

The title compound, C13H10N2O7S, was solved in the orthorhombic space group Pna21. The aromatic substituents on the sulfonate group are oriented gauche to one another with a C—O—S—C torsion angle of −62.0 (3)°. The supramolecular features that contribute to the crystal lattice are offset π-π and multiple C—H⋯O interactions.
Keywords: crystal structure, aryl sulfonate, π–π interactions, C—H⋯O interactions, p-toluenesulfonyl chloride
Abstract
The title compound, C13H10N2O7S, was synthesized via a nucleophilic substitution reaction between 2,4-dinitrophenol and p-toluenesulfonyl chloride. This crystal structure is a polymorph of CSD entry WUVYUH [Vembu et al. (2003). Acta Cryst, E59, o378–380]. The aromatic substituents on the sulfonate group are oriented gauche to one another with a C—O—S—C torsion angle of −62.0 (3)°. The supramolecular features that contribute to the crystal stability are offset π–π [centroid–centroid distance = 3.729 (2) Å] and multiple C—H⋯O interactions.
Chemical context
Nucleophilic substitution reactions at the carbonyl carbon atom are an important class of reactions in biological processes. Analogous to the carbonyl group, nucleophilic substitution reactions of sulfonyl derivatives have also been reported (Castro et al., 2003 ▸; Terrier et al., 2003 ▸; Um et al., 2004 ▸, 2013 ▸; Qrareya et al., 2014 ▸). The mechanism of nucleophilic substitution reactions at the carbonyl group is well understood (Stefanidis et al., 1993 ▸; Lee et al., 2002 ▸). However, the mechanism for nucleophilic substitution reactions at the sulfonyl group is not fully understood (Morales-Rojas & Moss, 2002 ▸; Um et al., 2013 ▸).
A review of the current literature lends credence to both a concerted mechanism and a non-concerted mechanism (Guthrie, 1991 ▸; Colthurst & Williams, 1997 ▸; Spillane et al., 2001 ▸; Um et al., 2003 ▸, 2004 ▸, 2013 ▸). Using primary and secondary amines as nucleophiles, the factors influencing regioselectivity of nucleophilic substitution reactions at the sulfonyl group have been reported (Um et al., 2004 ▸). It has been demonstrated that the regioselectivity and S—O bond-fission mechanism depends on the basicity of the amine and the electronic nature of the sulfonyl substituent. Based on the current state of knowledge in the field, we have sought to capitalize on the chemistry learned on the mechanistic insight of S—O vs C—O bond fission by investigating the effect of different substituents on the reactivity of sulfonates. In our work, we are interested in using various sulfonate analogues (Fig. 1 ▸) as electrophilic substrates in nucleophilic aromatic substitution (SNAr) reactions similar to those reported by others (Qrareya et al., 2014 ▸). As the title compound is of interest in our ongoing effort on probing the mechanism of SNAr reactions with sulfonate derivatives, we report here on the synthesis and crystal structure of a new polymorph of 2,4-dinitrophenyl 4-methylbenzenesulfonate (Fig. 2 ▸).
Figure 1.

General structure of sulfonate analogues. R represents electron-donating and electron-withdrawing substituents.
Figure 2.

(a) The asymmetric unit of the title compound along with the atom-numbering scheme, showing displacement ellipsoids at the 50% probability level; (b) the structure and atom-numbering scheme of a polymorph of the title compound WUVYUH (Vembu, et al., 2003a ▸). All hydrogen atoms have been omitted for clarity.
Structural commentary
The central sulfur atom (S1) is tetrahedral with S=O bond lengths of 1.415 (3) and 1.414 (3) Å, and an S—O bond length of 1.634 (3) Å (Fig. 2 ▸ a). The bond angle between the S=O groups (O1—S1—O2) is 121.20 (17)°, while that of the aromatic substituents (O3—S1—C5) is 103.27 (15)°. The two aromatic rings are in a gauche orientation about the O3—S1 bond with a torsion angle (C8—O3—S1—C5) of −62.0 (3)°.
For comparison, the polymorph WUVYUH (Vembu et al., 2003a ▸) has S=O bond lengths of 1.4204 (10) and 1.4246 (10) Å, and the S—O bond length is 1.6195 (9) Å (Fig. 2 ▸ b). While the bond lengths of the two polymorphs agree within 0.01 Å of each other, there are some differences between bond angles. The aromatic rings in WUVYUH are in an anti orientation along the S—O bond, with a torsion angle of 141.02 (9)°. The bond angle between the S=O groups (O1—S1—O2) is 119.80 (6)°, while that of the aromatic substituents (O3—S1—C5) is 98.17 (5)°.
Supramolecular features
There are no classical hydrogen-bonding interactions in this crystal. There are, however, several intermolecular C—H⋯O interactions with D⋯A distances less than 3.5 Å and D—H⋯A angles greater than 120° (Table 1 ▸, Fig. 3 ▸). An offset π–π stacking interaction is present between the relatively electron-poor ring C8–C13 and the relatively electron-rich ring C2–C7v (Fig. 4 ▸). The centroid–centroid distance is 3.729 (2) Å, the C2–C7 ring is offset by 1.529 (5) Å and tilted 5.74 (12)° out of the plane defined by the C8–C13 ring [symmetry code: (v)  − x, −
− x, − + y, − + z].
+ y, − + z].
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| C3H3O2i | 0.95 | 2.42 | 3.273(5) | 149 |
| C4H4O6ii | 0.95 | 2.57 | 3.486(5) | 162 |
| C7H7O7iii | 0.95 | 2.75 | 3.499(5) | 137 |
| C10H10O7iv | 0.95 | 2.51 | 3.233(5) | 133 |
| C12H12O4v | 0.95 | 2.34 | 3.087(5) | 135 |
Symmetry codes: (i)  ; (ii)
; (ii) 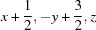 ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  .
.
Figure 3.

A drawing of a selection of the C—H⋯O interactions present in the crystal lattice using a ball and stick model. Symmetry codes: (i) −x + 1, −y + 1, z + ; (ii) −x + , y + , z − ; (iii) −x + 2, −y + 2, z − .
Figure 4.

A drawing of the intermolecular π–π stacking and nitro–sulfonic ester interactions present in the crystal using a ball and stick model. Symmetry code (v) −x + , y − , z + .
One nitro group (N2,O6,O7) is in proximity to the sulfonic ester of the C2–C7v ring of a nearby π–π dimer. Fig. 4 ▸ also shows that the atoms of these two functional groups are oriented to align the electron-poor N2(nitro) with the electron-rich O1(sulfonic ester), and the electron-poor S1(sulfonic ester) with the electron-rich O7(nitro). Interatomic distances are N2(nitro)⋯O17v(sulfonic ester) = 3.379 (4) Å, and O7(nitro)⋯S1v(sulfonic ester) = 3.877 (3) Å. The relatively short intermolecular distance between N2 and O17 suggests the presence of favorable N⋯O interactions in the crystal (Daszkiewicz, 2013 ▸).
Database survey
The Cambridge Structural Database (CSD, version 5.36, May 2015: Groom and Allen, 2014 ▸) contains 171 aromatic 4-toluenesulfonic esters. Of these, there are 14 structures where the aromatic ring bears substituents at both the 2- and 4-positions. One of these structures is quite similar to the title compound (GOFTIF: Ji et al., 2008 ▸) where the ortho-substituent is a nitro group and the para-position bears a second tosylate group. The remaining entries have various electron-rich groups in the ortho-position including methoxy (e.g. FEMROF: Ichikawa et al., 2004 ▸), ethoxy (e.g. HIRHOG: Ramachandran et al., 2007 ▸), chlorine (OJENEW: Vembu et al., 2003b ▸) and alkyl amine (PERFEZ: Zhao et al., 2013 ▸).
The CSD contains three additional structures where the position ortho to the sulfonic ester bears a nitro group. In FAYBAJ (Manivannan et al., 2005 ▸), this o-nitro group is the only substituent. The aromatic ring in XIYZIP is part of a naphthalene system and also bears a 4-nitro group (Ramachandran et al., 2008 ▸). The third structure in this set is a polymorph of the title compound (WUVYUH: Vembu et al., 2003a ▸) that was solved in the orthorhombic space group Pbca. One significant difference between WUVYUH and the title compound is the orientation of the groups around the S—O bond (see the Structural commentary section for more details).
Synthesis and crystallization
The title compound was prepared by stirring 2,4-dinitrophenol (5 mmol), p-toluenesulfonyl chloride (5 mmol) and pyridine (3 mmol) in 10 mL of dichloromethane for 30 minutes at room temperature. The reaction was heated to 353 K for 30 minutes in a microwave reactor, then cooled to room temperature and stirred overnight in a fume hood. The reaction mixture was transferred to a scintillation vial where the pale yellow product crystallized upon standing after several days and was filtered from the mother liquor (m.p. 393.4–394.7 K).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. Hydrogen atoms were placed in calculated positions and constrained to ride on their parent atoms, with U iso(H) = 1.2U eq(C) for CH groups and U iso(H) = 1.5 U eq(C) for methyl groups.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C13H10N2O7S |
| M r | 338.29 |
| Crystal system, space group | Orthorhombic, P n a21 |
| Temperature (K) | 173 |
| a, b, c () | 14.7716(12), 12.6403(11), 7.6734(6) |
| V (3) | 1432.8(2) |
| Z | 4 |
| Radiation type | Mo K |
| (mm1) | 0.27 |
| Crystal size (mm) | 0.27 0.21 0.20 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2014 ▸) |
| T min, T max | 0.686, 0.745 |
| No. of measured, independent and observed [I > 2(I)] reflections | 11432, 2624, 2326 |
| R int | 0.036 |
| (sin /)max (1) | 0.603 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.034, 0.088, 1.05 |
| No. of reflections | 2624 |
| No. of parameters | 209 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| max, min (e 3) | 0.18, 0.18 |
| Absolute structure | Flack x determined using 970 quotients [(I +)(I )]/[(I +)+(I )] (Parsons et al., 2013 ▸). |
| Absolute structure parameter | 0.02(4) |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015015650/pk2562sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015015650/pk2562Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015015650/pk2562Isup3.cml
CCDC reference: 1419864
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank the GVSU for financial support (Weldon Fund, CSCE), the NSF for a 300 MHz Jeol FT–NMR (CCLI-0087655) and Pfizer, Inc. for the donation of a Varian Inova 400 FT–NMR. The CCD-based X-ray diffractometers at Michigan State University were upgraded and/or replaced by departmental funds.
supplementary crystallographic information
Crystal data
| C13H10N2O7S | Dx = 1.568 Mg m−3 |
| Mr = 338.29 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pna21 | Cell parameters from 5781 reflections |
| a = 14.7716 (12) Å | θ = 2.8–25.4° |
| b = 12.6403 (11) Å | µ = 0.27 mm−1 |
| c = 7.6734 (6) Å | T = 173 K |
| V = 1432.8 (2) Å3 | Chunk, yellow |
| Z = 4 | 0.27 × 0.21 × 0.20 mm |
| F(000) = 696 |
Data collection
| Bruker APEXII CCD diffractometer | 2624 independent reflections |
| Radiation source: sealed tube | 2326 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.036 |
| Detector resolution: 8 pixels mm-1 | θmax = 25.4°, θmin = 2.1° |
| φ and ω scans | h = −17→17 |
| Absorption correction: multi-scan (SADABS; Bruker, 2014) | k = −15→15 |
| Tmin = 0.686, Tmax = 0.745 | l = −9→9 |
| 11432 measured reflections |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.034 | w = 1/[σ2(Fo2) + (0.0448P)2 + 0.2683P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.088 | (Δ/σ)max < 0.001 |
| S = 1.05 | Δρmax = 0.18 e Å−3 |
| 2624 reflections | Δρmin = −0.18 e Å−3 |
| 209 parameters | Absolute structure: Flack x determined using 970 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013). |
| 1 restraint | Absolute structure parameter: −0.02 (4) |
Special details
| Experimental. SADABS (Bruker, 2014) was used for absorption correction. wR2(int) was 0.0928 before and 0.0535 after correction. The Ratio of minimum to maximum transmission is 0.9202. The λ/2 correction factor is 0.00150. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.84591 (5) | 0.72917 (7) | 0.62435 (14) | 0.0377 (2) | |
| O1 | 0.92559 (18) | 0.7834 (2) | 0.5749 (4) | 0.0520 (8) | |
| O2 | 0.84576 (18) | 0.6573 (2) | 0.7657 (4) | 0.0480 (8) | |
| O3 | 0.82059 (18) | 0.6622 (2) | 0.4489 (3) | 0.0393 (6) | |
| O4 | 0.7331 (2) | 0.7555 (3) | 0.1741 (4) | 0.0698 (10) | |
| O5 | 0.6041 (2) | 0.8003 (2) | 0.2866 (5) | 0.0622 (9) | |
| O6 | 0.43774 (19) | 0.4573 (2) | 0.3474 (4) | 0.0569 (8) | |
| O7 | 0.5026 (2) | 0.3344 (2) | 0.4987 (4) | 0.0552 (8) | |
| N1 | 0.6675 (2) | 0.7407 (3) | 0.2684 (5) | 0.0447 (8) | |
| N2 | 0.5033 (2) | 0.4212 (3) | 0.4265 (4) | 0.0409 (7) | |
| C1 | 0.5319 (3) | 1.0307 (3) | 0.6779 (6) | 0.0526 (11) | |
| H1A | 0.4769 | 0.9950 | 0.6376 | 0.079* | |
| H1B | 0.5436 | 1.0927 | 0.6047 | 0.079* | |
| H1C | 0.5240 | 1.0531 | 0.7991 | 0.079* | |
| C2 | 0.6109 (2) | 0.9555 (3) | 0.6659 (5) | 0.0383 (9) | |
| C3 | 0.6881 (3) | 0.9821 (3) | 0.5730 (5) | 0.0391 (9) | |
| H3 | 0.6911 | 1.0490 | 0.5167 | 0.047* | |
| C4 | 0.7608 (2) | 0.9138 (3) | 0.5602 (4) | 0.0360 (8) | |
| H4 | 0.8130 | 0.9328 | 0.4950 | 0.043* | |
| C5 | 0.7559 (2) | 0.8168 (2) | 0.6448 (4) | 0.0299 (7) | |
| C6 | 0.6796 (2) | 0.7879 (3) | 0.7391 (4) | 0.0345 (8) | |
| H6 | 0.6770 | 0.7214 | 0.7966 | 0.041* | |
| C7 | 0.6077 (2) | 0.8575 (3) | 0.7480 (5) | 0.0373 (8) | |
| H7 | 0.5551 | 0.8380 | 0.8114 | 0.045* | |
| C8 | 0.7418 (2) | 0.6020 (3) | 0.4478 (5) | 0.0323 (8) | |
| C9 | 0.6651 (2) | 0.6401 (3) | 0.3637 (4) | 0.0307 (7) | |
| C10 | 0.5859 (2) | 0.5833 (3) | 0.3596 (5) | 0.0337 (8) | |
| H10 | 0.5329 | 0.6110 | 0.3061 | 0.040* | |
| C11 | 0.5861 (2) | 0.4846 (3) | 0.4361 (5) | 0.0323 (8) | |
| C12 | 0.6613 (2) | 0.4440 (3) | 0.5188 (5) | 0.0363 (8) | |
| H12 | 0.6593 | 0.3754 | 0.5693 | 0.044* | |
| C13 | 0.7395 (2) | 0.5035 (3) | 0.5279 (5) | 0.0362 (8) | |
| H13 | 0.7911 | 0.4774 | 0.5880 | 0.043* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0284 (4) | 0.0403 (4) | 0.0443 (5) | 0.0001 (4) | −0.0037 (5) | 0.0048 (5) |
| O1 | 0.0275 (12) | 0.0566 (16) | 0.072 (2) | −0.0066 (12) | 0.0026 (13) | 0.0053 (15) |
| O2 | 0.0478 (17) | 0.0453 (15) | 0.0507 (17) | 0.0058 (12) | −0.0094 (13) | 0.0130 (13) |
| O3 | 0.0330 (13) | 0.0407 (14) | 0.0444 (15) | 0.0004 (12) | 0.0077 (11) | −0.0014 (12) |
| O4 | 0.077 (2) | 0.069 (2) | 0.063 (2) | −0.0155 (17) | 0.0088 (18) | 0.0306 (17) |
| O5 | 0.073 (2) | 0.0364 (14) | 0.077 (2) | 0.0159 (15) | −0.0244 (17) | 0.0065 (15) |
| O6 | 0.0387 (16) | 0.0549 (17) | 0.077 (2) | −0.0012 (14) | −0.0106 (16) | −0.0068 (17) |
| O7 | 0.070 (2) | 0.0441 (15) | 0.0517 (17) | −0.0187 (14) | 0.0021 (15) | 0.0053 (14) |
| N1 | 0.058 (2) | 0.0326 (16) | 0.044 (2) | −0.0040 (16) | −0.0153 (17) | 0.0052 (14) |
| N2 | 0.044 (2) | 0.0391 (16) | 0.0391 (18) | −0.0051 (15) | 0.0050 (16) | −0.0065 (15) |
| C1 | 0.051 (2) | 0.047 (2) | 0.059 (3) | 0.0125 (18) | −0.009 (2) | −0.011 (2) |
| C2 | 0.0358 (19) | 0.0371 (18) | 0.042 (2) | 0.0007 (16) | −0.0077 (16) | −0.0076 (17) |
| C3 | 0.050 (2) | 0.0285 (17) | 0.039 (2) | −0.0011 (16) | −0.0090 (17) | 0.0033 (15) |
| C4 | 0.0358 (19) | 0.0388 (19) | 0.0335 (19) | −0.0086 (16) | 0.0015 (15) | 0.0028 (15) |
| C5 | 0.0268 (16) | 0.0322 (15) | 0.0307 (18) | −0.0046 (13) | −0.0045 (15) | 0.0029 (17) |
| C6 | 0.037 (2) | 0.0323 (18) | 0.0342 (19) | −0.0063 (15) | −0.0001 (16) | 0.0020 (16) |
| C7 | 0.0304 (18) | 0.042 (2) | 0.039 (2) | −0.0069 (16) | 0.0000 (16) | −0.0065 (17) |
| C8 | 0.0294 (18) | 0.0328 (18) | 0.0346 (17) | 0.0035 (14) | 0.0080 (15) | 0.0015 (16) |
| C9 | 0.0383 (19) | 0.0254 (16) | 0.0283 (16) | 0.0049 (15) | 0.0017 (15) | 0.0020 (14) |
| C10 | 0.036 (2) | 0.0325 (17) | 0.0324 (18) | 0.0090 (15) | −0.0008 (16) | −0.0015 (15) |
| C11 | 0.0328 (19) | 0.0326 (17) | 0.0315 (18) | −0.0003 (15) | 0.0062 (15) | −0.0024 (15) |
| C12 | 0.044 (2) | 0.0280 (17) | 0.0366 (19) | 0.0059 (15) | 0.0014 (17) | 0.0056 (15) |
| C13 | 0.0362 (19) | 0.0326 (17) | 0.0397 (19) | 0.0112 (15) | 0.0006 (16) | 0.0040 (16) |
Geometric parameters (Å, º)
| S1—O1 | 1.414 (3) | C3—H3 | 0.9500 |
| S1—O2 | 1.415 (3) | C3—C4 | 1.381 (5) |
| S1—O3 | 1.634 (3) | C4—H4 | 0.9500 |
| S1—C5 | 1.737 (3) | C4—C5 | 1.390 (4) |
| O3—C8 | 1.391 (4) | C5—C6 | 1.388 (5) |
| O4—N1 | 1.224 (4) | C6—H6 | 0.9500 |
| O5—N1 | 1.210 (4) | C6—C7 | 1.381 (5) |
| O6—N2 | 1.231 (4) | C7—H7 | 0.9500 |
| O7—N2 | 1.228 (4) | C8—C9 | 1.389 (5) |
| N1—C9 | 1.468 (5) | C8—C13 | 1.389 (5) |
| N2—C11 | 1.465 (4) | C9—C10 | 1.374 (5) |
| C1—H1A | 0.9800 | C10—H10 | 0.9500 |
| C1—H1B | 0.9800 | C10—C11 | 1.378 (5) |
| C1—H1C | 0.9800 | C11—C12 | 1.379 (5) |
| C1—C2 | 1.507 (5) | C12—H12 | 0.9500 |
| C2—C3 | 1.387 (5) | C12—C13 | 1.379 (5) |
| C2—C7 | 1.391 (5) | C13—H13 | 0.9500 |
| O1—S1—O2 | 121.20 (17) | C4—C5—S1 | 118.8 (3) |
| O1—S1—O3 | 102.73 (16) | C6—C5—S1 | 120.0 (2) |
| O1—S1—C5 | 110.64 (16) | C6—C5—C4 | 121.2 (3) |
| O2—S1—O3 | 107.40 (14) | C5—C6—H6 | 120.6 |
| O2—S1—C5 | 109.80 (17) | C7—C6—C5 | 118.8 (3) |
| O3—S1—C5 | 103.27 (15) | C7—C6—H6 | 120.6 |
| C8—O3—S1 | 118.7 (2) | C2—C7—H7 | 119.4 |
| O4—N1—C9 | 116.5 (3) | C6—C7—C2 | 121.3 (3) |
| O5—N1—O4 | 125.9 (3) | C6—C7—H7 | 119.4 |
| O5—N1—C9 | 117.6 (4) | C9—C8—O3 | 119.7 (3) |
| O6—N2—C11 | 118.6 (3) | C13—C8—O3 | 120.6 (3) |
| O7—N2—O6 | 123.2 (3) | C13—C8—C9 | 119.8 (3) |
| O7—N2—C11 | 118.3 (3) | C8—C9—N1 | 120.8 (3) |
| H1A—C1—H1B | 109.5 | C10—C9—N1 | 117.5 (3) |
| H1A—C1—H1C | 109.5 | C10—C9—C8 | 121.6 (3) |
| H1B—C1—H1C | 109.5 | C9—C10—H10 | 121.3 |
| C2—C1—H1A | 109.5 | C9—C10—C11 | 117.5 (3) |
| C2—C1—H1B | 109.5 | C11—C10—H10 | 121.3 |
| C2—C1—H1C | 109.5 | C10—C11—N2 | 118.2 (3) |
| C3—C2—C1 | 121.0 (3) | C10—C11—C12 | 122.3 (3) |
| C3—C2—C7 | 118.5 (3) | C12—C11—N2 | 119.5 (3) |
| C7—C2—C1 | 120.5 (3) | C11—C12—H12 | 120.2 |
| C2—C3—H3 | 119.2 | C11—C12—C13 | 119.7 (3) |
| C4—C3—C2 | 121.6 (3) | C13—C12—H12 | 120.2 |
| C4—C3—H3 | 119.2 | C8—C13—H13 | 120.5 |
| C3—C4—H4 | 120.7 | C12—C13—C8 | 119.1 (3) |
| C3—C4—C5 | 118.6 (3) | C12—C13—H13 | 120.5 |
| C5—C4—H4 | 120.7 | ||
| S1—O3—C8—C9 | 101.6 (3) | O7—N2—C11—C12 | 2.6 (5) |
| S1—O3—C8—C13 | −79.5 (4) | N1—C9—C10—C11 | 174.5 (3) |
| S1—C5—C6—C7 | −177.7 (3) | N2—C11—C12—C13 | 179.9 (3) |
| O1—S1—O3—C8 | −177.1 (2) | C1—C2—C3—C4 | −179.7 (3) |
| O1—S1—C5—C4 | 21.1 (3) | C1—C2—C7—C6 | −179.7 (4) |
| O1—S1—C5—C6 | −161.2 (3) | C2—C3—C4—C5 | −0.7 (5) |
| O2—S1—O3—C8 | 54.0 (3) | C3—C2—C7—C6 | 0.5 (5) |
| O2—S1—C5—C4 | 157.5 (3) | C3—C4—C5—S1 | 178.3 (3) |
| O2—S1—C5—C6 | −24.8 (3) | C3—C4—C5—C6 | 0.6 (5) |
| O3—S1—C5—C4 | −88.2 (3) | C4—C5—C6—C7 | 0.0 (5) |
| O3—S1—C5—C6 | 89.5 (3) | C5—S1—O3—C8 | −62.0 (3) |
| O3—C8—C9—N1 | 3.0 (5) | C5—C6—C7—C2 | −0.6 (5) |
| O3—C8—C9—C10 | −179.9 (3) | C7—C2—C3—C4 | 0.2 (5) |
| O3—C8—C13—C12 | −177.6 (3) | C8—C9—C10—C11 | −2.7 (5) |
| O4—N1—C9—C8 | 44.8 (5) | C9—C8—C13—C12 | 1.4 (5) |
| O4—N1—C9—C10 | −132.4 (4) | C9—C10—C11—N2 | −177.5 (3) |
| O5—N1—C9—C8 | −136.9 (4) | C9—C10—C11—C12 | 1.9 (5) |
| O5—N1—C9—C10 | 46.0 (5) | C10—C11—C12—C13 | 0.5 (5) |
| O6—N2—C11—C10 | 1.6 (5) | C11—C12—C13—C8 | −2.2 (5) |
| O6—N2—C11—C12 | −177.8 (3) | C13—C8—C9—N1 | −176.0 (3) |
| O7—N2—C11—C10 | −178.0 (3) | C13—C8—C9—C10 | 1.1 (5) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O2i | 0.95 | 2.42 | 3.273 (5) | 149 |
| C4—H4···O6ii | 0.95 | 2.57 | 3.486 (5) | 162 |
| C7—H7···O7iii | 0.95 | 2.75 | 3.499 (5) | 137 |
| C10—H10···O7iv | 0.95 | 2.51 | 3.233 (5) | 133 |
| C12—H12···O4v | 0.95 | 2.34 | 3.087 (5) | 135 |
Symmetry codes: (i) −x+3/2, y+1/2, z−1/2; (ii) x+1/2, −y+3/2, z; (iii) −x+1, −y+1, z+1/2; (iv) −x+1, −y+1, z−1/2; (v) −x+3/2, y−1/2, z+1/2.
References
- Bourhis, L. J., Dolomanov, O. V., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2015). Acta Cryst. A71, 59–75. [DOI] [PMC free article] [PubMed]
- Bruker (2013). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2014). SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Castro, E. A., Andújar, M., Toro, A. & Santos, J. G. (2003). J. Org. Chem. 68, 3608–3613. [DOI] [PubMed]
- Colthurst, M. J. & Williams, A. J. (1997). J. Chem. Soc. Perkin Trans. 2, pp. 1493–1498.
- Daszkiewicz, M. (2013). CrystEngComm, 15, 10427–10430.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Guthrie, J. P. (1991). J. Am. Chem. Soc. 113, 3941–3949.
- Ichikawa, M., Takahashi, M., Aoyagi, S. & Kibayashi, C. (2004). J. Am. Chem. Soc. 126, 16553–16558. [DOI] [PubMed]
- Ji, X., Cheng, B., Song, J. & Liu, C. (2008). Acta Cryst. E64, o1816. [DOI] [PMC free article] [PubMed]
- Lee, H. W., Guha, A. K., Kim, C. K. & Lee, I. (2002). J. Org. Chem. 67, 2215–2222. [DOI] [PubMed]
- Manivannan, V., Vembu, N., Nallu, M., Sivakumar, K. & Fronczek, F. R. (2005). Acta Cryst. E61, o118–o120.
- Morales-Rojas, J. & Moss, R. A. (2002). Chem. Rev. 102, 2497–2522. [DOI] [PubMed]
- Palmer, D. (2007). CrystalMaker. CrystalMaker Software, Bicester, England.
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Qrareya, H., Protti, S. & Fagnoni, M. (2014). J. Org. Chem. 79, 11527–11533. [DOI] [PubMed]
- Ramachandran, G., Kanakam, C. C. & Manivannan, V. (2008). Acta Cryst. E64, o873. [DOI] [PMC free article] [PubMed]
- Ramachandran, G., Kanakam, C. C., Manivannan, V., Thiruvenkatam, V. & Row, T. N. G. (2007). Acta Cryst. E63, o4638.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spillane, W. J., McGrath, P., Brack, C. & O’Byrne, A. B. (2001). J. Org. Chem. 66, 6313–6316. [DOI] [PubMed]
- Stefanidis, D., Cho, S., Dhe-Paganon, S. & Jencks, W. P. (1993). J. Am. Chem. Soc. 115, 1650–1656.
- Terrier, F., Le Guével, E., Chatrousse, A. P., Moutiers, G. & Buncel, E. (2003). Chem. Commun. pp. 600–601. [DOI] [PubMed]
- Um, I. H., Chun, S. M., Chae, O. M., Fujio, M. & Tsuno, Y. (2004). J. Am. Chem. Soc. 69, 3166–3172. [DOI] [PubMed]
- Um, I. H., Hong, J. Y., Kim, J. J., Chae, O. M. & Bae, S. K. (2003). J. Am. Chem. Soc 68, 5180–5185. [DOI] [PubMed]
- Um, I. H., Kang, J. S., Shin, Y. H. & Buncel, E. (2013). J. Org. Chem. 78, 490–497. [DOI] [PubMed]
- Vembu, N., Nallu, M., Garrison, J. & Youngs, W. J. (2003a). Acta Cryst. E59, o378–o380.
- Vembu, N., Nallu, M., Garrison, J. & Youngs, W. J. (2003b). Acta Cryst. E59, o939–o941.
- Zhao, J., Liu, X., Luo, W., Xie, M., Lin, L. & Feng, X. (2013). Angew. Chem. Int. Ed. 52, 3473–3477. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015015650/pk2562sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015015650/pk2562Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015015650/pk2562Isup3.cml
CCDC reference: 1419864
Additional supporting information: crystallographic information; 3D view; checkCIF report