

The title compound, [Cd(C5H2N5)2(C3H7NO)2]n, is a two-dimensional coordination polymer extending parallel to (100). Notably, both the primary amino group and the cyano groups are involved in hydrogen-bonding interactions with DMF ligands to direct the assembly and stabilize the crystal packing.
Keywords: crystal structure; 2-amino-4,5-dicyanoimidazole; metal–organic framework; cadmium coordination polymer; hydrogen bonding
Abstract
In the title structure, [Cd(C5H2N5)2(C3H7NO)2]n or [Cd(adci)2(DMF)2]n, the Cd2+ ion is located on a twofold rotation axis and is six-coordinated in a CdN4O2 manner by four imidazole N atoms of four symmetry-related 2-amino-4,5-dicyanoimidazolate (adci) anions in the equatorial plane and by two O atoms of symmetry-related N,N-dimethylformamide (DMF) ligands in axial positions. The adci− anions bridge adjacent Cd2+ ions [shortest Cd⋯Cd separation = 6.733 (3) Å] into a layered coordination polymer extending parallel to (001). The primary amino group and the non-coordinating cyano groups of adci− anions are involved in hydrogen-bonding interactions with DMF ligands to stabilize the crystal structure.
Chemical context
Porous materials such as metal-organic frameworks (MOFs) combining advantages of both organic and inorganic components have emerged as a unique class of crystalline solid-state materials today due to their potential applications in gas adsorption and separation (Collins & Zhou, 2007 ▸), catalysis (Gu et al., 2012 ▸) and analytical chemistry (Mondal et al., 2013 ▸). As a branch of MOFs, zeolitic imidazolate frameworks (ZIFs), which are topologically related to inorganic zeolites, commonly reveal high thermal and chemical stability (Eddaoudi et al., 2015 ▸). Bridging N-donor ligands such as 2-substituted 4,5-dicyanoimidazole (dci) molecules are often used to synthesize ZIFs (Sava et al., 2009 ▸; Mondal et al., 2014 ▸). In addition, the cyano group of dci can generate carboxylate- (Orcajo et al., 2014 ▸) or tetrazole-based (Xiong et al., 2002 ▸) ligands by in-situ ligand reactions.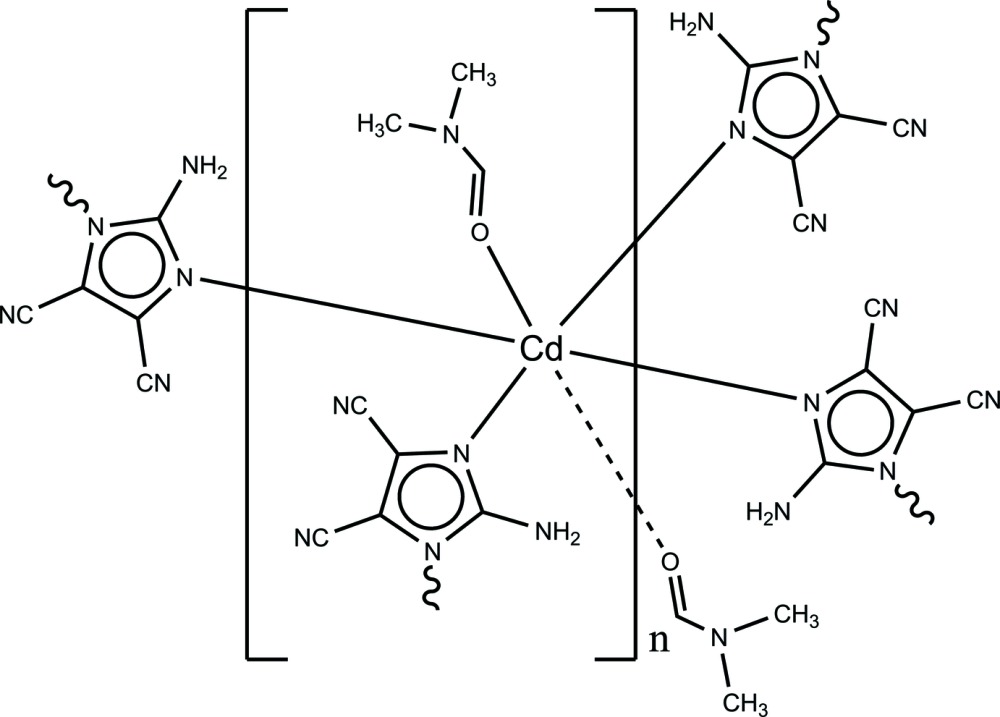
We chose a rigid planar ligand, viz. 2-amino-4,5-dicyanoimidazole (adci), and Cd2+ that exhibits strong coordination capabilities for imidazolates, to prepare new metal-organic polymers and report here the structure of the title compound, [Cd(C5H2N5)2(C3H7NO)2]n, or [Cd(adci)2(DMF)2]n (DMF is dimethylformamide), (I).
Structural commentary
Complex (I) is a mononuclear cadmium coordination polymer, in which the central Cd2+ ion exhibits a tetragonally distorted octahedral coordination environment (Fig. 1 ▸). The asymmetric unit of (I) comprises one Cd2+ ion located on a twofold rotation axis, one 2-amino-4,5-dicyanoimidazolate ion and one DMF ligand, both in general positions. The Cd2+ ion has an N4O2 coordination set defined by four N atoms of four symmetry-related adci− anions in the equatorial plane and by two oxygen atoms of two symmetry-related DMF ligands in axial positions. The Cd—N bond lengths [2.339 (4) and 2.353 (4) Å] and Cd—O bond length [2.322 (4) Å] fall in normal ranges (Groom & Allen, 2014 ▸). Each adci− anion bridges two adjacent Cd2+ ions in a bis-monodentate mode through two imidazole N atoms whereas the DMF molecules serve as terminal ligands. Thus, four Cd2+ ions and four bridging adci− ligands generate a square motif aligned parallel to (001), as shown in Fig. 2 ▸. The Cd⋯Cd distance along the edge of the square is 6.733 (3) Å, which is similar to previously reported structures (Li et al., 2010 ▸; Wang et al., 2010 ▸).
Figure 1.

The coordination sphere around Cd2+ in the structure of (I), with displacement ellipsoids drawn at the 30% probability level. H atoms bonded to C and N atoms have been omitted for clarity. [Symmetry code: (A) 2 − x, y,  − z.].
− z.].
Figure 2.

The two-dimensional network in the structure of (I), viewed perpendicular to the ab plane. Colour key: Cd steel, N blue, H grey, C light grey ande O red.
Supramolecular features
Complex (I) possesses various hydrogen-bonding interactions (Table 1 ▸). The amino group and the non-coordinating cyano N atoms are involved in hydrogen-bonding interactions with DMF ligands to stabilize the crystal structure. In the 2D metal-organic network, intermolecular N1—H1A⋯O1 hydrogen bonds between the primary amine group of adci− and the O atoms of an DMF ligand as well as C7—H7C⋯N5 interactions between the methyl C atoms of DMF and the non-coordinating N atoms of the cyano group of an adci− anion play a crucial role in directing and stabilizing the assembly of the supramolecular structure (Kim et al., 2015 ▸; Sava et al., 2009 ▸), as shown in Fig. 3 ▸ a. The layers are packed together by weak C7—H7B⋯N4 interactions, involving the methyl C atom of DMF and another N atom of a cyano group (Fig. 3 ▸ b). The lengths of these three hydrogen bonds fall in or approach the range (3.2–4.0 Å) of weak hydrogen-bonding interactions (Desiraju, 1996 ▸; Steed & Atwood, 2000 ▸).
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H1AO1i | 0.86 | 2.45 | 3.187(6) | 144 |
| C7H7CN5i | 0.96 | 2.68 | 3.429(12) | 135 |
| C7H7BN4ii | 0.96 | 2.65 | 3.496(11) | 148 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Figure 3.

(a) View of two kinds of hydrogen bonds in the layers. Dashed lines represent C—H⋯N (green) and N—H⋯O (red) hydrogen bonds, respectively. (b) The crystal packing between the layers in the title structure. C—H⋯N hydrogen-bonding interactions are drawn as red dashed lines. [Symmetry codes: (a) − x, − + y, z; (b) − x, + y, z; (c) x, −y, − + z].
+ y, z; (b) − x, + y, z; (c) x, −y, − + z].
Database survey
The cyano groups of the dci ligands exhibit a strong electron-withdrawing effect. Consequently, the formation of anionic species is relatively straightforward (Prasad et al., 1999 ▸) and 4,5-dicyanoimidazoles can be used in the preparation of coordination frameworks with different metal ions. However, reports on systems with 2-amino-4,5-dicyanoimidazole, a novel rigid planar ligand with five potential coordination sites, are rather scarce. A search in the Cambridge Structural Database (Version 5.27, May 2014; Groom & Allen, 2014 ▸) for 4,5-dicyanoimidazole revealed eleven complexes with 2-substituted 4,5-imidazoledicarbonitrile ligands. An unprecedented SHG-active silver-containing MOF with a rare 103 topology has been reported (Yang et al., 2013 ▸), as well as the synthesis and fluorescent properties of a 3D heterometallic polymeric complex {[K[Cd(dci)2(H2O)6]Cl]}n (Li et al., 2010 ▸), and of {[Zn2(IMDN)4(H2O)3]·3H2O3}n and [Co(IMDN)2(H2O)2]n (Hu et al., 2013 ▸) (IMDN is 2H-imidazole-4,5-dicarbonitrile) with chain structures. However, the coordination modes of the imidazoles in these complexes are different.
Synthesis and crystallization
Compound (I) was synthesized as follows: adci (0.0266 g, 0.2 mmol) and HNO3 (0.2 ml, 3.5 M in DMF) were mixed in 2 ml DMF. After stirring for 0.5 h, Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol) in 6 ml methanol was added dropwise. The mixture was further stirred for another hour and then filtrated. The filtrate was kept at ambient temperature. After about three weeks, yellow block-shaped crystals of (I) suitable for single X-ray diffraction were obtained. Yield: 0.0224 g (43% based on Cd). FT–IR (KBr, cm−1): 3436, 3346, 2930, 2217, 1658, 1525, 1486, 1444, 1385, 1328, 1305, 1115, 675.
Refinement
Crystal data, data collection and refinement details are summarized in Table 2 ▸. Hydrogen atoms of the organic ligands were placed in idealized positions, with d(C—H) = 0.93 Å for sp 2-bound H atoms and U iso(H) = 1.2U eq(C), and d(C—H) = 0.96 Å for methyl H atoms and U iso(H) = 1.5U eq(C). H atoms of the amino group were located from a difference map and were refined with d(N—H) = 0.86 Å and U iso(H) = 1.2U eq(N).
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | [Cd(C5H2N5)2(C3H7NO)2] |
| M r | 522.83 |
| Crystal system, space group | Orthorhombic, P b c n |
| Temperature (K) | 296 |
| a, b, c () | 9.8438(2), 9.1897(2), 22.8948(4) |
| V (3) | 2071.10(7) |
| Z | 4 |
| Radiation type | Mo K |
| (mm1) | 1.10 |
| Crystal size (mm) | 0.18 0.12 0.10 |
| Data collection | |
| Diffractometer | Bruker SMART APEX CCD area detector |
| Absorption correction | Multi-scan (SADABS; Bruker, 2008 ▸) |
| T min, T max | 0.854, 0.896 |
| No. of measured, independent and observed [I > 2(I)] reflections | 9497, 2386, 1741 |
| R int | 0.022 |
| (sin /)max (1) | 0.650 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.042, 0.159, 1.07 |
| No. of reflections | 2353 |
| No. of parameters | 143 |
| No. of restraints | 96 |
| H-atom treatment | H-atom parameters constrained |
| max, min (e 3) | 1.71, 0.66 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015014516/wm5187sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015014516/wm5187Isup2.hkl
CCDC reference: 1416545
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
This work was supported by the Nation Natural Science Foundation of China (grant Nos. 21173122 and 21006054).
supplementary crystallographic information
Crystal data
| [Cd(C5H2N5)2(C3H7NO)2] | F(000) = 1048 |
| Mr = 522.83 | Dx = 1.677 Mg m−3 |
| Orthorhombic, Pbcn | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2n 2ab | θ = 3.5–27.5° |
| a = 9.8438 (2) Å | µ = 1.10 mm−1 |
| b = 9.1897 (2) Å | T = 296 K |
| c = 22.8948 (4) Å | Block, yellow |
| V = 2071.10 (7) Å3 | 0.18 × 0.12 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX CCD area-detector diffractometer | 2386 independent reflections |
| Radiation source: fine-focus sealed tube | 1741 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.022 |
| phi and ω scans | θmax = 27.5°, θmin = 4.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −12→11 |
| Tmin = 0.854, Tmax = 0.896 | k = −11→11 |
| 9497 measured reflections | l = −29→26 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.159 | H-atom parameters constrained |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0999P)2 + 4.109P] where P = (Fo2 + 2Fc2)/3 |
| 2353 reflections | (Δ/σ)max < 0.001 |
| 143 parameters | Δρmax = 1.71 e Å−3 |
| 96 restraints | Δρmin = −0.65 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cd1 | 1.0000 | 0.11224 (5) | 0.7500 | 0.0180 (2) | |
| N1 | 0.8425 (4) | 0.4650 (5) | 0.71416 (18) | 0.0327 (10) | |
| H1A | 0.8043 | 0.5387 | 0.6978 | 0.039* | |
| H1B | 0.9243 | 0.4416 | 0.7050 | 0.039* | |
| N2 | 0.8289 (4) | 0.2699 (4) | 0.78205 (17) | 0.0269 (9) | |
| C4 | 0.7391 (6) | 0.1135 (5) | 0.8626 (2) | 0.0360 (12) | |
| O1 | 0.9090 (4) | 0.1297 (4) | 0.65667 (17) | 0.0449 (10) | |
| N3 | 1.1454 (4) | −0.0840 (4) | 0.72898 (18) | 0.0224 (8) | |
| C1 | 0.7732 (6) | 0.3852 (5) | 0.75474 (18) | 0.0235 (10) | |
| C2 | 0.7281 (5) | 0.2249 (5) | 0.81951 (18) | 0.0258 (9) | |
| C3 | 1.1166 (4) | −0.1874 (5) | 0.6876 (2) | 0.0234 (9) | |
| N6 | 0.9069 (5) | 0.1975 (7) | 0.5616 (2) | 0.0516 (13) | |
| N5 | 0.8982 (5) | −0.2271 (6) | 0.6291 (3) | 0.0571 (15) | |
| N4 | 0.7418 (7) | 0.0315 (6) | 0.9000 (2) | 0.0651 (17) | |
| C5 | 0.9921 (5) | −0.2038 (6) | 0.6565 (2) | 0.0308 (11) | |
| C6 | 0.9373 (9) | 0.2022 (10) | 0.6174 (3) | 0.0700 (18) | |
| H6 | 0.9931 | 0.2801 | 0.6272 | 0.084* | |
| C8 | 0.9492 (13) | 0.3011 (17) | 0.5189 (5) | 0.122 (3) | |
| H8A | 0.9115 | 0.3947 | 0.5282 | 0.183* | |
| H8B | 0.9178 | 0.2711 | 0.4811 | 0.183* | |
| H8C | 1.0465 | 0.3073 | 0.5187 | 0.183* | |
| C7 | 0.8193 (12) | 0.0771 (13) | 0.5425 (5) | 0.113 (3) | |
| H7A | 0.8668 | −0.0134 | 0.5472 | 0.169* | |
| H7B | 0.7959 | 0.0902 | 0.5022 | 0.169* | |
| H7C | 0.7381 | 0.0758 | 0.5657 | 0.169* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cd1 | 0.0143 (3) | 0.0181 (3) | 0.0217 (3) | 0.000 | 0.00059 (15) | 0.000 |
| N1 | 0.023 (2) | 0.032 (2) | 0.043 (2) | 0.0050 (18) | 0.0117 (18) | 0.0122 (19) |
| N2 | 0.0247 (19) | 0.029 (2) | 0.027 (2) | 0.0072 (17) | 0.0029 (15) | 0.0036 (16) |
| C4 | 0.043 (3) | 0.033 (3) | 0.032 (2) | 0.017 (2) | 0.010 (2) | 0.0043 (19) |
| O1 | 0.048 (2) | 0.060 (2) | 0.0270 (18) | 0.0003 (19) | −0.0062 (17) | 0.0052 (16) |
| N3 | 0.0155 (18) | 0.024 (2) | 0.0272 (18) | 0.0032 (16) | 0.0015 (16) | −0.0037 (17) |
| C1 | 0.022 (2) | 0.024 (2) | 0.024 (2) | 0.0054 (17) | −0.0030 (16) | −0.0004 (16) |
| C2 | 0.029 (2) | 0.026 (2) | 0.023 (2) | 0.0059 (19) | 0.0019 (18) | 0.0001 (17) |
| C3 | 0.021 (2) | 0.021 (2) | 0.028 (2) | −0.0009 (17) | 0.0007 (18) | −0.0001 (17) |
| N6 | 0.052 (3) | 0.076 (3) | 0.027 (2) | 0.014 (3) | −0.006 (2) | 0.005 (2) |
| N5 | 0.036 (3) | 0.069 (4) | 0.067 (4) | −0.004 (3) | −0.019 (3) | −0.021 (3) |
| N4 | 0.100 (5) | 0.054 (3) | 0.041 (3) | 0.030 (3) | 0.022 (3) | 0.018 (2) |
| C5 | 0.029 (3) | 0.028 (3) | 0.035 (3) | −0.0021 (19) | 0.001 (2) | −0.008 (2) |
| C6 | 0.072 (4) | 0.084 (4) | 0.054 (3) | 0.025 (4) | 0.004 (3) | 0.007 (3) |
| C8 | 0.143 (7) | 0.138 (8) | 0.085 (6) | 0.023 (7) | 0.016 (6) | 0.036 (6) |
| C7 | 0.116 (7) | 0.130 (7) | 0.092 (6) | 0.023 (6) | −0.024 (5) | −0.032 (6) |
Geometric parameters (Å, º)
| Cd1—O1i | 2.322 (4) | C1—N3iii | 1.342 (7) |
| Cd1—O1 | 2.322 (4) | C2—C3iii | 1.371 (6) |
| Cd1—N2i | 2.339 (4) | C3—C2ii | 1.371 (6) |
| Cd1—N2 | 2.339 (4) | C3—C5 | 1.426 (6) |
| Cd1—N3i | 2.353 (4) | N6—C6 | 1.311 (9) |
| Cd1—N3 | 2.353 (4) | N6—C8 | 1.427 (13) |
| N1—C1 | 1.366 (6) | N6—C7 | 1.469 (12) |
| N1—H1A | 0.8600 | N5—C5 | 1.136 (7) |
| N1—H1B | 0.8600 | C6—H6 | 0.9300 |
| N2—C1 | 1.347 (6) | C8—H8A | 0.9600 |
| N2—C2 | 1.375 (6) | C8—H8B | 0.9600 |
| C4—N4 | 1.141 (6) | C8—H8C | 0.9600 |
| C4—C2 | 1.426 (6) | C7—H7A | 0.9600 |
| O1—C6 | 1.154 (8) | C7—H7B | 0.9600 |
| N3—C1ii | 1.342 (7) | C7—H7C | 0.9600 |
| N3—C3 | 1.371 (6) | ||
| O1i—Cd1—O1 | 172.1 (2) | N3iii—C1—N1 | 123.0 (4) |
| O1i—Cd1—N2i | 88.18 (14) | N2—C1—N1 | 122.3 (5) |
| O1—Cd1—N2i | 86.91 (14) | C3iii—C2—N2 | 109.1 (4) |
| O1i—Cd1—N2 | 86.91 (14) | C3iii—C2—C4 | 124.4 (4) |
| O1—Cd1—N2 | 88.18 (14) | N2—C2—C4 | 126.3 (4) |
| N2i—Cd1—N2 | 103.5 (2) | C2ii—C3—N3 | 108.9 (4) |
| O1i—Cd1—N3i | 95.71 (15) | C2ii—C3—C5 | 124.5 (4) |
| O1—Cd1—N3i | 90.38 (15) | N3—C3—C5 | 126.6 (4) |
| N2i—Cd1—N3i | 167.68 (14) | C6—N6—C8 | 125.3 (9) |
| N2—Cd1—N3i | 88.42 (14) | C6—N6—C7 | 116.6 (8) |
| O1i—Cd1—N3 | 90.38 (15) | C8—N6—C7 | 118.0 (8) |
| O1—Cd1—N3 | 95.71 (15) | N5—C5—C3 | 173.8 (6) |
| N2i—Cd1—N3 | 88.42 (13) | O1—C6—N6 | 133.3 (9) |
| N2—Cd1—N3 | 167.68 (15) | O1—C6—H6 | 113.4 |
| N3i—Cd1—N3 | 79.89 (19) | N6—C6—H6 | 113.4 |
| C1—N1—H1A | 120.0 | N6—C8—H8A | 109.5 |
| C1—N1—H1B | 120.0 | N6—C8—H8B | 109.5 |
| H1A—N1—H1B | 120.0 | H8A—C8—H8B | 109.5 |
| C1—N2—C2 | 103.4 (4) | N6—C8—H8C | 109.5 |
| C1—N2—Cd1 | 129.4 (3) | H8A—C8—H8C | 109.5 |
| C2—N2—Cd1 | 122.0 (3) | H8B—C8—H8C | 109.5 |
| N4—C4—C2 | 174.4 (6) | N6—C7—H7A | 109.5 |
| C6—O1—Cd1 | 131.7 (6) | N6—C7—H7B | 109.5 |
| C1ii—N3—C3 | 103.9 (4) | H7A—C7—H7B | 109.5 |
| C1ii—N3—Cd1 | 132.5 (3) | N6—C7—H7C | 109.5 |
| C3—N3—Cd1 | 123.1 (3) | H7A—C7—H7C | 109.5 |
| N3iii—C1—N2 | 114.7 (4) | H7B—C7—H7C | 109.5 |
| O1i—Cd1—N2—C1 | 136.9 (5) | N2i—Cd1—N3—C3 | −116.7 (4) |
| O1—Cd1—N2—C1 | −36.8 (5) | N2—Cd1—N3—C3 | 78.0 (8) |
| N2i—Cd1—N2—C1 | 49.6 (4) | N3i—Cd1—N3—C3 | 59.4 (3) |
| N3i—Cd1—N2—C1 | −127.3 (5) | C2—N2—C1—N3iii | −0.9 (5) |
| N3—Cd1—N2—C1 | −145.5 (6) | Cd1—N2—C1—N3iii | 153.2 (3) |
| O1i—Cd1—N2—C2 | −73.1 (4) | C2—N2—C1—N1 | 178.5 (4) |
| O1—Cd1—N2—C2 | 113.1 (4) | Cd1—N2—C1—N1 | −27.4 (7) |
| N2i—Cd1—N2—C2 | −160.5 (4) | C1—N2—C2—C3iii | 1.1 (5) |
| N3i—Cd1—N2—C2 | 22.7 (3) | Cd1—N2—C2—C3iii | −155.5 (3) |
| N3—Cd1—N2—C2 | 4.4 (9) | C1—N2—C2—C4 | −174.0 (5) |
| O1i—Cd1—O1—C6 | 44.5 (6) | Cd1—N2—C2—C4 | 29.4 (6) |
| N2i—Cd1—O1—C6 | −7.3 (6) | N4—C4—C2—C3iii | −35 (8) |
| N2—Cd1—O1—C6 | 96.3 (6) | N4—C4—C2—N2 | 140 (7) |
| N3i—Cd1—O1—C6 | −175.3 (6) | C1ii—N3—C3—C2ii | −0.4 (5) |
| N3—Cd1—O1—C6 | −95.4 (6) | Cd1—N3—C3—C2ii | 172.2 (3) |
| O1i—Cd1—N3—C1ii | −34.6 (4) | C1ii—N3—C3—C5 | 179.5 (5) |
| O1—Cd1—N3—C1ii | 140.3 (4) | Cd1—N3—C3—C5 | −7.9 (7) |
| N2i—Cd1—N3—C1ii | 53.5 (4) | C2ii—C3—C5—N5 | −1 (6) |
| N2—Cd1—N3—C1ii | −111.8 (7) | N3—C3—C5—N5 | 179 (100) |
| N3i—Cd1—N3—C1ii | −130.4 (5) | Cd1—O1—C6—N6 | 164.9 (6) |
| O1i—Cd1—N3—C3 | 155.1 (4) | C8—N6—C6—O1 | 178.2 (9) |
| O1—Cd1—N3—C3 | −30.0 (4) | C7—N6—C6—O1 | −0.2 (13) |
Symmetry codes: (i) −x+2, y, −z+3/2; (ii) x+1/2, y−1/2, −z+3/2; (iii) x−1/2, y+1/2, −z+3/2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1iv | 0.86 | 2.45 | 3.187 (6) | 144 |
| C7—H7C···N5iv | 0.96 | 2.68 | 3.429 (12) | 135 |
| C7—H7B···N4v | 0.96 | 2.65 | 3.496 (11) | 148 |
Symmetry codes: (iv) −x+3/2, y+1/2, z; (v) x, −y, z−1/2.
References
- Brandenburg, K. (2006). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (2008). SMART, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Collins, D. J. & Zhou, H.-C. (2007). J. Mater. Chem. 17, 3154–3160.
- Desiraju, G. R. (1996). Acc. Chem. Res. 29, 441–449. [DOI] [PubMed]
- Eddaoudi, M., Sava, D. F., Eubank, J. F., Adil, K. & Guillerm, V. (2015). Chem. Soc. Rev. 44, 228–249. [DOI] [PubMed]
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Gu, Z.-Y., Yang, C.-X., Chang, N. & Yan, X.-P. (2012). Acc. Chem. Res. 45, 734–745. [DOI] [PubMed]
- Hu, T.-P., Liu, J.-F., Lu, X.-Y., Xiao, L.-Q. & Song, J.-F. (2013). Chin. J. Inorg. Chem. 29, 1928–1934.
- Kim, D.-W., Shin, J. W., Kim, J. H. & Moon, D. (2015). Acta Cryst. E71, 173–175. [DOI] [PMC free article] [PubMed]
- Li, J.-X., Du, Z.-X., Zhou, J., An, H.-Q., Zhu, B.-L., Wang, S.-R., Zhang, S.-M., Wu, S.-H. & Huang, W.-P. (2010). Inorg. Chem. Commun. 13, 127–130.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Mondal, S. S., Bhunia, A., Baburin, I. A., Jäger, C., Kelling, A., Schilde, U., Seifert, G., Janiak, C. & Holdt, H. J. (2013). Chem. Commun. 49, 7599–7601. [DOI] [PubMed]
- Mondal, S. S., Bhunia, A., Kelling, A., Schilde, U., Janiak, C. & Holdt, H.-J. (2014). J. Am. Chem. Soc. 136, 44–47. [DOI] [PubMed]
- Orcajo, G., Calleja, G., Botas, J. A., Wojtas, L., Alkordi, M. H. & Sánchez-Sánchez, M. (2014). Cryst. Growth Des. 14, 739–746.
- Prasad, B. L. V., Sato, H., Enoki, T., Cohen, S. & Radhakrishnan, T. P. (1999). J. Chem. Soc. Dalton Trans. pp. 25–30.
- Sava, D. F., Kravtsov, V. C., Eckert, J., Eubank, J. F., Nouar, F. & Eddaoudi, M. (2009). J. Am. Chem. Soc. 131, 10394–10396. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Steed, J. W. & Atwood, J. L. (2000). Supramolecular Chemistry, p. 26. Chichester: John Wiley & Son.
- Wang, S., Zhao, T.-T., Li, G.-H., Wojtas, L., Huo, Q.-S., Eddaoudi, M. & Liu, Y.-L. (2010). J. Am. Chem. Soc. 132, 18038–18041. [DOI] [PubMed]
- Xiong, R. G., Xue, X., Zhao, H., You, X. Z., Abrahams, B. F. & Xue, Z.-L. (2002). Angew. Chem. Int. Ed. 41, 3800–3803. [DOI] [PubMed]
- Yang, H., Sang, R.-L., Xu, X. & Xu, L. (2013). Chem. Commun. 49, 2909–2911. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015014516/wm5187sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015014516/wm5187Isup2.hkl
CCDC reference: 1416545
Additional supporting information: crystallographic information; 3D view; checkCIF report