

Abstract
Over the last several years we have synthesized and studied the in vitro and in vivo nAChR pharmacological properties of epibatidine (4) analogs. In this study we report the synthesis, nAChR in vitro and in vivo pharmacological properties of 3′-(substituted pyridinyl)-deschloroepibatidine analogs (5a–e and 6a–e). All of the analogs had high binding affinity for α4β2*-nAChRs. Several of the analogs were potent antagonists of α4β2-nAChRs in in vitro efficacy tests and were potent antagonists of nicotine-induced antinociception in the mouse tail-flick test. Compound 6b had a Ki = 0.13 nM in the binding assay, 25- and 46-fold selectivity for the α4β2*-nAChR relative to the α3β4- and α7-nAChR, respectively, in the in vitro efficacy test and an AD50 = 0.13 μg/kg in the tail-flick test. Combined with favorable calculated physiochemical properties compared to varenicline, our findings suggest that 6b should be considered for development as a potential pharmacotherapy for treating nicotine addiction and other CNS disorders.
Keywords: Nicotine receptor, epibatidine analogs, nAChR antagonist, varenicline, in vitro/in vivo study
Graphical abstract
1. Introduction
Tobacco use is the leading preventable cause of disease, disability, and death. Each year, smoking-related diseases are responsible for nearly 6 million premature deaths globally[1] and approximately 443,000 premature deaths in the United States.[2] The continued use of tobacco products is due to addiction to nicotine (1), which is present in the various tobacco products. Nicotine exerts its effects through action at nicotinic receptors (nAChRs), a family of acetylcholine gated ion channels. These receptors exist in the periphery and in the CNS and are expressed in various brain regions involved in neuronal development, learning and memory formation, pain and reward. Neuronal nicotinic acetylcholine receptors are pentamers of homomeric or heteromeric combinations of alpha (alpha2-alpha10) and beta (beta2-beta4) subunits, which have different pharmacological and biophysical properties and locations in the brain. The major contributor to high affinity nAChR binding in the brain is the α4β2-nAChR.[3] A primary function of nAChRs in the CNS is modulation of the release of various neurotransmitters, including dopamine, glutamate, GABA, norepinephrine and acetylcholine.[4–6] Even though nicotine (1) dependence has a huge impact on global health, drugs for treating tobacco use remain limited. At present, bupropion (2), varenicline (3), and nicotine replacement therapies (NRT) are the only Food and Drug Administration (FDA)-approved therapies. Since only about 20% of smokers are able to maintain long-term (12-months) abstinence with present therapies,[7, 8] new and improved pharmacotherapies are needed. Over the last several years we have synthesized and studied the nAChR properties of a number of epibatidine (4) analogs [9–16] as a way to identify compounds that have the potential as pharmacotherapies useful for treating smokers. In this study we report the synthesis, nAChR in vitro and in vivo pharmacological properties of 3′-(substituted pyridinyl)-deschloroepitatidine analogs (5a–e and 6a–e).
2. Chemistry
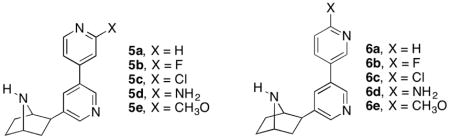
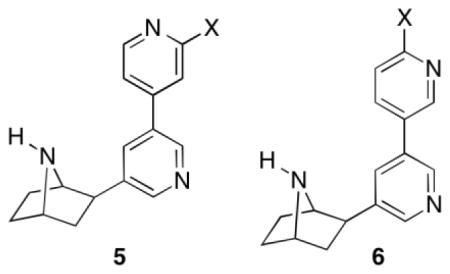
The synthesis of compounds 5a–e is given in Scheme 1. Suzuki cross-coupling of 7-tert-butoxycarbonyl-2-exo-(3′-bromo-5′-pyridinyl)-7-azabicyclo[2.2.1]heptane(7)[12] with 4-pyridine boronic acid in the presence of tetrakis(triphenylphosphine) palladium(0), potassium carbonate in a mixture of 1,2-dimethoxyethane and water, heated at 100 °C for 24 h in a sealed tube provided 8a. Suzuki cross-couplings of racemic 7 with commercially available 2-fluoropyridine-4-boronic acid or 2-chloropyridine-4-boronic acid in the presence palladium diacetate, tri-o-tolylphosphine, sodium carbonate in a 1,2-dimethoxyethane and water mixture heated at 80 °C for 5 h furnished the bipyridine derivatives 8b and 8c, respectively. Removal of the Boc-protection in compounds 8a–c was accomplished using trifluoroacetic acid in methylene chloride stirred at room temperature for 1 h to provide analogs 5a–c. In order to prepare 5d and 5e, compound 7 was first transformed to boronic ester 9 by cross-coupling using bis(pinacolato)diboron in the presence of potassium acetate, and palladium(II) dichloride [PdCl2(dppf)] in 1,4-dioxane, heated at 110 °C overnight. The resulting boronic ester 9 was further subjected to cross-coupling reactions with either 2-amino-4-bromopyridine or 4-bromo-2-methoxypyridine in the presence of tetrakis(triphenylphosphine) palladium(0), potassium carbonate in a 1,4-dioxane and water mixture heated at 110 °C in a sealed tube overnight to furnish the compounds 8d and 8e. Removal of the Boc-protecting group with trifluroacetic acid provided the amines 5d and 5e respectively.
Scheme 1.

Reagents and conditions: (a) 4-Pyridine boronic ester, Pd(PPh3)4, K2CO3, DME, H2O, 100 °C, 24 h (for 5a); (b) 2-fluoropyridine-4-boronic acid (for 5b) or 2-Chloropyridine-4-boronic acid (for 5c), Pd(OAc)2, P(o-tolyl)3, Na2CO3, DME, H2O, 80 °C, 5 h; (c) TFA, CH2Cl2, rt, 3 h; (d) Bis(pinacolato)diboron, KOAc, PdCl2(dppf), 1,4-dioxane, 110 °C, 18 h; (e) 3-amino-4-bromopyridine (for 8d), 4-bromo-2-methoxypyridine (for 8e), Pd(PPh3)4, K2CO3, 1,4-dioxane, H2O, 110 °C, overnight.
Compounds 6a–e were synthesized by procedures similar to those used to prepare 5a–e (Scheme 2). Thus, Suzuki cross-coupling reactions of 7 with either 2-flouro-5-boronic acid or 2-chloro-5-boronic acid in the presence of palladium diacetate, tri-o-tolylphosphine, sodium carbonate in a 1,2-dimethoxyethane and water mixture heated at 80 °C for 5 h furnished the bipyridine derivatives 10b and 10c. In a similar fashion, pyridine-3-boronic acid, 2-aminopyridine-5-boronic acid and 2-methoxypyridine-5-boronic acid were subjected to cross coupling reactions with compound 7 in the presence of tetrakis(triphenylphosphine) palladium (0), potassium carbonate, heated at reflux for 24 h in a 1,4-dioxane, and water mixture to provide the desired cross-coupled products 10a, d, and e in good yields. Removal of the Boc protecting group in compounds 10a–e was accomplished using trifluoroacetic acid in methylene chloride to provide analogs 6a–e.
Scheme 2.

Reagents and conditions: (a) 2-Fluoro-5-boronic acid (for 10b) or 2-Chloro-5-boronic acid (for 10c), Pd(OAc)2, P(o-tolyl)3, Na2CO3, DME, H2O, 80 °C, 5 h; (b) Pyridine-3-boronic acid (for 10a), or 2-methoxypyridine-5-boronic acid (for 10e), or 2-aminopyridine-5-boronic (for 10d), Pd(PPh3)4, K2CO3, dioxane, H2O, reflux, 24 h; (c) TFA, CH2Cl2, rt, 2 h.
3. Results and Discussion
The nAChR binding affinities and the in vitro functional nicotinic pharmacological properties of 3′-(substituted pyridinyl) deschloroepibatadine analogs 5a–e and 6a–e were determined. The Ki values for inhibition of [3H]epibatidine binding at the α4β2*-nAChR for compounds 5a–e and 6a–e along with reference compounds nicotine (1), nat-epibatidine (4), and varenicline (3) are given in Table 1. Compounds 5a–e and 6a–e all had sub-nanomolar Ki values. The Ki values for 5a–e ranged from 0.094 for 5e to 0.38 nM for 5d and for 6a–e, from 0.12 for 6a to 0.90 nM for 6d compared to 1.5, 0.026, and 0.12 for nicotine, epibatidine, and varenicline, respectively. The 4′-pyridyl and 3′-pyridyl analogs 5a and 6a, respectively, had Ki values of 0.22 and 0.81 nM, respectively. Substitution of 5a and 6a with 3′- and 4′-substituents, respectively, had only small effects on the α4β2*-nAChR binding affinity. Analyses of the data show that the presence of 3′- or 4′-substituents on the 4′-pyridyl (5b–e) and 3′-pyridyl ring (6b–e) did not show any clear structure binding affinity pattern to the Ki values. However, with the exception of the chloro substituent, each of the 3′-substituted 5b–e analogs with Ki values of 0.094 to 0.38 nM, were slightly more potent than the corresponding 4′-substituted 6b–e analogs, which had Ki values of 0.12 to 0.90 nM. In the case of the 6b–e series, the two highest affinity compounds were the electron-donating 3′-methoxy analogue 5e (Ki = 0.094 nM) and the electron-withdrawing 3′-fluoro analogue (Ki = 0.18 nM). For the 4′-substituted 3′-pyridyl analogs the 4′-chloro and 4′-fluoro electron-withdrawing analogs 6c and 6b (Ki = 0.12 and 0.13 nM, respectively) had lower Ki values than the 3′-amino and 3′-methoxy electron-donating analogs 6d and 6e (Ki = 0.90 and 0.27 nM, respectively).
Table 1.
Radioligand binding, antinociception, and efficacy profile data for 3′-(substituted pyridine)deschloroepibatidine analogs
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structurea | X | αβ [3H]Epibatidine (Ki, nM)b (Hill slope) |
mg/kg | AD50 (μg/kg) | Agonist Activity (% of max ACh at 100 μM)d |
Antagonist Activity (% EC50 ACh remaining at 100 μM)e |
||||||||
|
| ||||||||||||||
| ED50 Tail-Flick |
ED50 Hot-Plate |
ED50 Hypothermia |
ED50 Spontaneous Activity |
Tail-Flick | Hot-Plate | α4β2 | α3β4 | α7 | α4β2 | α3β4 | α7 | |||
| nicotinec | 1.50 ± 0.30 | 1.3 (0.5–1.8) | 0.65 (0.25–0.85) | 1.0 (0.6–2.1) | 0.5 (0.15–0.78) | 40 ± 1 | 37 ± 3 | 43 ± 5 | nd | nd | nd | |||
| nat epibatidine | 0.026 ± 0.002 | 0.006 (0.001–0.01) | 0.004 (0.001–0.008) | 0.004 (0.002–0.008) | 0.001 (0.0005–0.005) | 131 ± 13 | 97 ± 4 | 150 ± 8 | nd | nd | nd | |||
| varenicline | 0.12 ± 0.02 | 11% @ 10 | 10% @ 10 | 2.8 | 2.1 | 0.2 | 470 | 13 ± 0.4 | 66 ± 4 | 74 ± 5 | 38 ± 2 | nd | nd | |
| 5a | H | 0.22 ± 0.02 | 7% @ 10 | 4% @ 10 | 5 (2–13) | 1.76 (0.6–5.4) | 39 (4–63) | 20%1000 | 1.0 ± 0.2 | 9.5 ± 1.4 | 8.1 ± 1.3 | 3.9 ± 0.8 | 11 ± 2 | 69 ± 8 |
| 5b | F | 0.18 ± 0.04 | 13% @ 10 | 34% @ 10 | 5.2 (1.9–13.6) | 1.78 (0.7–4.3) | 1.2 (0.3–6.7) | 46% @ 1000 | 0 | 9.5 ± 1.8 | 5.7 ± 1.3 | 8.1 ± 2.3 | 18 ± 2 | 90 ± 11 |
| 5c | Cl | 0.29 ± 0.03 | 10% @ 10 | 30% @ 10 | 6.2 (3–12.5) | 1.6 (0.4–5.8) | 110 (20–645) | 36% @ 1000 | 0 | 6.5 ± 1.4 | 8.1 ± 2.4 | 5.7 ± 1.7 | 10 ± 1 | 47 ± 6 |
| 5d | NH2 | 0.38 ± 0.10 | 1% @ 10 | 5% @ 10 | 0% @ 10 | 20% @ 10 | 1 (0.13–1.4) | 32% @ 1000 | 0 | 1.2 ± 0.1 | 12 ± 2 | 5.2 ± 0.9 | 4.2 ± 0.8 | 44 ± 11 |
| 5e | CH3O | 0.094 ± 0.012 | 14% @ 10 | 50% @ 10 | 10 (1–114) | 4 (1.2–13.4) | 0.3 (0.04–20) | 900 (487–1687) | 0 | 6.1 ± 0.7 | 16 ± 2 | 4.0 ± 1.0 | 7.2 ± 0.9 | 47 ± 8 |
| 6a | H | 0.81 ± 0.08 | 3% @ 10 | 14% @ 10 | 7.0 (3.5–9.3) | 3 (0.3–2.7) | 0.68 (0.11–4) | 2000 (300–13700) | 0 | 4.5 ± 0.8 | 5.8 ± 1.1 | 5.3 ± 0.9 | 14 ± 0.5 | 59 ± 5 |
| 6b | F | 0.13 ± 0.02 | 10% @ 10 | 10% @ 10 | 2.78 (0.8–8.80) | 0.69 (0.08–6.5) | 2 (0.4–9) | 10% @ 1000 | 0 | 5.5 ± 0.8 | 0 | 7.9 ± 1.2 | 41 ± 10 | 36 ± 3 |
| 6c | Cl | 0.12 ± 0.01 | 8% @ 10 | 17% @ 10 | 5.5 (3.7–8) | 0.56 (0.1–1.9) | 0.5 (0.05–4.7) | 10% @ 1000 | 0 | 3.1 ± 0.9 | 4.8 ± 0.6 | 4.8 ± 0.6 | 12 ± 1 | 38 ± 3 |
| 6d | NH2 | 0.90 ± 0.02 | 3% @ 10 | 14% @ 10 | 0% @ 10 | 20% @ 10 | 1.5 (0.5–4.1) | 50% @ 10000 | 0 | 0 | 7.0 ± 1.5 | 5.7 ± 0.9 | 5.4 ± 0.7 | 46 ± 3 |
| 6e | CH3O | 0.27 ± 0.03 | 8% @ 10 | 17% @ 10 | 5.5 (3.7–8) | 0.56 (0.1–1.9) | 0.5 (0.05–4.7) | 10% @ 1000 | 0 | 2.4 ± 0.5 | 1.2 ± 0.3 | 4.5 ± 0.7 | 8.4 ± 2.3 | 18 ± 2 |
All compounds were tested as their (±)- isomers.
The Kd for (±)-epibatidine is 0.02 nM.
Data taken from ref 5.
Assessed by comparing the current response to 100 μM of each compound to the mean current response of three preceding applications of ACh, applied at an EC20 concentration (20 μM for α4β2, 100 μM for α3β4) or an EC50 concentration (300 μM for α7) and expressed as a percentage of the maximal response to ACh.
Assessed by comparing the current response to an EC50 concentration of ACh (70 μM for α4β2, 200 μM for α3β4, 300 μM for α7) in the presence of 100 μM of each compound to the mean current response of three preceding applications alone.
The receptor subtype selectivity of 5a–e and 6a–e was initially assessed in an electrophysiological assay using rat α4β2-, α3β4-, and α7-nAChRs expressed in Xenopus oocytes and assayed by a two-electrode voltage clamp. Compounds were compared to previously determined values for nicotine, [9] nat-epibatidine, [9] and varenicline [9] (Table 1). To assess potential agonist activity, current responses to a high concentration (100 μM) of each compound were compared to the maximum response that can be achieved with acetylcholine. Unlike nat-epibatidine, which is a full agonist at α4β2-, α3β4-, and α7-nAChRs, compounds 5a–e and 6a–e showed little or no agonist activity at α4β2-nAChR. The 3′- and 4′-amino analogs 5d and 6d, respectively, had little or no agonist activity at α3β4-nAChRs, while all the other 5 and 6 analogs displayed a low level of agonist activity at this subtype. At α7-nAChRs, compounds 6b and 6e had little or no agonist activity, compounds 5a–c, 6a, and 6c–d displayed low levels of agonist activity and compounds 5d and 5e, with 12 and 16% of the maximal ACh response, showed moderate levels of agonist activity. All of the compounds 5a–e and 6a–e showed substantially lower agonist activity at α4β2-, α3β4-, and α7-nAChRs than varenicline.
As an initial assessment of potential antagonist activity, a high concentration (100 μM) of each compound was tested for the ability to inhibit current responses to an EC50 concentration of ACh (Table 1). Compounds 5a–c, 5e, 6a and 6c displayed a similar selectivity profile, with substantial antagonist activity at α4β2-nAChRs, somewhat less antagonist activity at α3β4-nAChRs and much less antagonist activity at α7-nAChRs. Compound 6e had a similar rank order of activity (α4β2> α3β4 > α7), but the antagonist activity at α7-nAChRs was still substantial (> 80% inhibition). Compounds 5d and 6d showed similarly strong inhibition at α4β2- and α3β4-nAChRs, with less antagonist activity at α7-nAChRs. Compound 6b showed strong inhibition at α4β2-nAChRs, with similarly lesser antagonist activity at α3β4- and α7-nAChRs.
Compounds 5a–c, 5e, 6a–c and 5e were examined for subtype selectivity in more detail by generating IC50 values from concentration-inhibition curves (Table 2). Each of these compounds was a more potent antagonist at α4β2-nAChRs than at α3β4- and α7-nAChRs. In addition, each of the compounds was less potent at the α7- than the α3β4 and α4β2-nAChR. The most potent compounds at α4β2-nAChRs were 5b, 5e, 6b, and 6e, which had IC50 values of 2.9, 1.2, 1.3, and 1.8 μM, respectively. Compound 6b, with 25- and 46-fold selectively for α4β2-nAChRs, relative to the α3β4 and α7-nAChRs, respectively, was the most α4β2-selective compound. Compound 6e, with an IC50 value of 8.2 μM was the most potent compound at the α3β4-nAChR. All of the compounds showed IC50 values of 30 μM or greater at the α7-nAChR.
Table 2.
Comparison of antagonist potency values, 5a–c and 5e, and 6a–c and 5e at α4β2-, α3β4- and α7-nAChRsa
| Compounds | Antagonist Activity IC50, μM
|
||
|---|---|---|---|
| α4β2 | α3β4 | α7 | |
| varenicline | 0.20 ± 0.03b | na | na |
| 5a | 7.1 ± 1.0 | 23 ± 2 | 179 ± 34 |
| 5b | 2.9 ± 0.4 | 23 ± 3 | 233 ± 40 |
| 5c | 11 ± 1 | 20 ± 2 | 97 ± 17 |
| 5e | 1.2 ± 0.2 | 13 ± 3 | 114 ± 20 |
| 6a | 6.4 ± 0.8 | 22 ± 3 | 117 ± 22 |
| 6b | 1.3 ± 0.2 | 33 ± 8 | 60 ± 9 |
| 6c | 2.7 ± 0.6 | 24 ± 0.3 | 66 ± 9 |
| 6e | 1.8 ± 0.3 | 8.2 ± 1.5 | 30 ± 6 |
Current responses of rat nAChRs expressed in Xenopus oocytes were recorded under two-electrode voltage clamp. IC50 values for 5a–c and 5e and 6a–c and 5e inhibition of α4β2-, α3β4- and α7-nAChRs are derived from concentration-inhibition curves, in which the current response to an EC50 concentration of acetylcholine (70 μM for α4β2, 200 μM for α3β4, 300 μM for α7) in the presence of various concentrations of each compound are compared to the response to acetylcholine alone.
Data taken from ref 5.
The 3′-substituted 4′-pyridyl analogs 5a–e and the 4′-substituted 3-pyridyl analogs 6a–e were evaluated for their in vivo nAChR properties in mice, and the results were compared to the properties of varenicline (Table 1). None of the 5a–e or 6a–e compounds displayed any agonist activity in the mice tail-flick or hot-plate tests. Similar to varenicline all of the compounds except 5d had agonist activity in the hypothermia and spontaneous activity assays. In the hypothermia test the ED50 values ranged form 2.78 mg/kg for 6b to 10 mg/kg for 5e, compared to an ED50 = 2.8 mg/kg for varenicline. For the spontaneous activity the ED50 values varied from 0.56 mg/kg for both 6c and 6e to 4 mg/kg for 5e, compared to an ED50 = 2.1 mg/kg for varenicline. The discrepancy between the high binding affinity of 5a–e and 6a–e for α4β2*-nAChR and their absence of agonist effects in the tail-flick and hot-plate antinociceptive tests suggested that these compounds might act as functional nAChR antagonists in the two pain tests. As expected, all of the compounds were antagonists of nicotine-induced antinociception in the tail-flick test, with AD50 values of 0.3, 0.5, 0.5, 0.68, 1, 1.5, and 2 μg/kg for 5e, 6c, 6e, 6a, 5d, 6d, and 6b. Even 5a and 5c had AD50 values of 39 and 110 μg/kg in the tail-flick test. Varenicline has an AD50 = 0.2 μg/kg in the tail-flick test. Compounds 5a and 6a have AD50 values of 900 and 2,000 μg/kg in the hot plate test, compared to 470 μg/kg for varenicline.
Calculated lipophilicity (clogP), topological polar surface area (TPSA), and logBB derived values provide information as to the potential of a compound for development as a pharmacotherapy for treating CNS disorders. These molecular descriptors were calculated for compounds 5a–e and 6a–e as well as reference compounds nicotine, epibatidine, and varenicline (Table 3). Note that compounds 5a–e have the same molecular formula as the respective compounds 6a–e and thus, identical calculated values. In general, successful drugs used for treating CNS disorders have a clogP in the range 2–4, [17] TPSA less than 76Å, [18] and logBB greater than −1. [19] With the exception of possibly 5d and 6d, which have clogP values of 1.21, all the other compounds have clogP values within or close to the desirable range. All of the compounds have TPSA values of less than 76Å and logBB values between −0.62 and −0.08. For comparison varenicline has clogP, TPSA, and logBB values of 1.01, 37.81Å, and 0.27, respectively. The calculated values for 5a–e and 6a–e also compare favourably to the values of epibatadine.
Table 3.
Calculated physiochemical properties of 5a–e and 6a–e, nicotine, epibatidine, and varenicline
| Compd ID | LogPa | TPSAa | logBBb |
|---|---|---|---|
| nicotine | 1.16 | 16.13 | 0.08 |
| epibatidine | 1.84 | 24.92 | 0.05 |
| varenicline | 1.01 | 37.81 | −0.27 |
| 5a | 1.45 | 37.81 | −0.20 |
| 5b | 1.99 | 37.81 | −0.12 |
| 5c | 2.27 | 37.81 | −0.08 |
| 5d | 1.21 | 63.83 | −0.62 |
| 5e | 1.89 | 47.04 | −0.27 |
| 6a | 1.45 | 37.81 | −0.20 |
| 6b | 1.99 | 37.81 | −0.12 |
| 6c | 2.27 | 37.81 | −0.08 |
| 6d | 1.21 | 63.83 | −0.62 |
| 6e | 1.89 | 47.04 | −0.27 |
ChemAxon Calculator Plugins, Marvin 6.1.0, 2013.
logBB = −0.0148 * TPSA + 0.152 * LogP + 0.139 (from ref. 15).
4. Conclusions
In conclusion, the 3′-(substituted pyridinyl)deschloroepibatidine analogs 5a–e and 6a–e were synthesized and evaluated in vitro for their ability to inhibit [3H]epibatidine binding at nAChR and for agonist and antagonist efficacy at α4β2-, α3β4-, and α7-nAChR using an electrophysiology assay. In addition, the compounds were evaluated for agonist effects in the tail-flick, hot-plate, spontaneous activity, and hypothermia tests in the mouse and antagonists of nicotine-induced antinociception in the tail-flick and hot-plate tests in the mouse. All of the 3′-(substituted pyridinyl)deschloroepibatidine analogs had high binding affinity for α4β2* nAChRs. Several of the analogs were potent antagonists of α4β2-nAChRs in in vitro efficacy tests and were potent antagonists of nicotine-induced antinociception in the mouse tail-flick test. Compound 6b, with a Ki of 0.13 nM in the binding assay, had the greatest selectivity for the α4β2-nAChR in the electrophysiological assay, with an IC50 of 1.3 μM at this receptor, a 25- and 46-fold selectivity relative to the α3β4- and α7-nAChRs, respectively. Compound 6b also had an AD50 of 0.13 μg/kg in the tail-flick test. These data, combined with favorable calculated physicochemical properties compared to varenicline, suggest that 6b will be a valuable pharmacological tool for studying nAChRs and should be considered for development as a potential pharmacotherapy for treating nicotine addiction and other CNS disorders.
5. Experimental
Melting points were determined on a Mel-temp (Laboratory Devices, Inc.) capillary tube apparatus. NMR spectra were recorded on a Brunker Avance 300 or AMX 500 Spectrometer using tetramethylsilane as internal standard. Mass Specs were determined on a Perkin-Elmer Sciex API 150EX mass spectrometer outfitted with an APCI and ESI sources. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross GA. Analytical thin-layer chromatography (TLC) was carried out on plates pre-coated with silica gel (60 F254). TLC visualization was accomplished with a UV lamp or in an iodine chamber. Purifications by flash chromatography were performed on a Combiflash® Teledyne ISCO instrument.
General Procedure - Cross-coupling of boronic acids - Method A
To a resealable reaction vessel under nitrogen was added 1.0 equiv of compound 7, Pd(OAc)2 (0.1 equiv), P(o-tolyl)3 (0.2 equiv), sodium carbonate (2.0 equiv) and the respective pyridinyl boronic acid (1.6 equiv), DME (6 mL) and water (0.7 mL). The mixture was degassed through bubbling nitrogen for 40 min, sealed and heated in an oilbath at 80 °C for 5 h. The mixture was cooled, poured into 20 mL of a saturated aqueous solution of NaHCO3 (20 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered through Celite and the solvent removed under reduced pressure. The resultant residue was purified on a silica gel ISCO column and eluted with EtOAc/hexanes.
General Procedure - Cross-coupling of boronic acids – Method B
To a resealable reaction pressure vessel under nitrogen was added racemic 7 (1.0 equiv), Pd(PPh3)4 (5 mol %), potassium carbonate (2.0 equiv), the respective pyridine boronic acid (1.3 equiv), 1,4-dioxane (10 mL) and water (2 mL). The mixture was degassed through bubbling nitrogen for 30 min and heated at 110 °C for 24 h. After cooling to room temperature the mixture was poured into a 30 mL of aqueous solution of NaHCO3 and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered through Celite and the solvent removed in vacuo. The resultant residue was purified on silica gel eluted with hexanes/EOAc to furnish the respective products.
General Procedure C: Removal of the Boc-protecting group
The Boc-protected compound in methylene chloride (5 mL) was treated with TFA (1.5 mL) and stirred at room temperature overnight. In some cases the solution was heated at 40 °C for 2 h then stirred at room temperature overnight. The solvent was then removed in vacuo and the residue was treated with a solution of NH4Cl (20 mL) and extracted with CHCl3/MeOH (10%) (3 × 30 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the residue was purified by flash chromatography on a silica gel column eluted with MeOH/CH2Cl2 or CHCl3 (10/90) to provide the desired product.
General Procedure D: Conversion to the Fumarate Salts
A solution of the amine in ether (2 mL) was treated with a solution of fumaric acid (1.2 equivalent) in MeOH. The mixture was left to stand in a refrigerator overnight. Filtration and washing of the filter cake with ether, followed by recrystallization from MeOH-ether to provide the fumarate salt as a crystalline solid.
2-exo-[3′-(Pyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (5a) Fumarate
A solution of 8a (175 mg, 0.498 mmol) was treated according to the General Procedure C to provide 102 mg (84%) of compound 5a. 1H NMR (300 MHz, CDCl3) δ 1.50 – 1.88 (m, 7H), 1.93 – 2.00 (dd, J = 9.0, 11.2 Hz, 1H), 2.85 – 2.90 (dd, J = 3.9, 6.9 Hz, 1H), 3.65 (s, 1H), 3.82 (br s 1H), 7.53 (d, J = 5.6 Hz, 2H), 8.04 (t, J = 2.0 Hz, 1H), 8.60 – 8.71 (m, 4H); 13C NMR (CDCl3) δ 30.2, 31.5, 40.4, 45.2, 56.4, 62.8, 121.6, 133.0, 133.5, 142.5, 145.6, 149.6, 150.4; MS (ESI) m/z 252.3 (M+H)+.
A solution of 5a (102 mg, 0.404 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (92.4 mg) as white crystalline solid. M.p 182 – 186 °C: 1H NMR (500 MHz, METHANOL-d4) δ 8.85 (d, J = 1.74 Hz, 1H), 8.66 – 8.67 (m, 2H), 8.64 (d, J = 2.1 Hz, 1H), 8.21 – 8.22 (m, 1H), 7.83 – 7.84 (m, 2H), 6.69 (s, 2H), 4.63 (d, J = 3.83 Hz, 1H), 4.37 (br s, 1H), 3.57 (dd, J = 6.10, 9.59 Hz, 1H), 2.52 (dd, J = 9.59, 13.42 Hz, 1H), 2.15 – 2.26 (m, 1H), 1.97 – 2.15 (m, 4H), 1.82 – 1.97 (m, 1H); 13C NMR (500 MHz, METHANOL-d4) δ 27.03, 29.07, 37.72, 44.01, 60.44, 64.00, 123.57, 135.07, 135.33, 135.90, 139.60, 147.17, 147.30, 150.14, 151.06, 170.06; MS (ESI) m/z 252.3 [(M-fumarate)+, M=C16H17N3•C4H4O4]. Anal. calcd. for C20H21N3O4: C, 63.06; H, 5.95; N, 11.03. Found: C, 62.97; H, 5.74; N, 10.46.
2-exo-[3′-(2-Fluoropyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (5b) Fumarate
A solution of 8b (217 mg, 0.587 mmol) was treated according to the General Procedure C to provide 102 mg (65%) of compound 5b. 1H NMR (300 MHz, CDCl3) δ 1.50 – 1.75 (m, 6H), 1.82 (br s, 1 H), 1.94 – 2.01 (dd, J = 9.0, 11.2 Hz, 1H), 2.85 – 2.90 (dd, J = 3.9, 6.9 Hz, 1H), 3.65 (s, 1H), 3.84 (br s 1H), 7.17 (s, 1H), 7.43 (dt, J = 1.6, 5.2 Hz, 1H), 8.07 (t, J = 2.1 Hz, 1H), 8.29 (d, J = 5.2 Hz, 1H) 8.63 (d, J = 2.0 Hz, 1H), 8.71 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 30.3, 31.5, 40.1, 45.1, 56.4, 62.8, 107.0, 119.5, 133.1, 142.7, 145.6, 148.2, 148.4, 151.2, 162.8, 166; MS (ESI) m/z 270.4 (M+H)+.
A solution of 5b (85 mg, 0.314 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (80 mg) as white crystalline solid. Mp. 210 – 214 °C; 1H NMR (500 MHz, METHANOL-d4) δ 8.87 (d, J = 1.74 Hz, 1H), 8.65 (d, J = 1.74 Hz, 1H), 8.33 (d, J = 5.23 Hz, 1H), 8.22 – 8.23 (m, 1H), 7.72 (td, J = 1.70, 5.32 Hz, 1H), 7.52 (s, 1H), 6.671 (s, 2H), 4.63 (br s, 1H), 4.35 – 4.37 (br s, 1H), 3.55 – 3.57 (m, 1H), 3.57 (d, J = 5.93 Hz, 1H), 2.50 (d, J = 9.76 Hz, 3H), 2.52 (d, J = 9.76 Hz, 3H), 2.21 (s, 6H), 1.98 – 2.16 (m, 17H), 1.87 – 1.908 (m, 6H); 13C (500 MHz, METHANOL-d4) δ 27.3, 29.1, 37.7, 44.0, 60.4, 64.0, 108.6, 121.2, 135.1, 136.0, 139.7, 147.3, 149.5, 149.6, 150.6, 170.5; MS (ESI) m/z 270.2 [(M-fumarate)+, M=C16H16FN3•C4H4O4]. Anal. calcd. for C20H20FN3O4•0.5H2O: C, 60.92; H, 5.37; N, 10.65. Found: C, 60.72; H, 5.15; N, 10.49.
2-exo-[3′-(2-Chloropyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (5c) Fumarate
A solution of 8c (175 mg, 0.454 mmol) was treated according to the General Procedure C to provide 98.3 mg (76%) of compound 5c as a coloress oil. 1H NMR (300 MHz, CDCl3) δ 1.57 – 1.74 (m, 6H), 1.93 – 2.00 (dd, J = 9.1, 11.2 Hz, 1H), 2.85 – 2.89 (m, 1H), 3.65 (s, 1H), 3.83 (br s 1H), 7.45 (dd, J = 1.5, 5.2 Hz, 1H), 7.56 (s, 1H), 8.05 (t, J = 2.1 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H) 8.62 (d, J = 2.0 Hz, 1H), 8.68 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 30.3, 31.6, 40.4, 45.1, 56.4, 62.8, 120.5, 122.1, 132.3, 133.1, 142.7, 145.6, 148.8, 150.2, 152.4; MS (ESI) m/z 286.5 (M+H)+.
A solution of 5c free base (104 mg, 0.365 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide 95 mg of the fumarate salt as a white crystalline solid. M.p 205 – 208 °C. 1H NMR (500 MHz, METHANOL-d4) δ 8.85 (d, J = 1.95 Hz, 1H), 8.65 (d J = 1.85 Hz, 1H), 8.48 (d, J = 5.35 Hz, 1H), 8.20 – 8.23 (m, 1H), 7.90 – 7.93 (m, 1H), 7.77 (dd, J = 1.66, 5.27 Hz, 1H), 6.69 (s, 2H), 4.61 – 4.64 (br s, 1H), 4.35 – 4.38 (br s, 1H), 3.53 – 3.59 (m, 1H), 2.47 – 2.55 (m, 1H), 2.21 – 2.21 (m, 1H), 2.07 – 2.11 (m, 1H), 1.98 – 2.07 (m, 1H), 1.89 – 1.97 (m, 1H); 13C NMR (500 MHz, METHANOL-d4) δ 27.0, 29.1, 37.8, 44.0, 60.4, 64.0, 122.3, 123.7, 134.3, 135.1, 136.0, 139.7, 147.4, 150.0, 150.7, 151.5, 153.5, 170.5; MS (ESI) m/z 286.5 [(M-fumarate)+, M=C16H16ClN3•C4H4O4]. Anal. calcd. for C20H20ClN3O4: C, 59.78; H, 5.02; N, 10.46. Found: C, 59.76; H, 4.97; N, 10.40.
2-exo-[3′-(2-Aminopyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (5d) Hydrochloride
A solution of 8d (180 mg, 0.491 mmol) was treated according to the General Procedure C to provide 88 mg (67%) of compound 5d. 1H NMR (300 MHz, CDCl3) δ 1.50 – 1.76 (m, 6H), 1.86 (br s, 1H) 1.92 – 1.99 (dd, J = 9.0, 11.2 Hz, 1H), 2.84 – 2.88 (dd, J = 3.9, 6.9 Hz, 1H), 3.64 (s, 1H), 3.83 (br s 1H), 4.69 (br s, 2H) 6.70 (s, 1H), 6.85 (dd, J = 1.1, 5.3 Hz, 1H), 7.85 (d, J = 1.7 Hz, 1H), 8.13 (d, J = 5.3 Hz, 1H), 8.55 (d, J = 1.8 Hz, 1H), 8.65 (d, J = 2.0 Hz, 1H); 13C NMR (CDCl3) δ 30.2, 31.5, 40.4, 45.2, 56.5, 62.8, 106.2, 112.5, 132.9, 134.1, 142.2, 145.6, 147.4, 148.9, 149.3, 159.1; MS (ESI) m/z 267.1 (M+H)+.
A solution of 5d (88.4 mg, 0.332 mmol) in chloroform in a vial was treated with a 2.0 equiv solution of HCl in diethyl ether and allowed to stand at room temperature. The solid obtained was recrystallized from MeOH/diethyl ether to provide 80.2 mg (66%) of 5d•HCl as an off white solid. Mp. 209 – 214 °C; 1H NMR (300 MHz, CD3OD) δ 1.90 – 2.0 (m, 4H), 2.10 – 2.34 (m, 1H), 2.52 – 2.60 (dd, J = 9.0, 11.2 Hz, 1H), 3.64 – 3.69 (dd, J = 3.9, 6.9 Hz, 1H), 4.42 (s, 1H), 4.70 (br s 1H), 7.38 (dd, J = 1.6, 6.8 Hz, 1H), 7.49 (s, 1H), 8.01 (d, J = 6.7 Hz, 1H), 8.49 (s, 1H), 8.82 (s, 1H), 8.96 (s, 1H); 13CNMR (CD3OD) δ 26.8, 28.9, 37.4, 43.9, 60.5, 64.0, 112.5, 112.7, 137.3, 137.4, 140.3, 145.9, 149.9, 153.2; MS (ESI) m/z 267.2 [(M-HCl)+, M = C16H18N4•2HCl]; Anal. calcd. for C16H20Cl2N4•1.5H2O: C, 52.46; H, 6.33; N 15.30. Found: C, 52.81; H, 6.07; N, 15.17.
2-exo-[3′-(2-Methoxypyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (5e) Fumarate
A solution of 8e (270 mg, 0.708 mmol) was treated according to the General Procedure C to provide 138 mg (69%) of compound 5e. 1H NMR (300 MHz, CDCl3) δ 1.50 – 1.76 (m, 6H), 1.86 (br s, 1H) 1.92 – 1.99 (dd, J = 9.0, 11.2 Hz, 1H), 2.84 – 2.88 (dd, J = 3.9, 6.9 Hz, 1H), 3.64 (s, 1H), 3.83 (br s 1H), 4.69 (br s, 2H) 6.70 (s, 1H), 6.85 (dd, J = 1.1, 5.3 Hz, 1H), 7.85 (d, J = 1.7 Hz, 1H), 8.13 (d, J = 5.3 Hz, 1H), 8.55 (d, J = 1.8 Hz, 1H), 8.65 (d, J = 2.0 Hz, 1H); 13C NMR (CDCl3) δ 30.0, 31.3, 40.2, 45.5, 55.2, 56.5, 62.8, 107.3, 108.4, 133.1, 134.6, 141.8, 145.8, 149.4, 151.2, 156.8, 166.4; MS (ESI) m/z 282.5 (M+H)+.
A solution of 5e free base (117.4 mg, 0.417 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (156 mg) as white crystalline solid. Mp. 160 – 164 °C; 1H NMR (300 MHz, METHANOL-d4) δ (d, J = 1.74 Hz, 1H) 8.60 (d, J = 1.71 Hz, 1H), 8.51 (d, J = 5.85 Hz, 1H), 7.52 (d, J = 1.8 Hz, 1H), 7.04 (dd, J = 2.4, 5.82 Hz, 1H), 6.68 (s, 1H), 4.60 (d, J = 2.0 Hz, 1H), 4.37 (br s, 1H), 3.98 (s, 3H), 3.57 (dd, J = 3.3, 9.3 Hz, 1H), 2.45 – 2.53 (m, 1H), 1.86 – 2.26 (m, 6H); 13C NMR (300 MHz, METHANOL-d4) δ 26.99, 28.84, 37.33, 43.91, 56.32, 60.33, 64.15, 109.52, 110.58, 134.72, 135.87, 138.97, 147.41, 149.72, 152.18, 157.20, 170.89; MS (ESI) m/z 282.4 [(M-fumarate)+, M=C16H16FN3•C4H4O4]. Anal. calcd. for C20H20FN3O4•0.5H2O: C, 59.42; H, 6.17; N, 9.90. Found: C, 59.38, H, 5.92, N, 8.83.
2-exo-[3′-(Pyridin-3″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (6a) Fumarate
A solution of 10a (177 mg, 0.504 mmol) was treated according to the General Procedure C to provide 106 mg (84%) of compound 6a. 1H NMR (300 MHz, CDCl3) δ 1.55 – 1.76 (m, 6H), 1.93 – 2.00 (dd, J = 9.0, 11.2 Hz, 1H), 2.86 – 2.90 (dd, J = 3.9, 6.9 Hz, 1H), 3.66 (s, 1H), 3.83 (br s 1H), 7.37 – 7.42 (m, 1H), 7.89 (dt, J = 2.0, 8.0 Hz, 1H), 7.98 (t, J = 2.0 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.63 – 8.66 (m, 2H) 8.85 (d, J = 1.8 Hz, 1H); 13C NMR (CDCl3) δ 30.2, 31.4, 40.4, 45.2, 56.4, 62.8, 123.6, 133.1, 133.8, 134.4, 142.4, 145.7, 148.3, 148.8, 149.1; MS (ESI) m/z 252.2 (M+H)+.
A solution of 6a free base (106 mg, 0.430 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (103 mg) as white crystalline solid. M.p 169 – 172 °C; 1H NMR (300 MHz, METHANOL-d4) δ 1.91 – 2.22 (m, 5H), 2.47 – 2.55 (dd, J = 9.0, 11.2 Hz, 1H), 3.53 – 3.58 (dd, J = 3.9, 6.9 Hz, 1H), 4.35(s, 1H), 4.63 (br s 1H), 6.65 (s, 2H), 7.57 – 7.61 (dd, J = 3.0, 5.3 Hz, 1H), 8.17 (t, J = 1.9 Hz, 1H), 8.22 (dt, J = 2.0, 6.2 Hz, 1H), 8.60 – 8.63 (m, 2H), 8.77 (d, J = 1.9 Hz, 1H), 8.91 (d, J = 2.0 Hz, 1H); 13C NMR (300 MHz, METHANOL-d4) δ 27.1, 29.2, 37.8, 44.1, 60.4, 64.1, 125.8, 135.8, 136.3, 137.1, 139.6, 147.3, 148.8, 149.2, 150.1, 171.4; MS (ESI) m/z 252.2 [(Mfumarate)+, M=C16H17N3•C4H4O4]; Anal. calcd. for C20H21N3O4•0.5H2O: C, 63.82; H, 5.89; N 11.16. Found: C, 63.53; H, 5.89; N, 11.02.
2-exo-[3′-(2″-Fluoropyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (6b) Fumarate
A solution of 10b (170 mg, 0.461 mmol) was treated according to the General Procedure C to provide 76 mg (61%) of compound 6b. 1H NMR (300 MHz, CDCl3) δ 1.52 – 1.73 (m, 6H), 1.93 – 2.00 (m, 1H), 2.82 – 2.86 (m, 1H), 3.62 (s, 1H), 3.80 (br s 1H), 7.01 (dd, J = 3.0, 8.5 Hz, 1H), 7.92 – 8.00 (m, 2H), 8.39 (d, J = 1.9 Hz, 1H), 8.54 (d, J = 2.0 Hz, 1H), 8.59 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 30.2, 31.5, 40.4, 45.2, 56.4, 62.8, 109.4, 132.1, 133.1, 139.9, 142.5, 145.6, 145.9, 148.9, 161.8, 165.1; MS (ESI) m/z 270.3 (M+H)+.
A solution of 6b free base (141 mg, 0.525 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (142 mg) as white crystalline solid. M.p 205 – 207 °C; 1H NMR (300 MHz, METHANOL-d4) δ 1.85 – 2.24 (m, 5H), 2.47 – 2.55 (m, 1H), 3.53 – 3.58 (dd, J = 3.9, 6.9 Hz, 1H), 4.35 (s, 1H), 4.62 (br s 1H), 6.67 (s, 3H), 7.22 (dd, J = 2.6, 8.6 Hz, 1H), 8.13 (s, 1H), 8.31 (td, J = 2.6, 8.2 Hz, 1H), 8.57 (d, J = 2.4 Hz, 1H), 8.59 (d, J = 1.9 Hz, 1H), 8.76 (d, J = 1.8 Hz, 1H); 13C NMR (300 MHz, METHANOL-d4) δ 27.1, 29.1, 37.7, 44.1, 60.5, 64.1, 111.0, 135.0, 136.0, 139.5, 142.2, 147.2, 147.4, 149.1, 163.5, 167.0, 170.5; MS (ESI) m/z 270.4 [(M-fumarate)+, M=C16H16FN3•1.5C4H4O4]; Anal. calcd for C22H22FN3O6: C, 59.59; H, 5.00; N, 9.48. Found, C, 59.46; H, 5.00; N, 9.54.
2-exo-[3′-(2″-Chloropyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (6c) Fumarate
A solution of 10c (112 mg, 0.290 mmol) was treated according to the General Procedure C to provide 71 mg (86%) of compound 6c. 1H NMR (300 MHz, CDCl3) δ 1.53 – 1.76 (m, 6H), 1.93 – 2.00 (dd, J = 9.1, 11.2 Hz, 1H), 2.85 – 2.89 (m, 1H), 3.65 (s, 1H), 3.83 (br s 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.85 (dd, J = 2.1, 8.3 Hz, 1H), 7.97 (t, J = 2.1 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H) 8.62 (dd, J = 2.2, 5.0 Hz, 1H); 13C NMR (CDCl3) δ 30.3, 31.5, 40.4, 45.2, 56.4, 62.8, 124.4, 131.9, 132.9, 133.1, 137.3, 142.6, 145.6, 148.0, 149.2, 151.1; MS (ESI) m/z 286.6 (M+H)+.
A solution of 6c free base (71 mg, 0.247 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (70 mg) as white crystalline solid. M.p 199 – 202 °C; 1H NMR (300 MHz, METHANOL-d4) δ 1.88 – 2.24 (m, 5H), 2.47 – 2.54 (dd, J = 9.1, 11.2 Hz, 1H), 3.53 – 3.58 (dd, J = 3.9, 6.9 Hz, 1H), 4.37 (s, 1H), 4.63 (br s 1H), 6.67 (s, 3H), 7.59 (d, J = 8.3 Hz, 1H), 8.15 (s, 1H), 8.18 (dd, J = 2.6, 8.4 Hz, 1H), 8.6 (d, J = 2.0 Hz, 1H) 8.77 (dd, J = 2.0, 8.3 Hz, 1H); 13C NMR (300 MHz, METHANOL-d4) δ 27.1, 29.1, 37.7, 44.1, 60.4, 64.0, 126.1, 133.8, 134.1, 135.0, 136.0, 139.6, 147.2, 149.3, 149.4, 152.6, 170.6; MS (ESI) m/z 286.6 [(M-fumarate)+, M=C16H16ClN3•1.5C4H4O4]; Anal. calcd. for C22H22ClN3O6•0.75H2O: C, 55.82; H, 5.00; N 8.88. Found: C, 56.17; H, 4.84; N, 9.04.
2-exo-[3′-(2″-Aminopyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (6d) Fumarate
A solution of 10d (157 mg, 0.429 mmol) was treated according to the General Procedure C to provide 91.2 mg (80%) of compound 6d. 1H NMR (300 MHz, CDCl3) δ 1.52 – 1.84 (m, 6H), 1.92 – 1.99 (dd, J = 9.0, 10.9 Hz, 1H), 2.84 – 2.89 (dd, J = 3.9, 6.9 Hz, 1H), 3.65 (s, 1H), 3.82 (br s 1H), 4.58 (br s, 2H), 6.58 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 2.4, 8.5 Hz, 1H), 7.83 (t, J = 2.2 Hz, 1H), 8.31 (d, J = 2.3 Hz, 1H), 8.48 (d, J = 2.1 Hz, 1H), 8.59 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 30.2, 31.3, 40.3, 45.3, 56.5, 62.7, 108.6, 124.0, 132.1, 133.7, 136.5, 141.9, 145.1, 146.5, 147.5, 158.3; MS (ESI) m/z 267.1 (M+H)+.
A solution of 6d free base (91.2 mg, 0.342 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (72 mg) as white crystalline solid. M.p 157 – 160 °C; 1H NMR (300 MHz, METHANOL-d4) δ 1.84 – 2.20 (m, 5H), 2.45 – 2.53 (dd, J = 9.6, 9.7 Hz, 1H), 3.49 – 3.54 (dd, J = 3.3, 7.8 Hz, 1H), 4.34 (s, 1H), 4.59 (br s 1H), 6.68 (s, 2H), 6.74 (d, J = 8.8 Hz, 1H), 7.88 (dd, J = 2.4, 8.8 Hz, 1H), 8.02 (t, J = 1.9 Hz, 1H), 8.26 (d, J = 2.3 Hz, 1H), 8.46 (d, J = 1.8 Hz, 1H), 8.64 (d, J = 1.8 Hz, 1H); 13C NMR (300 MHz, METHANOL-d4) δ 27.1, 29.2, 37.8, 44.1, 53.8, 60.5, 64.1, 111.1, 123.3, 133.7, 136.2, 138.6, 139.3, 145.7, 146.2, 147.5, 160.7, 171.0; MS (ESI) m/z 267.2 [(M-fumarate)+, M=C16H18N4•1.5C4H4O4]; Anal. calcd. for C21H23N3O5•0.75H2O: C, 58.21; H, 5.66; N 12.34. Found: C, 57.85; H, 5.69; N, 12.73.
2-exo-[3′-(2″-Methoxypyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (6e)
A solution of 10e (111 mg, 0.291 mmol) was treated according to the General Procedure C to provide 71 mg (86%) of compound 6e. 1H NMR (300 MHz, CDCl3) δ 1.50 – 1.76 (m, 6H), 1.86 (br s, 1H) 1.92 – 1.99 (dd, J = 9.0, 11.2 Hz, 1H), 2.84 – 2.88 (dd, J = 3.9, 6.9 Hz, 1H), 3.65 (s, 1H), 3.82 (br s 1H), 3.98 (s, 3H) 6.83 (d, J = 8.6 Hz, 1H), 7.8 (dd, J = 2.6, 8.6 Hz, 1H), 7.88 (t, J = 2.1 Hz, 1H), 8.39 (d, J = 2.5 Hz, 1H), 8.51 (d, J = 2.0 Hz, 1H), 8.61 (d, J = 2.3 Hz, 1H); 13C NMR (CDCl3) δ 30.1, 31.4, 40.3, 45.3, 53.6, 56.5, 62.7, 111.0, 127.1, 132.6, 133.2, 137.4, 142.1, 145.1, 145.4, 148.0, 164.0; MS (ESI) m/z 282.5 (M+H)+.
A solution of 6e free base (78.5 mg, 0.279 mmol) in ether was converted to the fumarate salt according to the General Procedure D to provide the fumarate salt (50 mg) as white crystalline solid. M.p 96 – 100 °C; 1H NMR (300 MHz, METHANOL-d4) δ 1.85 – 2.22 (m, 5H), 2.46 – 2.53 (dd, J = 9.0, 11.2 Hz, 1H), 3.50 – 3.55 (dd, J = 3.9, 6.9 Hz, 1H), 3.97 (s, 3H) 4.34 (s, 1H), 4.60 (br s 1H), 6.65 (s, 2H), 6.92 (d, J = 8.8 Hz, 1H), 8.03 (dd, J = 2.6, 8.7 Hz, 1H), 8.08 (t, J = 2.1 Hz, 1H), 8.48 (d, J = 1.8 Hz, 1H), 8.52 (d, J = 2.5 Hz, 1H), 8.69 (d, J = 1.8 Hz, 1H); 13C NMR (300 MHz, METHANOL-d4) δ 27.1, 29.2, 37.8, 44.1, 54.4, 60.4, 64.1, 112.3, 127.5, 134.5, 136.3, 137.1, 139.3, 146.6, 146.7, 148.2, 166.0, 171.4; MS (ESI) m/z 282.4 [(M-fumarate)+, M=C17H19N3•C4H4O4]; Anal. calcd. for C21H23N3O5•H2O: C, 60.71; H, 6.07; N 10.11. Found: C, 60.88; H, 5.99; N, 10.05.
7-tert-Butoxycarbonyl-2-exo-[3′-(pyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (8a)
To a resealable reaction pressure vessel under nitrogen was added racemic 7 (215 mg, 0.607 mmol, 1.0 equiv), Pd(PPh3)4 (70 mg, 0.061 mmol, 10 mol %), potassium carbonate (168 mg, 1.22 mmol, 2.0 equiv), pyridine 4-boronic acid (104.4 mg, 0.849 mmol, 1.2 equiv), DME (15 mL) and water (2 mL). The mixture was degassed through bubbling nitrogen and heated at 100 °C for 24 h. After cooling to room temperature the mixture was poured into 30 mL of H2O and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered through Celite and the solvent removed in vacuo. The resultant residue was purified on a silica gel column eluted with hexanes/isopropanol to furnish 175 mg (82%) of 8a as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 1.43 (br s, 9H), 1.58 – 1.66 (m, 2H), 1.87 – 1.94 (m, 3H), 1.93 – 2.00 (m, 1H), 2.04 – 2.11 (dd, J = 9.0 Hz, 1H), 2.97 – 3.02 (m, 1 H), 4.29 (s, 1H), 4.43 (br s, 1H), 7.51 – 7.56 (m, 2H), 7.93 (d, J = 1.9 Hz, 1H), 8.56 (d, J = 1.9 Hz, 1H) 8.69 – 8.74 (m, 3H); 13C NMR (CDCl3) δ; 28.3 (3 C), 28.8, 29.8, 40.4, 45.4, 55.9, 61.8, 79.8, 121.5, 132.5, 133.5, 141.4, 145.3, 145.9, 149.4, 150.4, 154.8; MS (ESI) m/z 352.3 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2-flouropyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (8b)
Racemic compound 7 (386 mg, 1.09 mmol) and 2-fluoropyridine-4-boronic acid (200 mg, 1.42 mmol, 1.3 equiv) were cross-coupled according to the General Procedure A to provide 243 mg (60%) of 8b as an oil. 1H NMR (300 MHz, CDCl3) δ 1.44 (br s, 9H), 1.48 – 1.69 (m, 2H), 1.87 – 1.93 (m, 3H), 2.05 – 2.12 (dd, J = 9.0 Hz, 1H), 2.99 – 3.03 (m, 1H), 4.29 (s, 1H), 4.43 (br s, 1H), 4.54 (s, 2 NH), 7.16 (s, 1H), 7.42 – 7.44 (dt, J = 1.7, 5.2 Hz, 1H), 7.95 (t, J = 2.0 Hz, 1H), 8.30 (d, J = 5.3 Hz, 1H), 8.59 (d, J = 2.0 Hz, 1H), 8.73 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.7, 40.5, 45.3, 56.0, 61.8, 79.9, 107.4, 119.4, 132.5, 141.6, 145.8, 148.2, 150.0, 150.9, 154.9, 162.9, 166.0; MS (ESI) m/z 386.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2-chloropyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (8c)
Racemic compound 7 (194 mg, 0.549 mmol) and 2-chloropyridine-4-boronic acid (138 mg, 0.878, 1.6 equiv) were cross-coupled according to the General Procedure A to provide 175 mg (83%) of 8a as an oil. 1H NMR (300 MHz, CDCl3) δ 1.45 (br s, 9H), 1.48 – 1.69 (m, 2H), 1.87 – 1.91 (m, 3H), 2.05 – 2.12 (m, 1H), 2.98 – 3.03 (m, 1H), 4.29 (s, 1H), 4.43 (br s, 1H), 4.54 (s, 2 NH), 7.46 (dd, J = 1.5, 4.2 Hz, 1H), 7.55 (s, 1H), 7.94 (t, J = 2.0 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.71 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.7, 40.5, 45.3, 55.9, 61.7, 79.9, 120.3, 122.0, 132.4, 132.5, 141.7, 145.8, 148.5, 150.0, 150.2, 152.4, 154.9; MS (ESI) m/z 386.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2-aminopyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (8d)
To a resealable reaction pressure vessel under nitrogen was added 1.0 equiv of 9 (265 mg, 0.662 mmol), Pd(PPh3)4 (38 mg, 0.033 mmol, 5 mol %), K2CO3 (184 mg, 1.324 mmol, 2.0 equiv), 2-amino-4-bromopyridine (137 mg, 0.794 mmol, 1.2 equiv), dioxane (12 mL) and water (1 mL). The mixture was degassed through bubbling nitrogen for 40 min, sealed and heated at 110 °C for 20 h. After cooling to room temperature, water (10 mL) was added and the organic product was extracted using EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered through Celite and the solvent removed in vacuo. The residual was purified on a silica get column eluted with EtOAc/hexanes to provide 180 mg (74%) of 8d as colorless oil. 1H NMR (300 MHz, CDCl3) δ 1.43 (br s, 9H), 1.53–1.66 (m, 2H), 1.80 – 1.91 (m, 3H), 2.01 – 2.08 (m, 1H), 2.94 – 2.98 (m, 1H), 4.27 (s, 1H), 4.41 (br s, 1H), 4.76 (s, 2 NH), 6.70 (s, 1H), 6.85 (d, J = 4.3 Hz, 1H), 7.85 (s, 1H), 8.13 (d, J = 5.3 Hz, 1H), 8.52 (d, J = 1.7 Hz, 1H), 8.67 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.8, 40.4, 45.5, 55.9, 61.8, 79.8, 106.2, 112.3, 132.5, 134.2, 141.2, 145.9, 147.1, 148.8, 149.1, 154.9, 159.1; MS (ESI) m/z 367.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2-methoxypyridin-4-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (8e)
To a resealable reaction pressure vessel under nitrogen was added 1.0 equiv of 9 (266 mg, 0.665 mmol), Pd(PPh3)4 (38 mg, 0.033 mmol, 5 mol %), K2CO3 (184 mg, 1.33 mmol, 2.0 equiv), 2-methoxy-4-bromopyridine (137 mg, 0.732 mmol, 1.1 equiv), dioxane (20 mL) and water (2 mL). The mixture was degassed through bubbling nitrogen for 40 min and heated at 110 °C overnight. After cooling to room temperature, water (20 mL) was added and the organic product was extracted using EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered through Celite and the solvent removed in vacuo. The residual was purified on a silica gel column eluted with EtOAc/hexanes to provide 160 mg (63%) of 8e as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 1.42 (br s, 9H), 1.49 – 1.63 (m, 2H), 1.86 – 1.98 (m, 3H), 2.00 – 2.07 (m, 1H), 2.96 – 3.01 (m, 1H), 3.92 (s, 3H), 4.30 (s, 1H), 4.42 (br s, 1H), 6.81 (dd, J = 5.7, 2.4 Hz, 1H), 7.25 (d, J = 2.2 Hz, 1H), 8.22 (t, J = 1.9 Hz, 1H), 8.53 (d, J = 5.7 Hz, 1H), 8.56 (d, J = 2.0 Hz, 1H), 9.00 (d, J = 2.0 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.9, 40.2, 45.8, 55.9, 61.8, 79.7, 107.2, 108.6, 132.9, 134.7, 140.9, 146.2, 149.1, 151.1, 155.0, 156.7, 166.5; MS (ESI) m/z 382.7 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[5′-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3′-pyridnyl]-7-azabicyclo[2.2.1]heptane (9)
To a resealable reaction pressure vessel under nitrogen was added a solution of racemic 7 (209 mg, 0.590 mmol, 1.0 equiv), Bis(pinacolato)diboron (225 mg, 0.886 mmol, 1.5 equiv), PdCl2(dppf) (22 mg, 0.0295 mmol, 5 mol %), and KOAc (180 mg, 1.83 mmol, 3.0 equiv) in 1,4-dioxane (10 mL). The mixture was degassed through bubbling nitrogen for 40 min then heated at between 100 and 110 °C for 24 h. After cooling to room temperature the reaction was diluted in EtOAc and filtered through a plug of Celite and anhydrous sodium sulfate. The solvent was removed in vacuo and the residue was purified by flash chromatography on silica gel eluted with ethyl acetate to provide 199 mg (84%) of 9 as a brownish oil. 1H NMR (300 MHz, CDCl3) δ 1.34 (s, 12H) 1.44 (br s, 9H), 1.53 – 1.60 (m, 2H), 1.80 – 1.88 (m, 2H), 1.91 – 2.08 (m, 1H), 2.87 – 2.96 (m, 2H), 4.22 (s, 1H), 4.42 (br s, 1H), 8.02 (d, J = 5.3 Hz, 1H), 8.52 (d, J = 1.7 Hz, 1H), 8.67 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3) δ 24.6, 24.8, 24.9, 25.0, 28.3 (3 C), 28.8, 29.8, 39.9, 45.8, 55.7, 61.7, 79.6, 84.1, 140.1, 140.6, 147.4, 148.8, 151.0, 153.1, 162.5; MS (ESI) m/z 367.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(pyridin-3″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (10a)
Racemic compound 7 (334 mg, 0.944 mmol) and pyridine-3-boronic acid (150 mg, 1.23, 1.3 equiv) were cross-coupled according to the General Procedure B to provide 176 mg (53%) of 10a as an oil. 1H NMR (300 MHz, CDCl3) δ 1.43 (br s, 9H), 1.50 – 1.68 (m, 2H), 1.90 – 1.95 (m, 3H), 2.03 – 2.10 (m, 1H), 2.96 – 3.01 (m, 1H), 2.97 – 3.02 (m, 1 H), 4.29 (s, 1H), 4.43 (br s, 1H), 7.38 – 7.43 (m, 1H), 7.87 – 7.91 (m, 2H), 8.54 (d, J = 1.9 Hz, 1H), 8.65 (dd, J = 1.6, 4.8 Hz, 1H), 8.69 (d, J = 2.2 Hz, 1H), 8.85 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ; 28.3 (3 C), 28.8, 29.8, 40.4, 45.5, 55.9, 61.8, 79.8, 123.7, 133.3, 133.6, 134.4, 141.3, 146.0, 148.2, 148.6, 149.3, 154.9; MS (ESI) m/z 352.4 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2″-flouropyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (10b)
Racemic compound 7 (240 mg, 0.680 mmol) and 2-flouropyridine-5-boronic acid (124 mg, 0.883, 1.3 equiv) were cross-coupled according to the General Procedure A to provide 170 mg (68%) of 10b as an oil. 1H NMR (300 MHz, CDCl3) δ 1.43 (br s, 9H), 1.55 – 1.68 (m, 2H), 1.80 – 1.93 (m, 3H), 2.03 – 2.12 (m, 1H), 2.96 – 3.00 (m, 1H), 4.28 (s, 1H), 4.42 (br s, 1H), 7.03 – 7.07 (dd, J = 3.0, 8.5 Hz, 1H), 7.83 (t, J = 2.1 Hz, 1H), 7.98 (td, J = 7.6, 2.6 Hz, 1H), 8.42 (d, J = 2.6 Hz, 1H), 8.54 (d, J = 2.0 Hz, 1H), 8.65 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.8, 40.4, 45.5, 56.0, 61.8, 79.9, 109.5, 123.5, 132.6, 139.8, 141.5, 145.8, 148.7, 154.9, 161.9, 165.1; MS (ESI) m/z 370.4 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2″-chloropyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (10c)
Racemic compound 7 (213 mg, 0.602 mmol) and 2-chloropyridine-5-boronic acid (114 mg, 0.722, 1.2 equiv) were cross-coupled according to the General Procedure A to provide 69 mg (30%) of 10c as an oil. 1H NMR (300 MHz, CDCl3) δ 1.37 (br s, 9H), 1.50 – 1.68 (m, 2H), 1.87 – 2.03 (m, 3H), 2.05 – 2.10 (m, 1H), 2.96 – 3.00 (m, 1H), 4.28 (s, 1H), 4.42 (br s, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.83 – 7.87 (m, 2H), 8.55 (d, J = 1.8 Hz, 1H), 8.61 (d, J = 2.5 Hz, 1H), 8.66 (d, J = 2.1 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.7, 40.5, 45.5, 56.0, 61.8, 79.9, 124.4, 122.0, 132.1, 132.6, 137.2, 141.7, 145.9, 147.9, 149.0, 151.2, 155.0; MS (ESI) m/z 386.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2″-aminopyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (10d)
Racemic compound 7 (303 mg, 0.857 mmol) and 2-aminopyridine-5-pinacoboronic ester (245 mg, 1.11, 1.3 equiv) were cross-coupled according to the General Procedure B to provide 166 mg (53%) of 10d as an oil. 1H NMR (300 MHz, CDCl3) δ 1.42 (br s, 9H), 1.53 – 1.66 (m, 2H), 1.80 – 1.91 (m, 3H), 2.01 – 2.10 (m, 1H), 2.92 – 2.96 (m, 1H), 4.27 (s, 1H), 4.41 (br s, 1H), 4.70 (s, 2 NH), 6.58 (d, J = 8.6 Hz, 1H), 7.65 (dd, J = 2.3, 8.5 Hz, 1H), 7.76 (t, J = 2.0 Hz, 1H), 8.29 (d, J = 2.2 Hz, 1H), 8.43 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 28.3 (3 C), 28.8, 29.8, 40.3, 45.6, 56.0, 61.9, 79.7, 108.6, 112.3, 123.9, 131.7, 133.8, 136.4, 141.1, 145.3, 146.4, 147.3, 155.0, 158.3; MS (ESI) m/z 367.6 (M+H)+.
7-tert-Butoxycarbonyl-2-exo-[3′-(2″-methoxypyridin-5″-yl)-5′-pyridnyl]-7-azabicyclo[2.2.1]heptane (10e)
Racemic compound 7 (304 mg, 0.859 mmol) and 2-methoxypyridine-5-boronic acid (171 mg, 1.12, 1.3 equiv) were cross-coupled according to the General Procedure B to provide 118 mg (36%) of 10e as an oil. 1H NMR (300 MHz, CDCl3) δ 1.42 (br s, 9H), 1.49 – 1.63 (m, 2H), 1.86 – 1.98 (m, 3H), 2.00 – 2.07 (m, 1H), 2.96 – 3.01 (m, 1H), 3.92 (s, 3H), 4.30 (s, 1H), 4.42 (br s, 1H), 6.81 (dd, J = 5.7, 2.4 Hz, 1H), 7.25 (d, J = 2.2 Hz, 1H), 8.22 (t, J = 1.9 Hz, 1H), 8.53 (d, J = 5.7 Hz, 1H), 8.56 (d, J = 2.0 Hz, 1H), 9.00 (d, J = 2.0 Hz, 1H); 13C NMR (CDCl3) δ 27.8 (3 C), 28.3, 29.3, 39.8, 45.0, 55.5, 61.4, 79.3, 110.6, 126.4, 132.9, 136.9, 134.7, 140.7, 145.2, 147.3, 154.5, 163.6; MS (ESI) m/z 382.4 (M+H)+.
[3H]Epibatidine Binding Assay
The inhibition of [3H] binding at rat brain α4β2*-nAChRs was conducted as previously reported.[13]
Electrophysiology
The electrophysiology assays with rat α4β2-, α3β4-, and α7-nAChRs were conducted as previously described.[20]
In vivo testing
The antinociception (tail-flick and hot-plate), locomotor, and body temperature tests were all conducted as previously described.[13]
Supplementary Material
Chart 1.
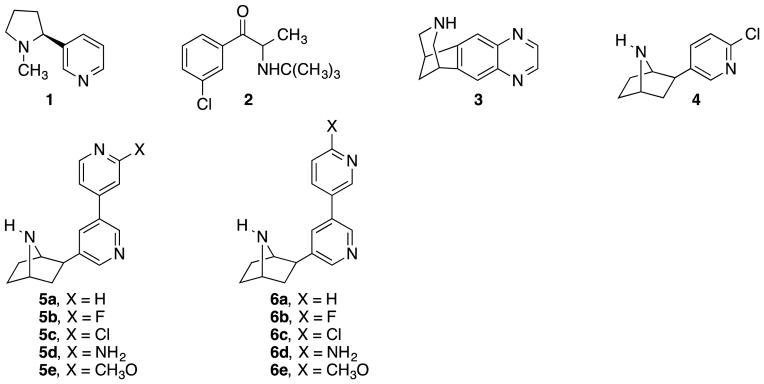
Structures of Compounds 1–4, 5a–e and 6a–e.
Acknowledgments
This research was supported by the National Institute on Drug Abuse Grant DA12001. We thank Mr. Keith Warner for conducting the [3H]epibatidine binding studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. 2011 [Google Scholar]
- 2.Center for Disease Control and Prevention. Morb Mortal Wkly Rep. 2010;59:1135. [Google Scholar]
- 3.Rahman S, Lopez-Hernandez GY, Corrigall WA, Papke RL. CNS & neurological disorders drug targets. 2008;7:422. doi: 10.2174/187152708786927831. [DOI] [PubMed] [Google Scholar]
- 4.Dani JA, Ji D, Zhou FM. Neuron. 2001;31:349. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 5.Role LW, Berg DK. Neuron. 1996;16:1077. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 6.Wonnacott S. Trends Neurosci. 1997;20:92. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 7.Johnston AJ, Ascher J, Leadbetter R, Schmith VD, Patel DK, Durcan M, Bentley B. Drugs. 2002;62(Suppl 2):11. doi: 10.2165/00003495-200262002-00002. [DOI] [PubMed] [Google Scholar]
- 8.Hesse LM, Venkatakrishnan K, Court MH, von Moltke LL, Duan SX, Shader RI, Greenblatt DJ. Drug metabolism and disposition: the biological fate of chemicals. 2000;28:1176. [PubMed] [Google Scholar]
- 9.Ondachi PW, Castro AH, Bartkowiak JM, Luetje CW, Damaj MI, Mascarella SW, Navarro HA, Carroll FI. J Med Chem. 2014;57:836. doi: 10.1021/jm401602p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll FI, Ma W, Deng L, Navarro HA, Damaj MI, Martin BR. J Nat Prod. 2010;73:306. doi: 10.1021/np9006124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll FI, Yokota Y, Ma W, Lee JR, Brieaddy LE, Burgess JP, Navarro HA, Damaj MI, Martin BR. Bioorg Med Chem. 2008;16:746. doi: 10.1016/j.bmc.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll FI, Ma W, Yokota Y, Lee JR, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J Med Chem. 2005;48:1221. doi: 10.1021/jm040160b. [DOI] [PubMed] [Google Scholar]
- 13.Carroll FI, Ware R, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. J Med Chem. 2004;47:4588. doi: 10.1021/jm040078g. [DOI] [PubMed] [Google Scholar]
- 14.Carroll FI, Lee JR, Navarro HA, Ma W, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2002;45:4755. doi: 10.1021/jm0202268. [DOI] [PubMed] [Google Scholar]
- 15.Carroll FI, Lee JR, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2001;44:4039. doi: 10.1021/jm015561v. [DOI] [PubMed] [Google Scholar]
- 16.Carroll FI, Liang F, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2001;44:2229. doi: 10.1021/jm0100178. [DOI] [PubMed] [Google Scholar]
- 17.Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. J Pharmacol Exp Ther. 2007;322:205. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- 18.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. ACS Chem Neurosci. 2012;3:50. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark DE. J Pharm Sci. 1999;88:815. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 20.Ondachi P, Castro A, Luetje CW, Damaj MI, Mascarella SW, Navarro HA, Carroll FI. J Med Chem. 2012;55:6512. doi: 10.1021/jm300575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.