

Abstract
Cellular translation is down-regulated by host antiviral responses. Picornaviridae and Flaviviridae including hepatitis C virus (HCV) evade this process using internal ribosomal entry sequences (IRESs). Although HCV IRES translation is a prerequisite for HCV replication, only few host factors critical for IRES activity are known and the global regulator network remains largely unknown. Since signal transduction is an import regulator of viral infections and the host antiviral response we combined a functional RNAi screen targeting the human signaling network with a HCV IRES-specific reporter mRNA assay. We demonstrate that the HCV host cell cofactors PI4K and MKNK1 are positive regulators of HCV IRES translation representing a novel pathway with a functional relevance for the HCV life cycle and IRES-mediated translation of viral RNA.
Hepatitis C virus (HCV) is a positive stranded RNA virus replicating in intracellular phospholipid-enriched membrane domains. Several unbiased RNAi screens identified a panel of host factors required for HCV entry, replication and assembly1,2,3,4 but none of these previous approaches discriminates effects on mRNA translation. Host protein translation is initiated with the recruitment of the 40S ribosomal subunit to mRNA. This process mostly involves the recognition of a 5′ m7GpppN cap structure by eIF4E of the cap binding complex eIF4F5. Most eukaryotic mRNAs also contain a 3′ poly(A) tail, which is acting synergistically with the cap structure to enhance translation6,7,8. Initiation of cap-dependent translation is susceptible to regulation via eIF4F by eIF4E inhibitory proteins by phosphoinositide 3-kinase (PI3K)/Akt and mammalian target of rapamycin (mTOR) signaling pathways9. Furthermore, MAP kinase pathways modulate cap-dependent translation by phosphorylation of ribosomal protein S610 and by eIF4E phosphorylation via MAP kinase interacting serine/threonine kinase 1 (MKNK1)11,12. Furthermore, cap translation is inhibited by heatshock proteins13 and by protein kinase R (PKR) and PKR-like endoplasmic reticulum kinase (PERK), which are activated by double stranded viral RNA intermediates and ER-stress, respectively14. PKR and PERK are thus triggered by a cell in despair trying to prevent viral RNA replication and to activate repair mechanisms that rely on an alternative translation initiation mechanism mediated by internal ribosomal entry sequences (IRESs). IRES translation is thus of particular physiological importance when cap-dependent translation is compromised15,16,17,18, but which is also used by some positive strand RNA viruses including HCV5,19,20 promoting viral protein synthesis21. It has been demonstrated that miR-122 stimulates HCV IRES translation20,22 and that RACK1 controls the IRES-mediated translation of viruses including HCV23 but additional host factors which are critical for HCV IRES activity remain largely to be determined. Since cellular signaling events regulate key aspects cap-dependent translation9, miRNA expression24 and the HCV life cycle2,25 we studied the role of host kinases and protein phosphatases in IRES-dependent translation.
Results
To analyze the impact of gene silencing on IRES- and cap-dependent translation, respectively, we co-transfected reporter mRNAs (100 ng/0.3 cm2) in gene silenced hepatoma cells 48 h post siRNA transfection as described previously26,27 (Fig. 1): Renilla luciferase mRNA initiated by a m7G cap structure and firefly luciferase mRNA containing a non-physiological adenosine cap structure (‘A-cap’) and the HCV IRES element. The A-cap maintains stability of the mRNA, but is not recognized by the cap binding complex. Luciferase expression was assessed by a Mithras LB 940 (Berthold Technologies) using Dual-Luciferase Reporter Assay or Bright-Glo (Promega). Toxicity of gene silencing was assessed using MTT (Sigma) and Presto Blue (Sigma) for the tertiary screen. In the primary screen targeting 893 genes we identified 46 candidates that predominant impact HCV IRES-dependent over cap-dependent translation (Supplementary table S1). In a secondary validation screen using side-by-side transfection of firefly reporter mRNAs of cap and HCV IRES (Fig. 1) we validated 11 hits of the primary screen (Supplementary table S2) and thus confirmed that these genes predominantly affect HCV IRES- rather than cap-dependent translation. As HCV IRES translation is a key step in the viral life cycle we assessed whether the identified genes confirm as positive regulators of HCV infection. We validated the results from the two foregoing screens (performed with siRNA pools) in a tertiary screen by at least two of four individual siRNAs per target (Fig. 1) to minimize off-target effects and validated mRNA knockdown specificity of the final hits by qPCR (Supplementary figure S1). As a result we confirmed that silencing of 3 genes from the secondary screening have a reproducible and significant impact on HCV infection: phosphatidylinositol 4-kinase catalytical subunit beta (PIK4CB), MAP kinase interacting serine/threonine kinase 1 (MKNK1), and tumor protein D52-like 3 (NYD-SP25) (Supplementary table S3). We were not able to validate a specific silencing of NYD-SP25 mRNA expression and therefore cannot rule out that off-target effects being responsible for the impact in IRES-dependent translation. Using a specific inhibitor of MKNK128 we demonstrate a significant (p < 0.01, t-test) and preferential inhibition of IRES-dependent translation over cap-dependent translation of luciferase reporter genes (Fig. 2a) at absent cell toxicity (Fig. 2b). Long-term treatment with the MKNK1 inhibitor over three days significantly (p < 0.01, t-test) block HCV infection (Fig. 2c) demonstrating that the molecular mechanism of action of MKNK1 involves its kinase activity.
Figure 1.

High-throughput RNAi screen identifying human kinases and protein phosphatases withpredominant impact on IRES-dependent translation and HCV infection.
Figure 2. Kinase activity of MKNK1 promotes IRES-dependent translation and HCV infection.
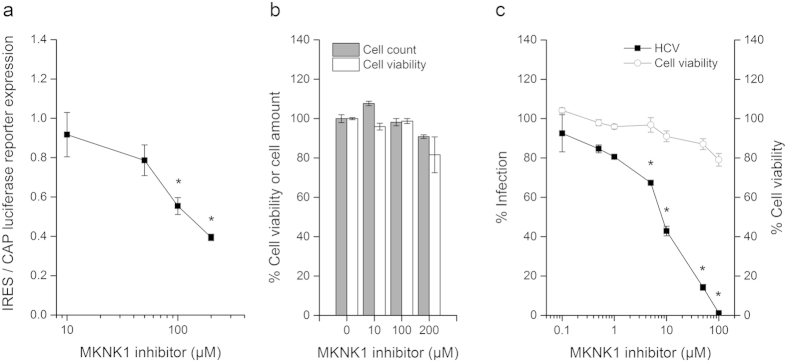
(a) A specific inhibitor of MKNK1 kinase activity preferentially impairs IRES-dependent over CAP-dependent luciferase reporter translation. Huh7.5 cells incubated for one hour with increasing doses of MKNK1 inhibitor. Treated cells were co-transfected with reporter mRNAs (IRES-firefly and cap-renilla) for four hours prior measuring of the firefly and renilla luciferase activity as described in the methods section. Data are expressed as means of the ratio of firefly/renilla luciferase activity +/− SEM. *p < 0.01 (Student’s t-test, n = 15 of three independent experiments). (b) MKNK1 inhibitor has only a minor impact on cell viability of Huh7.5 cells. After 5 h incubation with increasing concentrations of MKNK1 inhibitor the cell number was assessed by counting and the cell viability was assessed using Presto Blue. Data are expressed as means +/− SEM (n = 3 of one representative experiment). (c) MKNK1 inhibitor significantly and dose-dependently inhibits HCV infection at absent cell toxicity. Huh7.5.1 cells were pre-treated for one hour with increasing concentrations of MKNK1 inhibitor prior infection with cell culture-derived HCV (strain Luc-Jc1). Infected cells were maintained in the presence of the respective MKNK1 inhibitor concentration prior cell lysis and the measurement of the luciferase activity at day three. Data are expressed as means +/− SEM. *p < 0.01 (Student’s t-test, n = 3 of one representative experiment). All inhibitor dilutions and controls in this figure were prepared in a constant background of 1% DMSO.
Discussion
Understanding the IRES-mediated control of HCV protein synthesis is important for the understanding of HCV infection. Although much progress has been made in understanding HCV entry and replication translational control by the HCV IRES and the involved host factors remain largely unknown. For genes identified as HCV cofactors it was mostly not conclusive whether they affect HCV translation or -replication or both steps in the viral life cycle. An established unbiased multistep screening approach identified five genes modulating HCV IRES-dependent translation. Among the genes was MKNK1, a regulator of cap translation and recently described HCV entry factor29. MKNK1 regulates EIF4E affinity to cap mRNA in fibroblasts and is required for Herpes simplex virus (HSV-1) replication. For the first time we demonstrate that MKNK1 predominantly promotes IRES-dependent translation in hepatocytes (Supplementary table S1) that likely contributes to decreased core protein expression levels observed upon MKNK1 inhibition29. A specific MKNK1 inhibitor predominantly impairs IRES over cap-dependent translation demonstrating that the molecular mechanism of action of MKNK1 involves its kinase activity. Indeed, MKNK1 inhibitor specifically and dose-dependently impairs HCV infection highlighting its potential as antiviral target for IRES-dependent viruses. No phosphatase was identified suggesting that HCV IRES translation is mainly dependent on protein phosphorylation. Interestingly, the screen identified a phosphatidylinositol 4-kinase (PI4K), another key member of the phospholipid metabolism. PI4K generate membranes enriched in phosphatidylinositide 4-phosphate lipids, which serve as replication platforms for RNA viruses from the Picornaviridae and the Flaviviridae30. Strikingly, most members of these virus families replicate in such membranous platforms (poliovirus, coxsackievirus, Aichi virus, enterovirus, HCV)30,31 also rely on IRES-dependent translation. PI4K alpha and beta isoforms have been implicated in entry, replication and packaging of HCV1,2,3,4,32. Thus our data suggest an additional and previously unrecognized role of PI4K-beta for HCV IRES-mediated translation of the viral polyprotein and potentially also for other viruses that employ an IRES mechanism. Collectively, our data identify a novel pathway of HCV-host interactions with functional relevance for the HCV life cycle and IRES-mediated translation of viral RNA.
Methods
Transcription and transfection and RT-PCR
All transfections were performed at the High Throughput Screening platform of the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) in Illkirch, France. Gene silencing by RNAi for all screens was performed on 5000 cells/3.5 pmol siRNA/0.3 cm2 in 96 well plates by reverse transfection using Interferin (Polyplus Transfection) as described previously2,25. Non-targeting scrambled siRNA (Qiagen) and RISC-free siGlo RNA (Dharmacon) were transfected as mock controls. 100 ng/0.3 cm2 reporter mRNA was transfected 48 h post siRNA transfection using TransMessager reagent (Qiagen) as described previously26. In vitro transcription of mRNAs were described previously27. Reporter mRNAs were co-transfected in a ratio (IRES-firefly: cap-renilla) of 1:4 in the primary screen. In the secondary screen reporter mRNAs (IRES-firefly, cap-firefly) were transfected side-by-side. Luciferase expression was assessed by a Mithras LB 940 (Berthold Technologies) using Dual-Luciferase Reporter Assay System System or Bright-Glo (Promega). Toxicity was assessed using MTT assay (Sigma) and Presto Blue assay (Sigma) for the tertiary screen and the MKNK1 inhibitor experiments. RNA was extracted using RNeasy kit (Qiagen) and cDNA were generated using Maxima Reverse Transcriptase (Life Technologies). qPCR was performed using RT2 SYBR Green qPCR Mastermix (Qiagen) and a C1000 Touch Thermo Cycler (Bio-Rad).
Cells culture, virus, DNA, siRNA and small molecules
Cell growth conditions of hepatoma cell lines Huh7.5 and Huh7.5.1 were described33,34. Infection with HCVcc (strain LUC-Jc1) was described2. siRNAs for the primary and secondary screen comprised the Human Kinase RNAi Set V2.0 (pool of four siRNAs) targeting 691 kinases and associated proteins and siRNAs targeting 203 human phosphatases from the Human Druggable Genome siRNA Set Version 4.0 (pools of four siRNAs) from Qiagen. Individual siRNA (four individual siRNAs per target) for the tertiary screen were obtained from Qiagen. The plasmids encoding firefly luciferase pT3FireflyLuc(pA), the renilla luciferase control plasmid pT3RenillaLuc(pA) and pHCV-IRES-luc has been described previously27,35,36. qPCR primers were obtained from Qiagen (RT2 qPCR assays). MKNK1 inhibitor was obtained from Calbiochem (Merck Millipore) and solved in DMSO.
Screening hit selection
To evaluate the impact of gene silencing on IRES and cap-dependent translation, ratios of relative light units emitted by cells transfected with IRES firefly luciferase and capped renilla luciferase reporter mRNAs were formed and values normalized by the plate median of the relative light signals. Measurements were scored calculating z-scores using CellHTS2 software. Hits were assigned from the primary screen if z = <−1.29. Secondary screening results were normalized to % luciferase activity of control cells transfected with scrambled siRNAs. Hits were assigned if target silencing decreased IRES-dependent firefly luciferase activity and if the ratio of IRES- to cap-dependent firefly luciferase activities is <0.8. An impact of target-specific siRNA (four siRNA per target) on infection with HCVcc was identified in a tertiary screen using the method of strictly standardized mean differences (SSMD)37,38 using GUItars software39. A target gene with specific impact on HCVcc was considered a hit if at least two individual siRNAs impairs HCVcc infection with an SSMD < =−1.645 (“fairly strong” inhibition) and absent toxicity (cell viability higher than a threshold of 2x plate median of standard deviations). Tertiary screen hits were scored by products of SSMD values from individual siRNAs with SSMD < =−1.645.
Additional Information
How to cite this article: Lupberger, J. et al. PI4K-beta and MKNK1 are regulators of hepatitis C virus IRES-dependent translation. Sci. Rep. 5, 13344; doi: 10.1038/srep13344 (2015).
Supplementary Material
Acknowledgments
We thank Petra Binninger for excellent technical assistance. This work was supported by a grant from the Wilhelm Sander-Stiftung to C.T. and T.F.B. (Förderantrag 2010.023.1), the European Union (ERC-2008-AdG-233130-HEPCENT, INTERREG-IV-Rhin Supérieur-FEDER-Hepato-Regio-Net 2012, EU FP7 HEPAMAB; all T. F. B), ANRS 2013/108 to T.F.B, ARC TheraHCC (TheraHCC IHUARC IHU201301187 to T.F.B.), and Laboratoire d’excellence LabEx HEPSYS (Investissement d’Avenir; ANR-10-LAB-28). C.T. was a recipient of a Heisenberg-Fellowship from the Deutsche Forschungsgemeinschaft (TH788/2-1 and TH788/2-2). J.L. is a recipient of a grant from the French ANR and the University of Strasbourg (IDEX, Projet Attractivité 2014).
Footnotes
Author Contributions C.T. initiated and supervised the study. J.L., C.C., B.F., I.F., A.W., N.F., A.K., T.F., M.R. and L.B. designed and conducted experiments and analyzed data. J.L., T.F.B. and C.T. wrote the manuscript. C.S. reviewed and edited the manuscript.
References
- Li Q., Brass A. L. & Ng A. et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA. 106, 16410–16415 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J., Zeisel M. B. & Xiao F. et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17, 589–595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai A. W., Benita Y. & Peng L. F. et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5, 298–307 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotard M., Lepere-Douard C. & Regeard M. et al. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23, 3780–3789 (2009). [DOI] [PubMed] [Google Scholar]
- Hellen C. U. & Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15, 1593–1612 (2001). [DOI] [PubMed] [Google Scholar]
- Gallie D. R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 5, 2108–2116 (1991). [DOI] [PubMed] [Google Scholar]
- Preiss T. & Hentze M. W. Dual function of the messenger RNA cap structure in poly(A)-tail-promoted translation in yeast. Nature 392, 516–520 (1998). [DOI] [PubMed] [Google Scholar]
- Tarun S. Z. Jr. & Sachs A. B. A common function for mRNA 5′ and 3′ ends in translation initiation in yeast. Genes Dev. 9, 2997–3007 (1995). [DOI] [PubMed] [Google Scholar]
- Richter J. D. & Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480 (2005). [DOI] [PubMed] [Google Scholar]
- Roux P. P., Shahbazian D. & Vu H. et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 282, 14056–14064 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrogio A., Nagaoka K. & Richter J. D. Translational control of cell growth and malignancy by the CPEBs. Nat. Rev. Cancer 13, 283–290 (2013). [DOI] [PubMed] [Google Scholar]
- Waskiewicz A. J., Johnson J. C. & Penn B. et al. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell Biol. 19, 1871–1880 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta R., Laroia G. & Schneider R. J. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 14, 1460–1470 (2000). [PMC free article] [PubMed] [Google Scholar]
- Jackson R. J., Hellen C. U. & Pestova T. V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W. V., Zhou K., Butler T. K. & Doudna J. A. Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317, 1224–1227 (2007). [DOI] [PubMed] [Google Scholar]
- Kim J. H., Paek K. Y. & Choi K. et al. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol. Cell Biol. 23, 708–720 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoneley M., Chappell S. A. & Jopling C. L. et al. c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Mol. Cell Biol. 20, 1162–1169 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subkhankulova T., Mitchell S. A. & Willis A. E. Internal ribosome entry segment-mediated initiation of c-Myc protein synthesis following genotoxic stress. Biochem. J. 359, 183–192 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen C. U. IRES-induced conformational changes in the ribosome and the mechanism of translation initiation by internal ribosomal entry. Biochim. Biophys. Acta 1789, 558–570 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niepmann M. Activation of hepatitis C virus translation by a liver-specific microRNA. Cell Cycle 8, 1473–1477 (2009). [DOI] [PubMed] [Google Scholar]
- Schneider R. J. & Mohr I. Translation initiation and viral tricks. Trends Biochem. Sci. 28, 130–136 (2003). [DOI] [PubMed] [Google Scholar]
- Henke J. I., Goergen D. & Zheng J. et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 27, 3300–3310 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzoub K., Hafirassou M. L. & Meignin C. et al. RACK1 controls IRES-mediated translation of viruses. Cell 159, 1086–1095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov K. D., Boldin M. P., Chang K. J. & Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 103, 12481–12486 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona L., Lupberger J. & Sidahmed-Adrar N. et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 13, 302–313 (2013). [DOI] [PubMed] [Google Scholar]
- Casanova C. M., Sehr P. & Putzker K. et al. Automated high-throughput RNAi screening in human cells combined with reporter mRNA transfection to identify novel regulators of translation. PLoS One 7, e45943 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma C., Bergamini G., Galy B., Hundsdoerfer P. & Hentze M. W. Enhancement of IRES-mediated translation of the c-myc and BiP mRNAs by the poly(A) tail is independent of intact eIF4G and PABP. Mol. Cell 15, 925–935 (2004). [DOI] [PubMed] [Google Scholar]
- Worch J., Tickenbrock L. & Schwable J. et al. The serine-threonine kinase MNK1 is post-translationally stabilized by PML-RARalpha and regulates differentiation of hematopoietic cells. Oncogene 23, 9162–9172 (2004). [DOI] [PubMed] [Google Scholar]
- Kim S., Ishida H. & Yamane D. et al. Contrasting roles of mitogen-activated protein kinases in cellular entry and replication of hepatitis C virus: MKNK1 facilitates cell entry. J. Virol. 87, 4214–4224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N. & Balla T. Phosphatidylinositol 4-kinases: hostages harnessed to build panviral replication platforms. Trends Biochem. Sci. 37, 293–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A. L. & Jackson W. T. Behind closed membranes: the secret lives of picornaviruses? PLoS Pathog. 9, e1003262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S., Harak C. & Romero-Brey I. et al. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 9, e1003359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight K. J., McKeating J. A. & Rice C. M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76, 13001–13014 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J., Gastaminza P. & Cheng G. et al. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102, 9294–9299 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka N., Najita L., Franzusoff A. & Sarnow P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol. Cell Biol. 14, 7322–7330 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R. & Hentze M. W. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature 447, 875–878 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang X. D., Ferrer M. & Espeseth A. S. et al. The use of strictly standardized mean difference for hit selection in primary RNA interference high-throughput screening experiments. J. Biomol. Screen. 12, 497–509 (2007). [DOI] [PubMed] [Google Scholar]
- Zhou H., Xu M. & Huang Q. et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 (2008). [DOI] [PubMed] [Google Scholar]
- Goktug A. N., Ong S. S. & Chen T. GUItars: a GUI tool for analysis of high-throughput RNA interference screening data. PLoS One 7, e49386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.