In order to achieve a greater dynamic range, experiments were performed at two levels of added Bcl-xL protein: a low level resulting in a weaker inhibition (i.e. a greater ceramide-induced MOMP) to detect mutant proteins that had a stronger inhibition than the wild type, and a high level to detect mutants with a weaker inhibitory affect. Figs. 3A and 3B show the measured % inhibition for one set of experiments using 0.72 and 2.1μM Bcl-xL protein, respectively. Note that the stronger inhibitory effect of V126Q was only detected at the lower concentration and the weaker inhibitors were apparent at the higher protein concentration. Since experiments with wild-type and mutant Bcl-xL were performed in parallel, the level of inhibition of the ceramide channels was normalized to that of the wild-type protein (Fig. 3C) and then data from independent experiments was pooled and averaged. This normalization was necessary because the potency of the Bcl-xL varied from one batch of isolated mitochondria to another. The data for the enhanced inhibition came from the low Bcl-xL concentration experiments whereas the data for the reduced inhibition came from the high concentration experiments. The mutants are organized in a pattern reflecting their location on the docking site (Fig. 2A). The results follow a distinct pattern.
Fig. 3.

The inhibitory potency of Bcl-xL mutants on ceramide channels formed in isolated mitochondria. The final concentration of added Bcl-xL or mutant proteins was 0.72 μM (A) and 2.1 μM (B). C. Relative inhibition of ceramide induced MOMP by Bcl-xL mutants. The inhibitory ability is expressed relative to that of the wild-type so that if it is the same as the wild type the result would be 1. As indicated in the text, this corrects both for variability from experiment to experiment and for the use of different concentrations of protein to reveal changes in inhibitory potency of both the weak and strong mutants. The statistically significant differences from the potency of the wild-type are indicated by the asterisk.
All but one of the mutants (E96L) were able to inhibit the formation of ceramide channels, the difference being the amount of protein needed to achieve the same level of inhibition. Thus perhaps E96 is such a critical site that removal of the carboxyl group results in failure to bind ceramide. V126W was nearly as effective as the wild-type, as expected from the mild change in the structure of the side chain. The inhibition capability of F97Q, R100L, F105Q, F146A and Y195A was weaker than that of the wild type whereas the capability of V126Q was stronger. From the docking pose, R100 hydrogen bonds with the C3 hydroxyl of ceramide. The same was seen in the MD simulations (S2). The hydroxyl group of tyrosine 195 interacts with the carbonyl of ceramide in the docking experiment whereas the MD simulations show intimate dipole-dipole interactions between this hydroxyl and the C1 hydroxyl of ceramide (S1A). Thus the weaker interaction of these mutations correlates with the structural information provided by the MD simulations. F97Q, F105Q and F146A disrupted hydrophobic interactions suggesting that these residues interact with the hydrophobic tails of ceramide. These results agree with one of the Bcl-xL/ceramide binding poses identified in the docking studies. In the MD simulations these three residues form the major portion of the hydrophobic surface that interacts with the ceramide acyl chain again confirming the importance of retaining these apolar surfaces (S1B).
3.3 Correlation between the quantum yield of fluorescent ceramide bound to Bcl-xL mutants and their ability to inhibit MOMP
The Bcl-xL mutants were tested for their ability to bind fluorescently labeled ceramide as was performed in Fig. 1. Direct binding measurements did not yield satisfactory results for 2 reasons: 1) The variability in the ceramide dispersion process resulted in variable amounts of binding. 2) The unidirectional nature of the binding process in aqueous solution means that changes in the rates of dissociation could not affect the amount of binding. However, in attempting these experiments it was noted that fluorescently labeled ceramide bound to mutated Bcl-xL produced much less fluorescence than the same ceramide bound to wild-type Bcl-xL. Fig. 4A shows the reduction of ceramide fluorescence upon binding to Bcl-xL. The solid circles show the spectrum of the ceramide bound to the Bcl-xL, whereas the open circles show the spectrum after extraction with isopropanol. These relative values were adjusted for the volume of the solution. The reduction in fluorescence is probably the result of exposing the fluorophore to a polar environment that quenches the fluorescence. Fig. 4B shows the fluorescent intensity of the same amount of ceramide bound to either wild-type Bcl-xL or Bcl-xL mutants. This difference is most likely due to greater interaction of the fluorophore with water. In a tight-binding situation water will have less contact with the fluorophore and thus result in less quenching. There is a good correlation between the intensity of the measured fluorescence and the potency of the mutant (Fig. 4C) to destabilize ceramide channels. These results indicate that binding of Bcl-xL to the ceramide channel was the basis for the inhibitory effect and thus the ceramide channel was being directly controlled by this anti-apoptotic protein.
Fig. 4.

A. Fluorescence spectrum of C11 TopFlour bound to Bcl-xL (WT + Cer), C11 TopFlour extracted with isopropanol (WT extracted), and controls lacking Bcl-xL. B. Fluorescence spectrum of C11 TopFlour bound to wild-type Bcl-xL (WT) and to various Bcl-xL mutants. The curves were normalized by the fluorescence of the extracted fluorescent ceramide. Thus changes in fluorescence intensity were due to reductions in the quantum yield due to some quenching process. C. Correlation between the quenching of fluorescent ceramide bound to Bcl-xL mutant protein and the ability of the same protein to inhibit ceramide permeabilization of the MOM.
3.4 Molecular dynamic simulations of ceramide binding to Bcl-xL
Wild type Bcl-xL and 5 mutants (E96L, R100L, F105Q, V126Q and Y195A) that span the range of abilities to inhibit ceramide permeabilization of the MOM, were examined using MD simulations to explore their interaction with a C16-ceramide molecule. In the first few nanoseconds of simulation the ceramide found its optimal position in the Bcl-xL hydrophobic pocket and from then on the positions varied according to local interactions with the Bcl-xL protein and the water environment. All the introduced mutations are among the residues that are likely to interact with ceramide; they do not participate in any significant inter-helical interactions that maintain protein fold. Therefore, the mutants are not likely to affect the protein structure itself. Indeed, in simulations the structure of the ceramide-binding region of Bcl-xL changed very little in the mutants compared to the wild-type (0.15± 0.04 nm all-mutant mean deviation of the backbone from the WT position) indicating that the mutations acted locally, on the protein-ceramide interactions rather than globally, through a change in the protein structure. However the results showed considerable changes in the location and dynamics of the bound ceramide −1.0±0.3 nm all-mutant mean deviation of ceramide from its average position in WT Bcl-xL. The dynamic motion of the ceramide molecule (S3) correlates very well with the loss of ability to inhibit ceramide permeabilization (Fig 5, Table 1), i.e. ceramide channel formation. The highest correlation was observed when the distal half of the sphingosine acyl chain was excluded. The fluorophore label experiencing more quenching with reduced inhibitory ability of the Bcl-xL mutant agrees with the simulation findings of greater mobility. Greater mobility should result in more access to water dipoles and thus more water-induced quenching. Note that the fluorophore is on the fatty acyl chain whose mobility correlates well with loss of Bcl-xL inhibitory ability.
Fig. 5.

Correlation between the dynamic motion of the ceramide molecule and the ability of a Bcl-xL mutant to inhibit ceramide permeabilization of the MOM. The mobility (RMSF) of regions of the ceramide molecule (inset) were measured. Those regions surrounded by a solid line are plotted as a solid line in the main figure. Similarly for the dotted and dashed line. The inhibitory ability (horizontal axis) is expressed relative to that of the wild-type so that if it is the same as the wild type the result would be 1.
TABLE 1.
Correlation between the dynamic motion (RMSF) of different regions of the ceramide molecule and the ability of Bcl-xL to inhibit ceramide permeabilization of the MOM
| Mobility | Deviation from wild type | ||
|---|---|---|---|
| Location | Correlation | Location | Correlation |
| Ceramide without sphingosine tail after C10 | −0.96 | Ceramide amide oxygen | −0.63 |
| Ceramide up to C10 and C8′ | −0.96 | Ceramide polar atoms | −0.60 |
| Ceramide amide tail C9′–C16′ | −0.91 | Ceramide headgroup | −0.59 |
| Ceramide whole | −0.78 | Ceramide fatty acid residue | −0.57 |
| Ceramide tail C10–C8′ | −0.57 | Ceramide amide tail | −0.54 |
| Bcl-xL | −0.09 | Ceramide whole | −0.24 |
| Bcl-xL in contact with Ceramide | −0.07 | ||
The relative location of the ceramide on Bcl-xL compared to the wild-type also correlates, although more weakly, with the loss of inhibitory ability. This is reasonable as a change in location could influence the ability of the bound Bcl-xL to destabilize the channel.
3.5 Comparison of the binding footprint on Bcl-xL of Bax and the ceramide channel
Bcl-xL is known to inhibit the formation of Bax channels in the mitochondrial outer membrane. The hydrophobic pocket of Bcl-xL binds the BH3 domain of pro-apoptotic proteins such as Bax and Bid (27, 28, 51) and thus inhibits their pro-apoptotic activity, including channel formation. The Bcl-xL mutants were tested for their effects on Bax channel formation in the MOM to determine if their relative potency is similar to that observed for ceramide channels. A complicating factor is that truncated Bid (t-Bid) is necessary to activate Bax resulting in MOMP and thus one cannot distinguish between an inhibitory effect on Bax or on t-Bid. Regardless of which protein binds to Bcl-xL, the mutants will probe whether the same region of Bcl-xL is used in both cases. Once again, in order to achieve a greater dynamic range, the amount of wild type protein added was adjusted to obtain a weaker (Fig. 6A) or a stronger (Fig. 6B) inhibition. The results show that V126Q and Y195A have a stronger inhibitory effect than the wild-type. F146A, R100L, V126W, F105Q, F97Q and E96L have a weaker inhibitory effect. As in the case of the ceramide channel, E96L has no effect. A comparison of the inhibitory effects of mutated Bcl-xL on Bax (Fig. 6C) and ceramide channels (Fig. 3C) reveals that two mutations have distinctly different effects on the two channels. A plot of one set of data vs the other (Fig. 7) shows that Y195A has stronger inhibition of Bax channels as compared to the wild-type protein but a weaker inhibitory effect on ceramide channels. V126W was not able to inhibit Bax channels as well as the wild type, but had the same ability to inhibit ceramide channels as the wild type. This information could be used to distinguish between apoptosis resulting from MOMP due to activated Bax or ceramide.
Fig. 6.

The inhibitory potency of Bcl-xL mutants on Bax permeabilization of the MOM. The final concentration of added Bcl-xL or mutants was 0.72 μM (A) and 0.86 μM (B). C. Relative inhibition of Bax permeabilization of MOM by Bcl-xL mutants. The inhibitory ability is expressed relative to that of the wild-type so that if it is the same as the wild type the result would be 1. The statistically significant difference from the potency of the wild-type is indicated by the asterisks.
Fig. 7.

Correlation between the ability of mutant Bcl-xL to inhibit MOM permeabilization by Bax and by ceramide.
3.6 Probing the relative importance of ceramide and Bax channels in apoptosis in intact cells
Both ceramide and Bax form channels that release proteins from the MIS. These two agents also act synergistically to induce MOMP (17). However it is unclear which channel-former is used in vivo. The Bcl-xL mutant proteins were expressed in cells lacking Bcl-xL in order to obtain some insight into this question. Cells expressing either wild type or one of three mutants (Y195A, V126Q, V126W) were stimulated to undergo apoptosis using 7 different chemical stimuli (cisplatin, gemcitabine, hydrogen peroxide, thapsigargin, doxorubicin, etoposide, or bortezomib) that activate different apoptotic pathways. A comparable level of expression of mutant proteins was achieved and a dose response curve was performed for all of the above stimuli. Fig. 8 shows an example of the dose-response curves. Two doses were selected for detailed statistical analysis, one in the transition region between no drug effect and the full effect and another immediately after a maximal effect of the chemical agent was achieved.
Fig. 8.

Relative cell viability after treatment with the indicated amount of bortezomib. Bcl-xL deficient MEF cells were engineered to stably express either wild type Bcl-xL (WT) or Bcl-xL point mutants V126W, V126Q, or Y195A.
V126W inhibits ceramide channels as well as wild-type Bcl-xL (Fig. 9) but has only half the potency on Bax channels. In most cases this mutant was less effective than the wild-type in protecting cells from apoptosis, consistent with the important role of Bax channels in the apoptotic process. V126Q is more effective at inhibiting both Bax and ceramide channels (Figs. 3 and 6); cells expressing V126Q were often more resistant to death stimuli than the wild type (gemcitabine low dose, bortezomib low dose, hydrogen peroxide, and thapsigargin high dose). Y195A is also more potent at inhibiting Bax channels but only half as potent on ceramide channels (Figs. 3 and 6). Cells expressing Y195A were more sensitive to gemcitabine (low dose), doxorubicin (high dose), and bortezomib (low dose) than cells expressing V126Q even though Y195A is a more potent inhibitor of Bax channels (Figs. 9 and 6). In the case of the high dose of bortezomib, cells expressing Y195A are more sensitive than cells expressing the wild-type Bcl-xL (Fig. 9B). The results with Y195A provide evidence for the role of ceramide channels in cells treated with these compounds. In other cases, such as thapsigargin treatment, the weaker ability of Y195A to inhibit ceramide channels had no effect as the cell viability observed with cells expressing V126Q and Y195A were not different. Thus ceramide channels may play a role in apoptosis induced by specific stimuli.
Fig. 9.

A Relative viability of cells expressing the indicated Bcl-xL and induced to undergo apoptosis with the indicated amount of chemical agent. The significant differences are indicated as follows: *, compared to empty vector; #, compared to V126W; $, compared to wild type (WT); +, compared to V126Q. B Additional apoptosis inducers are listed with details as in A.
4. DISCUSSION
The surprising ability of a lipid, ceramide, to form large, stable channels in phospholipid membranes was initially met with skepticism. Now the evidence supporting the existence of ceramide channels is overwhelming. This includes recording their properties in defined planar phospholipid membrane systems (23, 52), visualization by electron microscopy (22), demonstration of the stability of the ceramide channel model by molecular dynamic simulations (24), and examination of the elastic properties of these channels by microfluidic methods (25). The role of these channels in apoptosis was indicated by the ability of ceramide to permeabilize the MOM to proteins even when the source of the mitochondria was the yeast, S. cerevisiae, an organism lacking the Bcl-2 family of proteins (26). The evidence for this role was substantially increased by the findings that Bcl-2, Bcl-xL and CED-9 all inhibit ceramide channel formation in isolated mitochondria (26) whereas Bax acts synergistically with ceramide to permeabilize the MOM (17). Further support of a specific regulatory role of ceramide channels by Bcl-2 family proteins was the finding that Bcl-xL acts by interacting with the hydrophobic region of ceramide whereas Bax preferentially interacts with the polar regions (29). Furthermore, the results demonstrated that the ability of Bcl-xL to inhibit ceramide channel formation cannot be simply attributed to hydrophobic interactions between these entities. There is strong specificity. Bcl-xL strongly inhibits ceramide channels formed by ceramide with an acyl chain that is 16, 18 and 20 carbons long but inhibition of channels formed by ceramide with a 24 carbon chain is much weaker and when that chain is only 2 carbons long there is no inhibition (8). This chain length dependence is physiologically relevant (e.g. ref. 53). These were not binding experiments but nevertheless indicated strong specificity. Here we actually identify the region of Bcl-xL that binds ceramide and it turns out to be a similar region to the one that binds Bax.
The direct binding experiments used fluorescently-labeled ceramide as a surrogate for ceramide channels. The ability of Bcl-xL to bind ceramide monomers raises the question of whether Bcl-xL might be acting by depleting the free ceramide concentration, rather than binding to the ceramide channel. However, the amount of ceramide added to the mitochondrial suspension was 25 to 70 fold higher on a molar basis that Bcl-xL, making this possibility unlikely. Given that fact, one might wonder why the excess ceramide does not simply occupy all the sites on Bcl-xL and act as a competitive inhibitor, interfering with Bcl-xL binding to the ceramide channel. The answer might reside in the observation that Bcl-xL binds to the acyl chains of ceramide and typically these are buried either in the membrane bilayer or in the core of ceramide micelles and are thus unavailable for binding to Bcl-xL. However, according to the working model of the ceramide channel (S1), the ceramide molecules that form the channel are oriented parallel to the plane of the membrane and organized in columns that span the membrane. The ceramides at the ends of these columns would have their hydrophobic tails exposed to the water phase were it not for these being covered by the surrounding phospholipids to minimize the unfavorable interactions with water. However these phospholipids are in a distorted, high-energy conformation (24). MD simulations (Andriy Anishkin unpublished results) show that the ceramide tails at the end of the columns become transiently exposed, indicating the stressed state of this interface. If Bcl-xL were to displace these lipids it would bind the hydrophobic tails of the end ceramide and by doing so could change the structure of the channel. According to one hypothesis that describes how Bcl-xL binding destabilizes ceramide channels (29), the binding changes the curvature of the ceramide columns. MD simulations (24) show that the columns have a positive curvature resulting in the channel having a somewhat hourglass shape. This is due to the way the channel interfaces with the surrounding phospholipids in the bilayer. The neighboring phospholipids that curve towards the channel to cover the hydrophobic ends of the ceramide columns are under stress and thus pull on the ceramide columns, bending them. Bcl-xL binding removes this bending force by displacing the phospholipids. This results in mismatch with adjacent columns still interacting with phospholipids and thus strain develops that propagates to the rest of the structure through the hydrogen-bonded network, thus destabilizing the entire structure. Mutations that weaken the binding of Bcl-xL to ceramide (fluorescence and MD simulations) should reduce the ability of Bcl-xL to destabilize ceramide channels and that was demonstrated in their reduced ability to inhibit ceramide permeabilization of the MOM. Altering the position of the ceramide-Bcl-xL interface, as indicated in the MD simulation, may alter the above hypothesized curvature change and thus the amount of strain and the degree of destabilization of the channel structure. This explains the correlation between a shift in the location of ceramide binding to mutant Bcl-xL and a reduction in the Bcl-xL’s ability to inhibit the ceramide-induced MOMP. In addition to the proposed structural mismatch between adjacent ceramide columns, the displacement of the phospholipids by Bcl-xL also unbalances forces on the channel from the phospholipids in the two membrane leaflets. This should drive ceramide columns to the opposite side of the membrane and out of the channel, thus reducing channel size. The Bcl-xL could move from column to column catalyzing the loss of columns from the channel. There are other possible mechanisms all of which would require experimentation to include or exclude these possibilities.
The pro-apoptotic role of ceramide is well established (20, 21) whereas the in-vivo pro-apoptotic role of ceramide channels is not. Not only do these channels have the right properties, but mitochondrial ceramide levels have been shown to increase early in apoptosis to levels sufficient to form ceramide channels. However, these finding do not demonstrate that cells actually use ceramide channels during apoptosis. Indeed, ceramide metabolites were reported (54) to activate Bax and Bak and induce MOMP. In view of this one might question the role of ceramide in vivo especially considering the fact that cells lacking both Bax and Bak are very resistant to apoptosis. But here again the simple conclusion is misleading because cells lacking Bax and Bak also fail to increase mitochondrial ceramide levels (16), unlike the parental cells. It turns out that Bak is required for ceramide synthase-mediated long-chain ceramide generation during apoptosis. Thus the situation is far more complex and a more direct approach is needed. Here we report experiments with mutants of Bcl-xL that have an altered ability to inhibit MOMP induced by either ceramide or Bax. The results show that the ability of Bcl-xL to inhibit Bax channels is indeed, as expected, important for Bcl-xL to promote cell survival. However, when the ability of Bcl-xL to inhibit ceramide channels is reduced, the ability of Bcl-xL to protect cells from particular death stimuli was also reduced. This lack of protection from cell death in the mutant Bcl-xL occurred despite it being 15-fold more potent at inhibiting Bax channels. Thus it seems that inhibiting Bax channels alone is insufficient for Bcl-xL to protect from apoptosis and that under certain conditions ceramide channels take over the role of releasing proteins from mitochondria. Of course there are alternative explanations that seem less likely. For instance if Bcl-xL were to facilitate the transfer of ceramide between membranes and this transfer were important to the progress of apoptosis then mutations that weaken the binding of Bcl-xL to ceramide would also weaken the propensity to undergo apoptosis. This hypothetical function would not explain the results with isolated mitochondria but cannot be excluded as a possibility in the in vivo experiments. Regardless role of ceramide channels needs to be considered when interpreting experimental results.
Supplementary Material
{kind=link}
{kind=link}
Highlights.
The hydrophobic pocket on Bcl-xL binds ceramide.
Point mutations in the apolar pocket of Bcl-xL affect its ability to inhibit ceramide channels.
Similar regions on Bcl-xL are important in the inhibition of both Bax and ceramide channels.
Ceramide channels are involved in the onset of apoptosis in vivo.
Mutant Bcl-xL is a useful tool to distinguish between Bax and ceramide channels.
Acknowledgments
We thank Shang H. Lin for purification of Bax. We also thank Justin M. Wang for purification of mutated Bcl-xL. We thank Timothy Troppoli for helping with the cytochrome c accessibility assay. We thank Timothy Walsh for helping with the adenylate kinase assay. We also thank Chris Worth for assistance with cell sorting and Douglas Saforo and Cameron Conway for their technical assistance.
Footnotes
This work was supported by a grant from the National Science Foundation MCB-1023008 (to M.C.), the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institute of Health (R01DK093462 to L.J.S.), and the Kosair Pediatric Cancer Research Program Award (to L.J.B.).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basañez G, Meda P, Martinou JC. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16:2424–32. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6:118–25. doi: 10.1016/j.mito.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, Basañez G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem. 2004;279:30081–91. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- 7.Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–84. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Colombini M. Membrane channels formed by ceramide. Handb Exp Pharmacol. 2013;215:109–126. doi: 10.1007/978-3-7091-1368-4_6. [DOI] [PubMed] [Google Scholar]
- 9.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345(Pt 2):271–278. [PMC free article] [PubMed] [Google Scholar]
- 10.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 11.Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- 12.Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115:632–640. doi: 10.1002/jcb.24709. [DOI] [PubMed] [Google Scholar]
- 13.Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res. 2009;50(Suppl):S91–S96. doi: 10.1194/jlr.R800080-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HJ, Mun JY, Chun YJ, Choi KH, Kim MY. Bax-dependent apoptosis induced by ceramide in HL-60 cells. FEBS Lett. 2001;505:264–268. doi: 10.1016/s0014-5793(01)02836-8. [DOI] [PubMed] [Google Scholar]
- 15.Von Haefen C, Wieder T, Gillissen B, Stärck L, Graupner V, Dörken B, Daniel PT. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–19. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- 16.Siskind LJ, Mullen TD, Rosales KR, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, Obeid LM. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Chem. 2010;285:11818–11826. doi: 10.1074/jbc.M109.078121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesan V, Perera MN, Colombini D, Datskovskiy D, Chadha K, Colombini M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis. 2010;15:553–62. doi: 10.1007/s10495-009-0449-0. [DOI] [PubMed] [Google Scholar]
- 18.Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, Yin X, Ragupathi G, Ehleiter D, Gulbins E, Zhai D, Reed JC, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Mitochondrial ceramide-rich macrodomains functionalize bax upon irradiation. PLoS One. 2011;6(6):e19783. doi: 10.1371/journal.pone.0019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bleicken S, Classen M, Padmavathi PVL, Ishikawa T, Zeth K, Steinhoff HJ, Bordignon E. Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem. 2010;285:6636–6647. doi: 10.1074/jbc.M109.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodcock J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life. 2006;58:462–466. doi: 10.1080/15216540600871118. [DOI] [PubMed] [Google Scholar]
- 21.Siskind LJ. Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr. 2005;37:143–153. doi: 10.1007/s10863-005-6567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samanta S, Stiban J, Maugel TK, Colombini M. Visualization of ceramide channels by transmission electron microscopy. Biochim Biophys Acta - Biomembr. 2011;1808:1196–1201. doi: 10.1016/j.bbamem.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siskind LJ, Davoody A, Lewin N, Marshall S, Colombini M. Enlargement and contracture of C2-ceramide channels. Biophys J. 2003;85:1560–1575. doi: 10.1016/S0006-3495(03)74588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anishkin A, Sukharev S, Colombini M. Searching for the molecular arrangement of transmembrane ceramide channels. Biophys J. 2006;90:2414–26. doi: 10.1529/biophysj.105.071977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao C, Sun B, DeVoe DL, Colombini M. Dynamics of Ceramide Channels Detected Using a Microfluidic System. PLoS One. 2012;7(9):e43513. doi: 10.1371/journal.pone.0043513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siskind LJ, Feinstein L, Yu T, Davis JS, Jones D, Choi J, Zuckerman JE, Tan W, Hill RB, Hardwick JM, Colombini M. Anti-apoptotic Bcl-2 Family Proteins Disassemble Ceramide Channels. J Biol Chem. 2008;283:6622–30. doi: 10.1074/jbc.M706115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding J, Mooers BHM, Zhang Z, Kale J, Falcone D, McNichol J, Huang B, Zhang XC, Xing C, Andrews DW, Lin J. After Embedding in membranes antiapoptotic bcl-xl protein binds both bcl-2 homology region 3 and helix 1 of proapoptotic bax protein to inhibit apoptotic mitochondrial permeabilization. J Biol Chem. 2014;289:11873–11896. doi: 10.1074/jbc.M114.552562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perera MN, Lin SH, Peterson YK, Bielawska A, Szulc ZM, Bittman R, Colombini M. Bax and Bcl-xL exert their regulation on different sites of the ceramide channel. Biochem J. 2012;445:81–91. doi: 10.1042/BJ20112103. [DOI] [PubMed] [Google Scholar]
- 30.Wendt MD, Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang H, Nimmer PM, Ng SC, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH, Elmore SW. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J Med Chem. 2006;49:1165–81. doi: 10.1021/jm050754u. [DOI] [PubMed] [Google Scholar]
- 31.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 32.Manion MK, O’Neill JW, Giedt CD, Kim KM, Zhang KYZ, Hockenbery DM. Bcl-XL mutations suppress cellular sensitivity to antimycin A. J Biol Chem. 2004;279:2159–65. doi: 10.1074/jbc.M306021200. [DOI] [PubMed] [Google Scholar]
- 33.Parsons DF, Williams GR, Chance B. Characteristics of isolated and purified preparations of the outer and inner membranes of mitochondria. Ann N Y Acad Sci. 1966;137:643–666. doi: 10.1111/j.1749-6632.1966.tb50188.x. [DOI] [PubMed] [Google Scholar]
- 34.Basañez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, Zimmerberg J. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem. 2001;276:31083–91. doi: 10.1074/jbc.M103879200. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–54. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 36.Lin SH, Perera MN, Nguyen T, Datskovskiy D, Miles M, Colombini M. Bax forms two types of channels, one of which is voltage-gated. Biophys J. 2011;101:2163–9. doi: 10.1016/j.bpj.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perera MN, Ganesan V, Siskind LJ, Szulc ZM, Bielawski J, Bielawska A, Bittman R, Colombini M. Ceramide channels: influence of molecular structure on channel formation in membranes. Biochim Biophys Acta. 2012;1818:1291–301. doi: 10.1016/j.bbamem.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganesan V, Walsh T, Chang KT, Colombini M. The dynamics of Bax channel formation: influence of ionic strength. Biophys J. 2012;103:483–91. doi: 10.1016/j.bpj.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–31. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 44.Rostkowski M, Olsson MHM, Søndergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol. 2011;11:6. doi: 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 46.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 47.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 48.Eno CO, Eckenrode EF, Olberding KE, Zhao G, White C, Li C. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell. 2012;23:2605–18. doi: 10.1091/mbc.E12-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stiban J, Fistere D, Colombini M. Dihydroceramide hinders ceramide channel formation: Implications on apoptosis. Apoptosis. 2006;11:773–80. doi: 10.1007/s10495-006-5882-8. [DOI] [PubMed] [Google Scholar]
- 50.Minn AJ, Vélez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–7. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 51.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–35. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siskind LJ, Colombini M. The lipids C2- and C16-ceramide form large stable channels: Implications for apoptosis. J Biol Chem. 2000;275:38640–38644. doi: 10.1074/jbc.C000587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stiban J, Perera M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim Biophys Acta. 2015;1848:561–567. doi: 10.1016/j.bbamem.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 54.Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.