

Abstract
Common Variable Immunodeficiency (CVID) is a primary immunodeficiency of unknown etiology characterized by low serum IgG, a decreased ability to make specific antibodies, and variable T cell defects. Approximately 20% of patients with CVID develop clinical evidence of a diffuse parenchymal lung disease with a constellation of histopathological findings termed granulomatous and lymphocytic interstitial lung disease (GLILD). In this study, we characterized the histological and immunohistochemical features in a series of 16 cases diagnosed by open lung biopsy. Peribronchiolar and interstitial lymphocytic infiltration, granulomatous inflammation and organizing pneumonia were consistent features; interstitial fibrosis with architectural remodeling was also found in a subgroup of patients. By immunohistochemistry, a predominance of CD4+ T lymphocytes with variable numbers of CD8+ T cells and B cells were present, with a striking absence of FOXP3 positive T regulatory cells. This heretofore unrecognized immunohistochemical finding, needs further investigation for a potential role in the pathogenesis of the condition. The presence of interstitial fibrosis with or without architectural remodeling in a subset of patients also needs additional study, for effect on prognosis.
Keywords: Granuloma, lymphoid interstitial lung disease, common variable immunodeficiency, immunohistochemistry
1. Introduction
Common variable immunodeficiency (CVID) is a primary immunodeficiency disorder characterized by low serum IgG, IgA and/or IgM, and poor specific antibody production. It is the most common serious primary immunodeficiency with a prevalence of 1: 25000 – 50000 [1]. In a small minority of patients, single gene mutations resulting in B and T lymphocyte dysfunction have been identified [2]. Thus, the molecular pathogenesis of the disease is not known in the vast majority of cases. Although the age of onset of symptoms is variable, the diagnosis of CVID is usually made between the second and fourth decades of life. Upper and lower respiratory tract infections are the most common presenting manifestations, occurring in a vast majority of patients [3]. In CVID, there is an undefined defect in the differentiation of B cells into mature antibody producing plasma cells that leads to hypogammaglobulinemia and poor specific antibody production. T cell abnormalities occur in up to 50% patients and likely contribute to the heterogeneous clinical manifestations of this disorder [4,5].
With introduction of high dose intravenous immunoglobulin (IVIG) or subcutaneous gammaglobulin (SCIG), the morbidity and mortality due to infection has markedly decreased [6]. In contrast, the noninfectious complications of CVID, such as autoimmunity, inflammatory bowel disease, enteropathy, hepatitis, structural lung disease, and lymphoproliferation, including B cell lymphomas have increased, occurring in nearly 70% of patients [7–9]. Many of these noninfectious complications are associated with increased mortality in CVID.
In the course of evaluating patients for the noninfectious complications of CVID, a high resolution CT scan of the chest is routinely obtained, which may demonstrate diffuse lung parenchymal disease (DLPD). The differential diagnosis of DLPD in the context of CVID is diverse and includes infection, hypersensitivity pneumonitis, cryptogenic organizing pneumonia, lymphoma and a constellation of radiological-pathological findings known as granulomatous and lymphocytic interstitial lung disease (GLILD) [1,8,9]. Although GLILD is the most common cause of DLPD in CVID, the histopathological features of this disorder are incompletely characterized. Consequently, it is frequently confused with other granulomatous lung diseases, such as sarcoidosis. A prior retrospective study of patients with GLILD found histopathologic abnormalities, including granulomata, and lymphoid hyperplasia resembling the pattern of LIP and FB [9]. While recognizing that GLILD consists of a rather heterogeneous collection of pathological findings, and that there is insufficient prospective data on the natural history of this complication of CVID, the purpose of this study was to fill a void in the available literature, by attempting a systematic characterization of the spectrum of histopathologic and immunohistochemical features of GLILD. We also endeavored to see if there were any histologic features that may potentially identify patients at increased risk for disease progression.
2. Materials and methods
All components of the study were performed in compliance with relevant laws and institutional guidelines, with approval from the Institutional Review Board (IRB).
2.1 Demographics
16 patients with the common variable immunodeficiency (CVID) constitute the study. These patients were diagnosed with CVID, using standard diagnostic criteria [10,11]. The patients [females (n=10) and males (n=6)] ranged in age from 19 – 57 years (mean age – 37) and underwent either video-assisted thoracoscopy (VATS) guided or open lung wedge biopsies.
2.2 Histology
Histological examinations were performed on standard 4.0 microns thick hematoxylin and eosin (H & E) stained sections of formalin-fixed, paraffin embedded specimens. The following features were assessed: (1) airway inflammation including follicular bronchitis/bronchiolitis and interstitial inflammation/lymphoid interstitial pneumonia, (2) epithelioid cell granulomata including their frequency, distribution and appearance, (3) organizing pneumonia, characterized by Masson bodies(spherical proliferations of fibroblastic tissue set in a pale, myxoid stroma located within airspaces and adjacent interstitium), (4) interstitial fibrosis, and (5)alveolar remodeling characterized by microscopic honeycomb cysts lined with metaplastic bronchiolar epithelium and traction bronchiectasis. The pathologic features were evaluated by low power examination of several representative slides from each case. Assessment of these features was done semiquantitatively using the following scores: absent (0), mild (1+), moderate (2+) and severe (3+). The histologic grading was performed by two pathologists (NR and ACM) independently and subsequently in tandem with consensus achieved. Special stains for microorganisms (Ziehl-Neelsen stain for acid fast bacilli; and Gomori Methenamine stain for fungal organisms) were performed on all cases using established staining protocols.
2.3 Immunohistochemistry
Paraffin blocks for immunohistochemical studies were available in all cases. Four micron sections were stained using a Dako Autostainer Plus according to the manufacturer’s protocol. Slides were dried at 60°C for one hour and deparaffinized. Heat induced epitope retrieval was performed with Dako Envision FLEX target retrieval solution (high pH Tris/EDTA) at 100°C for 20 minutes. Primary antibodies (DAKO, Carpinteria, California) for: CD3 (Rabbit polyclonal), CD4 (4B12), CD8 (C8/144B), CD19 (LE-CD19), CD20 (L26), CD138 (MI15), and PAX5 (DAK-Pax5) were obtained from DAKO, Carpinteria, California. FoxP3 (236A-E7) was obtained from Abcam, Cambridge, Massachusetts. All antibodies were incubated at room temperature for 60 minutes. Signals were detected using a Dako FLEX detection kit. Counterstaining was performed with Envision FLEX hematoxylin for 7 minutes at room temperature. Appropriate positive and negative controls were run concurrently for all antibodies tested.
2.4 Immunohistochemical Analysis
Cytoplasmic and membranous expression of immunohistochemical staining was quantified for each case using an Automated Cellular Imaging System III (ACIS III, DAKO) as previously described [12]. For each CVID patient, a portion of lung was quantitatively analyzed by scanning a representative whole slide for each patient. ACIS software collects individual, overlapping images at 400X and then tiles these images to create a montage of the entire scanned lung specimen. The software evaluates each individual 400X image and combines the results into an aggregate quantitative measurement corresponding to the entire tissue specimen. The ACIS system measures the intensity of the staining based on three related color parameters: the color defined by hue, the “darkness” defined as luminosity, and the density of the color defined as saturation. ACIS software for the analysis was programmed by an experienced user-pathologist (ACM) by setting the color-specific thresholds to determine and calculate staining intensity and the ratio of positively stained cells to the entire area of selection. This was used to determine the approximately percentage of positive staining tissue in each specimen.
3. Results
3.1 Histopathology of GLILD
The histologic findings are summarized in Table 1. Nodular peribronchiolar inflammation, consisting of mature appearing lymphocytes predominantly was present in all cases (100%) (Fig A). The extent of peribronchiolar inflammation ranged from mild (1+) in 4/16 cases (25%), moderate (2+) in 9/16 cases (56 %), to severe (3+) in 3/16 cases (19%). Dense nodular and diffuse interstitial chronic was present in all 16 cases as well (Fig B). The intensity of interstitial chronic inflammation ranged from mild (1+) in 3/16 cases (19 %), moderate (2+) in 4/16 cases (25%) to severe (3+) in 9/16 cases (56 %). In cases with marked (3+) interstitial inflammatory infiltrate, there was homogeneous and uniform expansion of the interstitium by the chronic inflammatory infiltrate. Marked peribronchiolar and interstitial chronic inflammation (2+ or 3+) was present in 10/16 cases (62.5 %). It was unusual to see an isolated peribronchiolar or interstitial pattern, for example 3/16 cases (18.75 %) demonstrated significant interstitial inflammation without accompanying peribronchiolar infiltrate, whereas 2/16 cases (12.5%) showed the reverse pattern of inflammation. In one case (6.25%), there was a paucity of an inflammatory component, and neither patterns were observed in significant amounts. This case had rather advanced interstitial fibrosis with evidence of remodeling (case #3).
Table 1.
Histologic Features of GLILD
| Patient Age/Sex | Inflammation | Granulomas | Organizing Pneumonia | Fibrosis | |||
|---|---|---|---|---|---|---|---|
| Bronchiolar | Interstitial | Interstitial Fibrosis | Remodeling | ||||
| 1. | 19/F | 3 | 1 | 1 | 0 | 0 | 0 |
| 2. | 24/F | 3 | 3 | 1 | 2 | 1 | 0 |
| 3. | 53/F | 1 | 1 | 1 | 0 | 2 | 2 |
| 4. | 49/F | 2 | 3 | 3 | 1 | 3 | 2 |
| 5. | 49/M | 1 | 2 | 2 | 1 | 3 | 2 |
| 6. | 30/M | 2 | 3 | 2 | 3 | 1 | 0 |
| 7. | 28/F | 1 | 3 | 2 | 2 | 0 | 0 |
| 8. | 30/F | 2 | 3 | 0 | 2 | 1 | 0 |
| 9. | 49/M | 2 | 3 | 1 | 1 | 2 | 2 |
| 10. | 57/M | 1 | 2 | 1 | 1 | 3 | 1 |
| 11. | 39/F | 2 | 3 | 3 | 2 | 2 | 0 |
| 12. | 35/F | 3 | 2 | 2 | 2 | 0 | 0 |
| 13. | 34/M | 2 | 2 | 2 | 1 | 1 | 0 |
| 14. | 43/M | 2 | 1 | 1 | 1 | 0 | 0 |
| 15. | 33/F | 2 | 3 | 1 | 1 | 2 | 2 |
| 16. | 27/F | 2 | 3 | 1 | 2 | 1 | 0 |
Figure A, B.
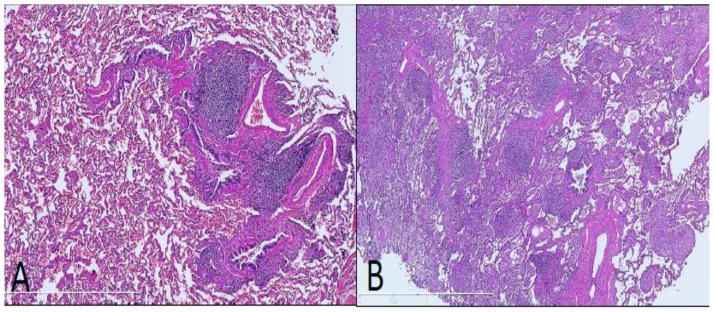
Peribronchiolar and interstitial lymphocytic infiltration in GLILD. There are nodular lymphoid aggregates associated with bronchioles (A), and dense, nodular and diffuse interstitial lymphocytic infiltration (B). (H&E, 100x)
Granulomata, ranging from well to poorly formed, were present in 15/16 cases (93.75 %). In some areas, poorly formed granulomas were associated closely with diffuse interstitial inflammation. The granulomas were nonnecrotizing and comprised of epithelioid histiocytes with occasional multinucleate giant cells (Fig C). They were randomly distributed throughout the lung parenchyma, both in association with the lymphocytic infiltration, as well as away from the inflammatory infiltrate. They were seen within the interstitium, along bronchovascular bundles, and occasionally within alveolar spaces. The frequency of granulomata ranged from mild (1+) in 8/16 cases (50 %), moderate (2+) in 5/16 cases (31.25%), and severe (3+) in 2/16 cases (12.5%). AFB and GMS stains were negative for microorganisms in all cases.
Figure C, D.
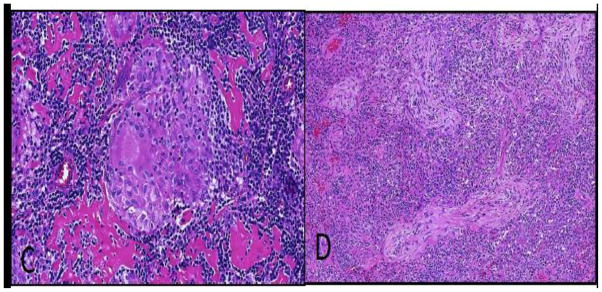
Granulomata and organizing pneumonia in GLILD. A well-formed epithelioid cell granuloma (C), and organizing pneumonia characterized by prominent Masson bodies within airspaces (D). (H&E, 200x (C), 100x (D))
Organizing pneumonia was present in 14/16 cases (87.5%) and characterized by Masson bodies composed of focal spherical proliferations of fibroblastic tissue set in a pale, myxoid stroma located within alveoli and adjacent interstitium (Fig D). Organizing pneumonia was mild (1+) in 7/16 cases (43.75 %), moderate (2+) in 6/16 cases (37.5 %), and severe (3+) in 1/16 cases (6.25%). In some areas, the organizing pneumonia was intimately associated with granulomas.
Interstitial fibrosis, characterized by patchy to expansive areas of collagenized fibrosis, was present in 12/16 cases (75%). It was mild (1+) in 5/16 cases (31.25 %), moderate (2+) in 4/16 cases (25%), and severe (3+) in 3/16 cases (18.75%). Five of the seven cases with significant interstitial fibrosis (2+ or 3+) displayed significant accompanying alveolar remodeling characterized by irregularly shaped air spaces (microscopic honeycomb cysts) lined by metaplastic bronchiolar epithelium and associated traction bronchiectasis (Fig E). Alveolar remodeling always accompanied significant interstitial fibrosis (2+ or 3+). In other cases mild (1+) interstitial fibrosis unaccompanied by alveolar remodeling was observed.
Figure E.

Interstitial fibrosis in GLILD. There is expansile interstitial fibrosis with remodeled airspaces lined by metaplastic bronchiolar epithelium. (H&E, 200x)
3.2 Immunohistochemical analysis
Characterization of the inflammatory infiltrate by immunohistochemistry (IHC) demonstrated T cells in all cases and B cells in 15/16 cases. CD3+ CD4+ T cells were the predominant lymphocyte in all but 2 cases, where CD20+ B cells predominated (Table 2). Strikingly, regulatory T cells, characterized by nuclear FoxP3 expression, were not found in the lungs of any patient with GLILD. The inflammatory infiltrate in areas of FB was predominantly CD3+ (Fig G), CD4+ (Fig H) T cells, with smaller numbers of CD8+ T cells (Fig I), and CD20+ (Fig J), PAX5+ (Fig K) B cells. In one case of FB, a small nodule of CD20+ cells was identified. Similar nodules were more commonly observed in the interstitium and demonstrated a positive correlation with the extent of interstitial inflammation. These nodules ranged from poorly formed and few in numbers to well-formed and rather more frequent. The nodules were typically composed of CD20+, PAX5+ positive B cells cuffed by a small ring of CD3+, CD4+ cells. Cases with alveolar remodeling demonstrated strong CD138 staining within the epithelium.
Table 2.
Ratio of lymphocyte subtypes by immunohistochemical staining in GLILD
| Pt | CD4/CD3 | CD8/CD3 | CD4/CD8 | FOXP3/CD3 | CD138/PAX5 | CD3/CD20+CD3 | CD138/CD20 | PAX5/CD20 | CD3/CD20 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.04 | 0.52 | 2.01 | 0.03 | 0.49 | 0.59 | 0.27 | 0.54 | 1.44 |
| 2 | 1.03 | 0.37 | 2.79 | 0.02 | 0.77 | 0.61 | 0.39 | 0.51 | 1.58 |
| 3 | 0.81 | 0.6 | 1.36 | 0.07 | 6.39 | 0.92 | 3.84 | 0.6 | 10.84 |
| 4 | 0.98 | 0.55 | 1.78 | 0 | 0.06 | 0.58 | 0.04 | 0.56 | 1.35 |
| 5 | 0.82 | 0.59 | 1.38 | 0.02 | 2.27 | 0.77 | 0.71 | 0.31 | 3.33 |
| 6 | 0.86 | 0.25 | 3.52 | 0.02 | 2.7 | 0.85 | 0.58 | 0.21 | 5.5 |
| 7 | 1.02 | 0.4 | 2.53 | 0.01 | NA | 0.45 | NA | 0.58 | 0.8 |
| 8 | 0.31 | 0.29 | 1.07 | 0.01 | NA | 0.4 | NA | 0.56 | 0.68 |
| 9 | 0.78 | 0.63 | 1.24 | 0.03 | 0.1 | 0.44 | 0.06 | 0.65 | 0.79 |
| 10 | 1.28 | 0.53 | 2.43 | 0.03 | 95.57 | 0.99 | 127.49 | 1.33 | 149.15 |
| 11 | 1.42 | 0.14 | 10.14 | NA | NA | 0.68 | NA | NA | 2.08 |
| 12 | 0.63 | 0.12 | 5.3 | NA | NA | 0.82 | NA | NA | 4.55 |
| 13 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 14 | 1.62 | 0.37 | 4.44 | NA | NA | 0.68 | NA | NA | 2.11 |
| 15 | 1.96 | 0.26 | 7.48 | NA | NA | 0.52 | NA | NA | 1.07 |
| 16 | 2.69 | 2.25 | 1.2 | NA | NA | 0.56 | NA | NA | 1.29 |
| Median | 1.02 | 0.4 | 2.43 | 0.02 | 1.52 | 0.61 | 0.485 | 0.56 | 1.58 |
| Average | 0.92 | 0.42 | 2.96 | 0.02 | 13.55 | 0.67 | 16.67 | 0.59 | 15.18 |
| Standard Deviation | 0.59 | 0.5 | 2.62 | 0.02 | 33.21 | 0.18 | 44.79 | 0.3 | 37.91 |
NA: Analysis not performed.
Figure F–K.

T lymphocyte predominance in GLILD. FB pattern infiltrate in GLILD (F) with predominantly CD3 positive (G), CD4 positive T cells (H), with smaller numbers of CD8 positive T cells (I) and CD20 positive (J), PAX-5 positive B cells (K). This pattern of T lymphocyte predominance was commonly observed.
4. Discussion
GLILD is the pulmonary component of multisystemic lymphoproliferative disease in CVID, which includes diffuse adenopathy, splenomegaly, and granulomatous inflammation in a variety of organs. It is the most common cause of DLPD in patients with CVID, occurring in approximately 10–30% of all patients [8,9,13–16]. Prior retrospective studies have suggested that the presence of GLILD in CVID is associated with increased mortality, although granulomatous disease per se in any organ may not be associated with poorer outcome [8,9,17].
Lymphoid proliferation is a cardinal manifestation of GLILD [8,9,17]. Our series highlights peribronchiolar and interstitial lymphoid infiltration as the predominant histologic patterns of lymphoid proliferation. A mixed inflammatory pattern, consisting of moderate to severe peribronchiolar and interstitial inflammation, was typically present. Only one case (which was characterized predominantly by interstitial fibrosis) demonstrated a minor/insignificant inflammatory component. Prior to the current study, there was not a quantitative analysis of the lymphocyte subtypes present in GLILD. By immunohistochemistry and the use of an Automated Cellular Imaging System we demonstrated that the lymphoid infiltrate typically had a predominance of CD4+ T cells, both in areas of FB and LIP. Nodules of CD20+ B cells ringed by CD4+T cells were also noted, mainly within the interstitium. With the exception of one patient, all of our cases showed the presence of B cells in the affected lungs. Detailed analysis of pulmonary lymphoid populations in CVID patients with lymphoid hyperplasia has shown the presence of distinct B-cell follicles and T-cell zones, which resembles the findings in our cases [18]. This feature has been termed “tertiary lymphoid neogenesis” and has been suggested to occur in various chronic inflammatory states, including those affecting the lung.
The importance of recognizing discrete B and T cell lymphoid populations in these situations lies in the implications for treatment. Combination immunosuppressive therapy targeting both B cells (rituximab) and T cells (azathioprine) has been successfully employed in in the treatment of GLILD [13]. The patient without the presence of B cells in the lung biopsy also lacked B cells in the peripheral blood. Interestingly, 8 years previous to the current lung biopsy, she had B cells both in the periphery and in the lung biopsy. The lack of B cells was clinically relevant as rituximab therapy in this case would have been an ineffective and unnecessary medication. Follow-up of this patient indicates that the pulmonary disease is stable on mycophenolate treatment. This case illustrates the utility of obtaining a repeat open lung biopsy to confirm the histological findings and diagnosis when the prior biopsy is several years in the past. It also emphasizes the importance of immunohistochemical characterization of the lymphoid infiltrate for planning treatment regimen in these patients.
Another novel immunohistochemical finding of the current study was the striking absence of regulatory T cells in the lungs in our cases with GLILD. Prior studies indicated that there is a decrease in the number and function of regulatory T cells in the peripheral blood in CVID compared to age and sex matched healthy controls [19–22]. Low numbers of regulatory T cells in the peripheral blood correlates with autoimmunity and granulomatous disease, including granulomatous lung disease, in patients with CVID [19–22]. Azathioprine and rituximab, which are used to treat GLILD, have been shown to increase the numbers of regulatory T cells [23,24]. We therefore hypothesize that the lack of regulatory T cells in the lungs of patients with GLILD is a contributing factor in the underlying pathogenesis of this disorder. To our knowledge, this is the first report describing a lack of regulatory T cells in the lung lesions of patients with GLILD.
Organizing pneumonia, in some cases a prominent histological feature, was present in a vast majority of our cases of GLILD. This finding has not been previously emphasized in GLILD. It is important to recognize that idiopathic or cryptogenic organizing pneumonia (COP) is also a common cause of DLPD in CVID patients [9,25,26]. Limited bronchoscopic lung biopsies in GLILD patients, where organizing pneumonia may be the predominant histological abnormality, may result in a mistaken diagnosis of COP. This was exemplified in one of our cases (patient #16), where a diagnosis of COP was made based on transbronchial biopsy findings. This led to the use of high dose corticosteroid therapy, which failed to result in radiographic improvement of the DLPD. A subsequent VATS biopsy established the diagnosis of GLILD, with resolution of pulmonary findings with rituximab and azathioprine therapy. The case emphasizes the importance of suggesting the diagnosis of GLILD on open lung or VATS biopsy whenever it can safely be performed, to avoid sampling error and incorrect characterization of DLPD in these patients.
Interstitial fibrosis is also a previously unappreciated feature that is present in many patients with GLILD. Overall, 12/17 patients had interstitial fibrosis, and in 7 of these patients the fibrosis was severe. In examining the presence of interstitial fibrosis in our cases, we evaluated its extent and distribution, as well as the presence or absence of architectural remodeling including microscopic honeycombing and traction bronchiectasis. A majority of our patients with severe interstitial fibrosis (5/7) also had architectural remodeling. The presence of interstitial fibrosis in a proportion of patients with GLILD is of interest. It emphasizes the need for prospective clinical studies, lacking thus far, to address the possibility that it may be a marker for disease progression, and perhaps provide a rationale for early diagnosis and treatment.
The histologic differential diagnosis of GLILD includes a group of interstitial lung diseases displaying granulomatous, inflammatory and fibrotic features in varying proportions. The various entities and their histologic differential diagnostic features are listed in Table 3. The granulomatous process raises the possibility of both sarcoidosis and hypersensitivity pneumonitis (HP) [14,27,28]. Chronic HP can be particularly difficult to distinguish from those cases of GLILD with significant interstitial fibrosis [29], and the importance of clinical correlation in separating lesions of GLILD from HP cannot be overemphasized. Lymphoproliferation and interstitial fibrosis in GLILD may resemble idiopathic interstitial pneumonia patterns such as LIP, NSIP and UIP [30–34]. LIP is characterized by dense, diffuse chronic inflammatory interstitial infiltrate, and significant granulomas or organizing pneumonia are variable, but generally inconspicuous [35,36]. Fibrotic NSIP and UIP may prove difficult to distinguish from GLILD with significant interstitial fibrosis, as seen in a subset of our cases. Finally, infection should always be considered in the differential diagnosis. Granulomatous infectious processes need to be excluded by appropriate special stains and microbiological studies. In summary, distinguishing various other granulomatous, inflammatory and fibrotic interstitial lung diseases from GLILD requires a high index of suspicion and significant clinical and microbiologic correlation.
Table 3.
Entities and Patterns In The Histologic Differential Diagnosis of GLILD
| Microscopic features | ||||
|---|---|---|---|---|
| GLILD | Well, moderate or poorly circumscribed; May be cuffed by lymphocytes or associated with lymphoid infiltration; random distribution | Lymphocytic infiltration of variable density – peribronchiolar and interstitial | Consistently present; associated with interstitial inflammation | Present in a subset of patients; expansile and associated with interstitial inflammation |
| Sarcoidosis | Well circumscribed; “naked” – unassociated with lymphocytic cuffing; lymphatic distribution | Significant interstitial inflammation is absent | Usually absent | Present in a subset of patients; with a distribution similar to granulomatous inflammation |
| Hypersensitivity pneumonitis | Well, moderate or poorly circumscribed; May be cuffed by lymphocytes or associated with lymphoid infiltration; bronchiolocentric or random distribution | Bronchiolocentric or diffuse interstitial chronic inflammation | Present, associated with peribronchiolar or interstitial inflammation and granulomas | May be present in chronic HP; and may follow a pattern similar to UIP |
| Lymphoid interstitial pneumonia | Granulomas are usually absent; isolated multinucleate giant cells with cholesterol clefts may be present | Diffuse interstitial chronic inflammatory infiltrate, with lymphoid aggregate formation | May be present focally (minor feature) | Usually absent |
| Nonspecific interstitial pneumonia | Granulomas are usually absent; isolated multinucleate giant cells with cholesterol clefts may be present | Homogeneous interstitial expansion by a variable intensity chronic inflammatory infiltrate | May be present focally (minor feature) | May be present in fibrosing NSIP; fibrosis follows a homogeneous and diffuse pattern similar to interstitial inflammation in cellular NSIP |
| Usual interstitial pneumonia | Granulomas are usually absent; isolated multinucleate giant cells with cholesterol clefts may be present | Scant and patchy chronic interstitial inflammation | Usually absent (if present; may raise the possibility of acute exacerbation of UIP) | Characteristic feature; patchwork fibrosis which is temporally heterogeneous (coexistent “young and old” fibrosis |
GLILD – Granulomatous and Lymphocytic Interstitial Lung Disease
HP – Hypersensitivity Pneumonitis
NSIP – Nonspecific Interstitial Pneumonia
UIP – Usual Interstitial Pneumonia
Recent studies show that pulmonary lesions similar to GLILD can also occur in primary immunodeficiencies other than CVID, for example, cytotoxic T lymphocyte antigen-4 (CTLA4) deficiency, lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency and hypomorphic mutations in the recombinase-activating gene 1 (Rag1) [37–40]. Importantly, hypogammaglobulinemia may be one of the immunological abnormalities found in these primary immunodeficiencies. There are also several primary immunodeficiencies that have granulomatous inflammation in extrapulmonary locations [41]. As more patients with these rare diseases are diagnosed and additional monogenic cause of primary immunodeficiencies are described, we believe that the number of primary immunodeficiencies where GLILD is found will increase.
5. Conclusions
In summary, GLILD is a conglomeration of pulmonary histopathological abnormalities seen in a subset of patients with CVID. Recent descriptions of similar findings in patients with other immune abnormalities, emphasizes that GLILD may represent a pulmonary reaction pattern that serves as a morphological endpoint in patients with these uncommon conditions. Granulomas, peribronchiolar and interstitial lymphocytic infiltration are present in nearly all patients with GLILD. Extensive organizing pneumonia and pulmonary interstitial fibrosis are also seen in a significant proportion of patients with GLILD. T cells, particular CD4+ T cells, are the predominant lymphocytes in the lung, with B cells present to a lesser extent. B cell predominance is seen in a minority of cases of GLILD. We also observed a near total absence of regulatory T cells, a finding that warrants further investigation. To the best of our knowledge, our description of significant interstitial fibrosis and large areas of organizing pneumonia, quantification of lymphocytic subsets, and observation of an absence of regulatory T cells in lung biopsy specimens, are unique findings that have not been described. Finally, our findings emphasize that GLILD is best assessed on open lung or VATS biopsies, rather than limited transbronchial biopsies.
GLILD Highlights for Review.
This study characterizes histological and immunohistochemical findings in GLILD in patients with CVID.
FB, LIP, granulomatous inflammation and organizing pneumonia were consistent findings.
Organizing pneumonia may be the dominant feature and has potential for misdiagnosis on limited transbronchial biopsies.
Interstitial fibrosis, previously not emphasized, can be present in a proportion of cases, likely carries worse prognosis and requires additional study.
We demonstrate predominance of CD4+ T cells, and hithertofore unreported striking absence of FOXP3 positive regulatory T cells, which needs investigation for a role in pathogenesis of GLILD.
Acknowledgments
Research in this manuscript was supported by NIH grant R01CA122539091 (JMR). Experiments were performed in the Department of Pathology Clinical and Translational Research Lab (CTRL).
Footnotes
Disclosures:
The authors do not have any conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nagarjun Rao, Email: arao@mcw.edu.
A. Craig Mackinnon, Email: amackinnon@mcw.edu.
John M. Routes, Email: jroutes@mcw.edu.
References
- 1.Prasse A, Kayser G, Warnatz K. Common variable immunodeficiency-associated granulomatous and interstitial lung disease. Curr Opin Pulm Med. 2013;19:503–9. doi: 10.1097/MCP.0b013e3283642c47. [DOI] [PubMed] [Google Scholar]
- 2.Salzer U, Warnatz K, Peter HH. Common variable immunodeficiency - an update. Arthritis Res Ther. 2012;14:223. doi: 10.1186/ar4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinti I, Soresina A, Guerra A, Rondelli R, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31:315–2. doi: 10.1007/s10875-011-9511-0. [DOI] [PubMed] [Google Scholar]
- 4.Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- 5.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 6.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel Hl. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–60. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 8.Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID) Clin Immunol. 2010;134:97–103. doi: 10.1016/j.clim.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114:415–21. doi: 10.1016/j.jaci.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 10.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–7. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 11.Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203–11. doi: 10.1111/cei.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma PC, Tretiakova MS, MacKinnon AC, Ramnath N, Johnson C, Dietrich S, et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer. 2008;47:1025–37. doi: 10.1002/gcc.20604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID) J Clin Immunol. 2013;33:30–9. doi: 10.1007/s10875-012-9755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–7. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep. 2005;5:370–5. doi: 10.1007/s11882-005-0008-x. [DOI] [PubMed] [Google Scholar]
- 16.Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM, Lammers JW, Bloem AC, Hoepelman AI, et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol. 2014;34:642–54. doi: 10.1007/s10875-014-0068-6. [DOI] [PubMed] [Google Scholar]
- 17.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maglione PJ, Ko HM, Beasley MB, Strauchen JA, Cunningham-Rundles Cl. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;133:535–42. doi: 10.1016/j.jaci.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arandi N, Mirshafiey A, Jeddi-Tehrani M, Abolhassani H, Sadeghi B, Mirminachi B, et al. Evaluation of CD4+CD25+FOXP3+ regulatory T cells function in patients with common variable immunodeficiency. Cellular immunology. 2013;28:129–33. doi: 10.1016/j.cellimm.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Fevang B, Yndestad A, Sandberg WJ, Holm AM, Muller F, Aukrust P, et al. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol. 2007;147:521–5. doi: 10.1111/j.1365-2249.2006.03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu GP, Chiang D, Song SJ, Hoyte EG, Huang J, Vanishsarn C, et al. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin Immunol. 2009;131:240–53. doi: 10.1016/j.clim.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao X, Miao Q, Chang C, Gershwin ME, Ma X. Common variable immunodeficiency and autoimmunity - an inconvenient truth. Autoimmunity reviews. 2014;13:858–64. doi: 10.1016/j.autrev.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Fattorossi A, Battaglia A, Bonanno G, Ciaraffa F, Scambia G, Evoli A. Megakaryocytopoiesis induced by PEG-rHu megakaryocyte growth and development factor (MGDF) Eur J Histochem. 1997;41(Suppl 2):13–4. [PubMed] [Google Scholar]
- 24.Catzola V, Battaglia A, Buzzonetti A, Fossati M, Scuderi F, Fattorossi A, et al. Changes in regulatory T cells after rituximab in two patients with refractory myasthenia gravis. J Neurol. 2013;260:2163–5. doi: 10.1007/s00415-013-6987-y. [DOI] [PubMed] [Google Scholar]
- 25.Wislez M, Sibony M, Naccache JM, Liote H, Carette MF, Oksenhendler E, et al. Organizing pneumonia related to common variable immunodeficiency. case report and literature review. Respiration. 2000;67:467–70. doi: 10.1159/000029552. [DOI] [PubMed] [Google Scholar]
- 26.Kaufman J, Komorowski R. Bronchiolitis obliterans organizing pneumonia in common variable immunodeficiency syndrome. Chest. 1991;100:552–3. doi: 10.1378/chest.100.2.552. [DOI] [PubMed] [Google Scholar]
- 27.Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J. 2013;41:115–22. doi: 10.1183/09031936.00189011. [DOI] [PubMed] [Google Scholar]
- 28.Leen CL, Bath JC, Brettle RP, Yap PL. Sarcoidosis and primary hypogammaglobulinaemia: a report of two cases and a review of the literature. Sarcoidosis. 1985;2:91–5. [PubMed] [Google Scholar]
- 29.Churg A, Muller NL, Flint J, Wright JL. Chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2006;30:201–8. doi: 10.1097/01.pas.0000184806.38037.3c. [DOI] [PubMed] [Google Scholar]
- 30.Kradin RL, Mark EJ. Benign lymphoid disorders of the lung, with a theory regarding their development. Hum Pathol. 1983;14:857–67. doi: 10.1016/s0046-8177(83)80161-0. [DOI] [PubMed] [Google Scholar]
- 31.Fishback N, Koss M. Update on lymphoid interstitial pneumonitis. Curr Opin Pulm Med. 1996;2:429–33. [PubMed] [Google Scholar]
- 32.Nicholson AG, Wotherspoon AC, Diss TC, Hansell DM, Du Bois R, Sheppard MN, et al. Reactive pulmonary lymphoid disorders. Histopathology. 1995;26:405–12. doi: 10.1111/j.1365-2559.1995.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 33.Gal AA, Staton GW., Jr Current concepts in the classification of interstitial lung disease. Am J Clin Pathol. 2005;123(Suppl):S67–81. doi: 10.1309/562DF88VC6G6QJU1. [DOI] [PubMed] [Google Scholar]
- 34.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liebow AA, Carrington CB. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med Clin North Am. 1973;57:809–43. doi: 10.1016/s0025-7125(16)32278-7. [DOI] [PubMed] [Google Scholar]
- 36.Koss MN, Hoccholzer L, Langloss JM, Wehunt JM, Lazarus AA. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology. 1987;19:178–85. doi: 10.3109/00313028709077131. [DOI] [PubMed] [Google Scholar]
- 37.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20:1410–6. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE5, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345:1623–7. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchbinder D1, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. 35:119–24. doi: 10.1007/s10875-014-0121-5. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose CD, Neven B, Wouters C. Granulomatous inflammation: The overlap of immune deficiency and inflammation. Best Pract Res Clin Rheumatol. 2014;28:191–212. doi: 10.1016/j.berh.2014.03.006. [DOI] [PubMed] [Google Scholar]