

Constitutive and ligand-dependent GHSR1a activity attenuates CaV2 current and hypothalamic GABA release through distinct mechanisms and signaling pathways.
Abstract
The growth hormone secretagogue receptor type 1a (GHSR1a) has the highest known constitutive activity of any G protein–coupled receptor (GPCR). GHSR1a mediates the action of the hormone ghrelin, and its activation increases transcriptional and electrical activity in hypothalamic neurons. Although GHSR1a is present at GABAergic presynaptic terminals, its effect on neurotransmitter release remains unclear. The activities of the voltage-gated calcium channels, CaV2.1 and CaV2.2, which mediate neurotransmitter release at presynaptic terminals, are modulated by many GPCRs. Here, we show that both constitutive and agonist-dependent GHSR1a activity elicit a strong impairment of CaV2.1 and CaV2.2 currents in rat and mouse hypothalamic neurons and in a heterologous expression system. Constitutive GHSR1a activity reduces CaV2 currents by a Gi/o-dependent mechanism that involves persistent reduction in channel density at the plasma membrane, whereas ghrelin-dependent GHSR1a inhibition is reversible and involves altered CaV2 gating via a Gq-dependent pathway. Thus, GHSR1a differentially inhibits CaV2 channels by Gi/o or Gq protein pathways depending on its mode of activation. Moreover, we present evidence suggesting that GHSR1a-mediated inhibition of CaV2 attenuates GABA release in hypothalamic neurons, a mechanism that could contribute to neuronal activation through the disinhibition of postsynaptic neurons.
INTRODUCTION
Growth hormone secretagogue receptor type 1a (GHSR1a) is a G protein–coupled receptor (GPCR) highly expressed in the hypothalamus (Zigman et al., 2006). Ghrelin, the natural GHSR1a agonist, is a potent growth hormone secretagogue, and it is the only known orexigenic peptide hormone (Nakazato et al., 2001). Ghrelin-induced GHSR1a activation in soma and dendrites regulates gene transcription and increases electrical activity in neurons (Nakazato et al., 2001; Cowley et al., 2003; Diano et al., 2006; Andrews et al., 2008; Shi et al., 2013; Ribeiro et al., 2014). In addition, GHSR1a activation modulates neurotransmitter release at presynaptic terminals (Cowley et al., 2003; Cui et al., 2011; Yang et al., 2011). The modulation of presynaptic voltage-gated calcium channels (CaV2.1 and CaV2.2) is a well-established mechanism by which GPCRs regulate neurotransmitter release at central synapses (Catterall and Few, 2008; Zamponi and Currie, 2013). However, the potential role of GHSR1a regulating CaV2 channels has not been tested.
GHSR1a displays two uncommon features: a high constitutive activity and multiple signaling cascades that greatly increase the complexity of this receptor’s function. Constitutive GHSR1a activity is ∼50% of the maximum activity triggered by saturating concentrations of ghrelin (Holst et al., 2003), and it is proposed to contribute to the physiological roles of the ghrelin–GHSR1a system (Petersen et al., 2009). However, the mechanisms by which constitutive GHSR1a activity regulates neuronal activity in hypothalamic neurons remain poorly understood. On the other hand, despite the fact that it is well known that GHSR1a activates Gq proteins (Howard et al., 1996; Holst et al., 2003), recent reports indicate that other G proteins, such as Gi/o and G12/13, as well as G protein–independent pathways, such as β-arrestin recruitment, can mediate GHSR1a actions (Bennett et al., 2009; Evron et al., 2014).
Here, we used a combination of genetic and pharmacological manipulations of GHSR1a activity to show that both constitutive and ghrelin-dependent GHSR1a activities inhibit CaV2.1 and CaV2.2. We found fundamental differences in the mechanisms of CaV2 inhibition by constitutive and agonist-dependent modes of GHSR1a activation, including the signaling cascades involved and the fact that constitutive activity reduces membrane channel protein levels. Moreover, we found that GHSR1a impairs GABA release in hypothalamic neurons. Based on our data, we propose that this GHSR1a-depedent regulation of CaV2 could modify presynaptic function in the hypothalamus, particularly under fasting, when GHSR1a expression is increased.
MATERIALS AND METHODS
Animals
Sprague-Dawley rats and GHSR-eGFP reporter mice (STOCK Tg(Ghsr-EGFP)KZ65Gsat/Mmucd; 030942-UCD; Mouse Mutant Regional Resource Center, University of California, Davis, Davis, CA) were bred at the animal facility of the Instituto Multidisciplinario de Biología Celular (IMBICE). We housed the animals in a 12-h light–dark cycle in a climate-controlled room (22°C) with ad libitum access to water and food, except when indicated. We performed this study in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Research Council, and all efforts were made to minimize suffering. All experimentation received approval from the Institutional Animal Care and Use Committee of the IMBICE. GHSR-eGFP reporter mice were provided by J. Zigman (University of Texas Southwestern Medical Center, Dallas, TX).
Rat neuronal primary cultures
Neuronal cultures were obtained from Sprague-Dawley rats at embryonic days 16–17. The procedure protocol was similar to the one described in Raingo et al. (2012). In brief, we anesthetized pregnant rats with chloral hydrate (500 mg/kg) and removed the embryos. We exposed the embryo brains and placed them on the dorsal face to remove the hypothalamus with forceps. We placed the blocks of tissue in sterile Hanks’ solution, and, after two washes, we dissociated the cells at 37°C for 20 min with Hanks’ solution containing 0.25 mg/ml trypsin (Microvet) and 0.28 mg/ml deoxyribonuclease I from bovine pancreas (Sigma-Aldrich). Next, we added 300 µl FBS (Internegocios) to stop the enzyme digestion. We mechanically dissociated the cells using several glass pipettes with consecutive smaller-tip diameters. We plated 70,000 cells on 12-mm diameter glass coverslips treated previously with poly-l-lysine (Sigma-Aldrich) and laid over 24-well plates. We incubated the cells at 37°C in a 95% air and 5% CO2 atmosphere with Dulbecco’s modified Eagle’s medium (Microvet)/F12 1:1 medium supplemented with 10% FBS, 0.25% glucose, 2 mM glutamine (Gibco), 3.3 µg/ml insulin (Novo Nordisk Pharmaceutical Industries, Inc.), 5 U/ml penicillin G sodium salt (Richet), 5 µg/ml streptomycin (Richet), 40 µg/ml gentamicin sulfate salt (Richet), and 1% vitamin solution (Microvet). On the fourth day in culture, we replaced half of the incubation medium with fresh medium containing cytosine β-d-arabinofuranoside (Sigma-Aldrich) to reach a final concentration of 5 µM.
Mouse neuronal primary cultures
We used GHSR-eGFP reporter mice at embryonic days 16–17. The procedure protocol was similar to the one described above. The culture conditions were the same except for the addition of B27 supplement (1:50; Gibco) to the incubation medium.
Rat neuron transfections
At the sixth day in culture, we transfected rat neurons with GHSR1a-YFP–, GHSR1a-A204E-YFP–, or eGFP-containing plasmids using a calcium phosphate technique (Jiang and Chen, 2006). We performed patch-clamp recordings on green fluorescent and nongreen neurons after 48 h in culture.
Human embryonic kidney (HEK) 293T cell culture and transfection
For patch-clamp experiments, we cotransfected 80% confluent HEK 293T cells with GHSR1a or GHSR1a-A204E and CaV2.1 (GenBank accession no. AY714490) or CaV2.2 (GenBank accession no. AF055477), and the calcium channel auxiliary subunits CaVα2δ1 (GenBank accession no. AF286488) and CaVβ3 (GenBank accession no. M88751) or CaVβ2a (GenBank accession no. M80545) in a 1:1:1:1 molar ratio using Lipofectamine 2000 (Invitrogen). Also, for GHSR1a constitutive activity studies, we decreased the amount of GHSR1a cDNA transfected as indicated in the Results. For some experiments, we cotransfected empty pcDNA3.1(+), the C-terminal GPCR kinase 2 (MAS-GRK2-ct) (Kammermeier and Ikeda, 1999; Raingo et al., 2007) or a Gαq dominant-negative mutant (Gαq-Q209L/D277N; Missouri S&T cDNA Resource Center, Rolla, MO). For imaging experiments, we replaced CaV2.1 and CaV2.2 with these proteins tagged with GFP. GHSR1a and GHSR1a-A204E clones were provided by J. Marie (Université de Montpellier, Montpellier, France). Untagged CaV clones used for this study were provided by D. Lipscombe (Brown University, Providence, RI), and CaV2.1-GFP was provided by E.S. Piedras-Renteria (Loyola University Chicago, Chicago, IL).
Fluorescein-ghrelin (F-ghrelin) binding
We transfected HEK 293T cells with 0.15 or 0.6 µg GHSR1a or 0.6 µg of empty pcDNA3.1+ plasmid as a control. We incubated the cells with 0.4 mM F-ghrelin (McGirr et al., 2011) in binding buffer (50 mM HEPES, pH 7.4, 5 mM MgCl2, 1 mM CaCl2, and 0.2% BSA, passed through a 0.45-µm filter). After 30 min, we washed the cells with PBS, fixed them with 4% formaldehyde, and covered them with mounting media. We observed the cells with a microscope (Eclipse 50i; Nikon) with a 500-nm filter. We took photomicrographs with a camera (DS-Ri1; Nikon) and analyzed the photomicrographs with ImageJ software (1.48v).
Electrophysiology
We recorded ionic currents with an amplifier (Axopatch 200; Molecular Devices). We sampled data at 20 kHz and filtered it at 10 kHz (−3 dB) using pClamp8.2 software (Molecular Devices). We used recording electrodes with resistances between 2 and 4 MΩ when filled with internal solution. We admitted series resistances of <6 MΩ and compensated 80% with a 10-µs lag time. We subtracted current leak online using a P/−4 protocol (except for the measure of evoked and miniature postsynaptic currents). We performed all recordings at room temperature.
Sodium and barium currents of primary neuronal cultures.
We patch-clamped 7–10 d in vitro rat- and mouse-cultured neurons in voltage-clamp whole-cell mode. Internal pipette solution contained (mM): 134 CsCl, 10 EGTA, 1 EDTA, 10 HEPES, and 4 MgATP, pH 7.2 with CsOH. We measured sodium currents with high sodium external solution containing (mM): 135 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES, and 10 glucose, pH 7.4 with NaOH. To measure CaV currents, we replaced the external solution by a high barium solution with the neurons clamped at negative potentials. High barium solution contained (mM): 10 BaCl2, 110 choline chloride, 20 tetraethyl-ammonium chloride, 1 MgCl2, 10 HEPES, 10 glucose, and 0.001 tetrodotoxin (TTX; Sigma-Aldrich), pH 7.4 with CsOH. We held neurons at −80 mV and applied test pulses to 0 mV for 20 ms every 10 s. I-V protocol: we applied increasing square test pulses of 20-ms duration ranging from −60 to 50 mV every 5 s (Raingo et al., 2007).
Postsynaptic currents of primary neuronal cultures.
We patch-clamped 10–17 d in vitro mouse neurons. Internal pipette solution contained (mM): 115 Cs-methanesulfonate, 10 CsCl, 5 NaCl, 10 HEPES, 20 tetraethylammonium chloride, 4 Mg-ATP, 0.3 NaGTP, 0.6 EGTA, and 10 lidocaine N-ethyl bromide, pH 7.2 with CsOH. The external solution used was the high sodium solution described above, containing 10 µM 6-cyano-7-nitroquinoxaline-2,3-dione (Alomone Labs) to isolate inhibitory postsynaptic currents (IPSCs). To elicit evoked responses, we delivered electrical stimulation through parallel platinum electrodes (1-ms duration; amplitude, 20 mA) while neurons were held at −80 mV. We added 1 µM TTX to record miniature IPSCs (mIPSCs).
Calcium currents of transiently transfected HEK 293T cells.
We performed whole-cell patch-clamp recordings on transfected HEK 293T cells. The internal solution was the same as that described for sodium and barium currents. External solution contained (mM): 2 CaCl2, 140 choline chloride, 1 MgCl2, 10 HEPES, pH 7.4 with CsOH. Test pulses protocol: we applied square pulses from −100 to 10 mV for 15 or 20 ms every 10 s. Prepulse protocol: we applied a prepulse of 15 ms at 80 mV 12.5 ms before the test pulse to effectively remove all voltage-dependent inhibition of CaV (Raingo et al., 2007; Lopez Soto and Raingo, 2012). This protocol has no effect on both CaV2.1 and CaV2.2 control current densities (CaV2.1 currents without prepulse = −54.60 ± 16.97 pA/pF vs. with prepulse = −54.27 ± 16.92 pA/pF; n = 13; P > 0.05; and CaV2.2 currents without prepulse = −46.28 ± 14.53 pA/pF vs. with prepulse = −45.38 ± 14.23 pA/pF; n = 8; P > 0.05; paired t tests). I-V protocol: we applied increasing square test pulses of 20-ms duration ranging from −60 to 50 mV every 5 s (Raingo et al., 2007).
Imaging
We transfected HEK 293T cells with CaV2.1-GFP (provided by E.S. Piedras-Renteria; Cao et al., 2004) or CaV2.2-GFP, both obtained by subcloning in pEGFP-C1 vector, its auxiliary subunits (CaVα2δ1 and CaVβ3), and GHSR1a or GHSR1a-A204E as described previously. 48 or 24 h after transfection for CaV2.1-GFP or CaV2.2-GFP, respectively, we replaced the culture medium by 1 ml of 1 µg/ml of membrane marker solution (CellMask orange plasma membrane stain; Molecular probes) and kept the cells at 37°C for 1 min. Next, we washed three times with PBS. Finally, we removed almost all of the PBS volume and placed a clean coverslip over the cell layer. We obtained fluorescence photomicrographs with an optical fluorescence microscope (Eclipse 50i; Nikon), equipped with B2A and G2A filters and with a camera (DS-Ri1; Nikon). We performed the analysis on photomicrographs with FIJI free software, using the CellMask red signal to mark out the plasma membrane and quantify green fluorescence intensity in both the internal area (excluding plasma membrane) and the total area of each cell as integrated density. We calculated the fluorescence intensity corresponding to the membrane (membrane fluorescence) as the difference between the fluorescence corresponding to the total (total fluorescence) and the internal area. Then we calculated the percentage of CaV2.1-GFP or CaV2.2-GFP membrane fluorescence as = (membrane fluorescence/total fluorescence) × 100 for each cell.
Western blots
We used Lipofectamine Plus reagent (Invitrogen) to transfect HEK 293T cells with CaV2.1 or CaV2.2, the CaV channel auxiliary CaVα2δ1 and CaVβ3 subunits, as well as the GHSR1a and the GHSR1a-A204E or the pCDNA3.1 empty vector. We extracted membrane protein using a membrane protein extraction kit (BioVision) as reported elsewhere (Gandini et al., 2014). For each condition, we used six 100-mm culture plates of HEK 293T cells expressing CaV channels. We determined cytosolic and plasma membrane protein concentration using the bicinchoninic acid assay. In brief, 40/50 µg of protein samples were boiled for 5 min in protein-loading buffer (1.7% SDS, 0.1 M 2-mercaptoethanol, 5% glycerol, 58 mM Tris-Cl, and 0.002% bromophenol blue, pH 6.8), resolved in 7% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes (Immobilon; EMD Millipore). After blocking with 5% nonfat dry milk in Tris-buffered saline Tween 20 (TBST; 100 mM Tris-Cl, 0.9% [wt/vol] NaCl, and 0.2% Tween 20, pH 7.5), we incubated membranes overnight with primary antibodies: anti-CaV2.1 (1:300 in TBST 2.5% milk; Alomone Labs), anti-CaV2.2 (1:200 in TBST; EMD Millipore), anti-cadherin (1:200 in TBST; Invitrogen), anti-AKT (1:15,000 in TBST; Santa Cruz Biotechnology, Inc.), and anti-Hsp90 (1:2,000 in TBST; Cell Signaling Technology). Next, we washed membranes and incubated them with horseradish peroxidase (HRP)- conjugated secondary antibodies (anti–rabbit HRP; 1:5,000 in TBST; Jackson InmunoResearch Laboratories, Inc.; anti–mouse HRP; 1:5,000 in TBST; Jackson InmunoResearch Laboratories) diluted in TBST-5% nonfat dry milk. For semiquantitative analysis, we normalized CaV channel signal to the cadherin signal. We used ImageJ software (National Institutes of Health) for densitometry analysis.
Ex vivo determination of [3H]GABA release in arcuate nucleus (ARC)
We euthanized ad libitum–fed and 48 h–fasted mice by live decapitation. We extracted brains, placed them briefly in cold PBS, and sectioned them into 1-mm coronal slices by using a stainless steel mouse brain matrix. We excised small punches of tissue corresponding to the known location of the ARC, as compared with the mouse brain atlas (Paxinos and Franklin, 2001), using a 15-gauge needle. We incubated the ARC microdissections on Krebs-Ringer bicarbonate buffer (KRBB) saturated with 95% O2 and 5% CO2 for 10 min at 37°C. Next, we incubated with [3H]GABA (367 cpm/ml, with a specific activity of 92.1 Ci/mmol; PerkinElmer) in KRBB for 20 min to fill synaptic vesicles with the tracer, and we performed two washes with KRBB. After that, we incubated ARC microdissections for 10 min with fresh KRBB containing 56 mM KCl. Finally, we collected the medium, mixed it with 150 µl scintillator (Ecolite), and measured the radioactivity in a β-counter (Tracor Analytic).
Quantitative real-time (qRT)-PCR analysis
We euthanized ad libitum–fed and 48 h–fasted mice by live decapitation. We extracted brains, placed them briefly in cold diethylpyrocarbonate-PBS, and excised small punches of tissue corresponding to the ARC, as explained above. We then performed qRT-PCR analysis as described in Chuang et al. (2011a). In brief, we isolated total RNA from these punches using RNA STAT-60 (Tel-Test, Inc.). We treated the RNA with RNase-free DNase (Roche) and reverse-transcribed it into cDNA with SuperScript II reagents (Invitrogen). We performed a quantitative PCR using a sequence detection system (7900HT; Applied Biosystems) and SYBR green chemistry (Applied Biosystems). Primers were mGHSR-QF1, 5′-ACCGTGATGGTATGGGTGTCG-3′, and mGHSR-QR1, 5′-CACAGTGAGGCAGAAGACCG-3′; they amplified a product within exon 2 of the ghsr gene. We confirmed these results by a second set of specific primers, one of which is located in exon 1 of the ghsr gene (mGHSR-QF3, 5′-ATCTCCAGTGCCAGGCACTGCT-3′) and the other of which is located in exon 2 (mGHSR-GR3, 5′-AATGGGCGCGAGCAGCAGGAA-3′) of the ghsr gene. The mRNA relative levels were expressed relative to the housekeeping gene 36B4 and calculated by the comparative threshold cycle (ΔΔCt) method (Kurrasch et al., 2004). The data are presented as a percentage of levels observed in wild-type ARC punches.
Drugs
We used ghrelin esterified with n-octanoic acid (Global Peptide); a GHSR1a inverse agonist, [d-Arg1,d-Phe5,d-Trp7,9,Leu11]–substance P (SPA; Santa Cruz Biotechnology, Inc.); the inhibitor of Gs protein, cholera toxin (ChTx; Sigma Aldrich); a specific inhibitor of Gi/o protein, pertussis toxin (PTx; Sigma-Aldrich); the CaV2.1 blocker, ω-agatoxin-IVA (Peptides International); and the CaV2.2 blocker, ω-conotoxin-GVIA (Alomone Labs).
Statistics
We analyzed and plotted the data using the OriginPro 8 (OriginLab) and GraphPad Prism 5 (GraphPad Software, Inc.) software. We examined the normal distribution of data by the Kolmogorov–Smirnov test. Based on the normality test results, we established statistical significance (P < 0.05) by one- or two-sample t test (normal distributed data) or Mann–Whitney test (no normal distributed data), and multiple comparison ANOVA with Dunnett’s or Tukey’s post-test (normal distributed data) or nonparametric Kruskal–Wallis test with Dunn’s post-test (no normal distributed data). The specific statistical test used is detailed in each case. We expressed data as mean ± SE with the number of observations in brackets.
RESULTS
Constitutive and ghrelin-dependent GHSR1a activities differentially inhibit CaV2 currents in a heterologous expression system
We first examined the effect of GHSR1a and GHSR1a-A204E, a mutant lacking constitutive activity (Pantel et al., 2006), on cloned CaV2.1 and CaV2.2 channels. We coexpressed GHSR1a and CaV2 in a 1:1 molar ratio (0.6 µg of receptor cDNA per transfection) and measured calcium currents in HEK 293T cells. Calcium currents were significantly smaller in cells coexpressing GHSR1a and CaV2, as compared with those expressing CaV2 alone. In contrast, CaV2 currents in cells coexpressing GHSR1a-A204E were not different from control recordings. We next tested if agonist-independent GHSR1a-induced inhibition of CaV2 varies with GHSR1a expression levels. Thus, we measured CaV2 currents in cells transfected with CaV2.1 or CaV2.2 channels and different amounts of GHSR1a plasmid and found that CaV2 current amplitudes were inversely proportional to GHSR1a cDNA amount per transfection (Fig. 1 A). Moreover, we found that F-ghrelin binding positively correlated with the level of GHSR1a cDNA used in the transfection (Fig. 1 B). Next, we measured basal CaV2 currents in cells expressing GHSR1a with or without preincubation with the GHSR1a inverse agonist SPA (1 µM) for 16 h, and we found no significant difference with currents recorded in cells coexpressing GHSR1a-A204E (Fig. 1 C). To assess ghrelin-dependent GHSR1a action on CaV2 currents, we used GHSR1a-A204E, which lacks constitutive activity but is prone to activation by agonist binding. The application of a saturating dose of ghrelin (0.5 µM; Pantel et al., 2006) inhibited both CaV2 subtypes, but it was more effective on CaV2.2 compared with CaV2.1 currents (44.5 ± 6.9%, n = 8 vs. 15.4 ± 3.9%, n = 5, respectively, at 0.6 µg of receptor cDNA; t test; P = 0.01). Moreover, to test if the ghrelin-mediated effect was independent on GHSR1a expression levels and A204E mutation, we applied ghrelin in cells transfected with 0.15, 0.3, and 0.6 µg of GHSR1a- or 0.6 µg of GHSR1a-A204–containing cDNA per well and CaV2.1 or CaV2.2 and its auxiliary subunits. When 0.6 µg GHSR1a cDNA was used for transfections, we tested the effect of ghrelin only in cells that showed a measurable amount of current. We found that ghrelin-evoked GHSR1a activation inhibits CaV2.1 and CaV2.2 independently of A204E mutation and at the same extent in all the GHSR1a cDNA amounts tested (Fig. 1 D).
Figure 1.

Constitutive and ghrelin-dependent GHSR1a activity inhibit CaV2 currents. (A) Representative ICa traces from HEK 293T cells transfected with CaV2.1 or CaV2.2, CaVα2δ1, CaVβ3, and 0.6 µg GHSR1a, GHSR1a-A204E, or empty pcDNA3.1+ (control), and averaged ICa at different amounts of GHSR1a plasmid transfected. (B) Representative microphotographs (left) and average fluorescent signal intensity (right) for the F-ghrelin binding in cells transfected with increasing amounts of GHSR1a plasmid. (C) Representative ICa traces from cells cotransfected with CaV2.1 and CaV2.2, CaVα2δ1, CaVβ3, and 0.6 µg GHSR1a or GHSR1a-A204E with or without 1 µM SPA preincubation (left), and the averaged ICa for each condition (right). (D) Time courses and representative traces of ghrelin effect on ICa from cells expressing CaV2.1 or CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a-A204E (left), and the averaged percentage of CaV2.1 and CaV2.2 current inhibition at different amounts of GHSR1a plasmid transfected. ANOVA with Dunnett’s (C) and Tukey’s post-test (D). *, P < 0.05. Error bars represent mean ± SE.
We obtained two more sets of evidence toward two differential inhibitory mechanisms of constitutive and ghrelin-evoked GHSR1a’s effects on CaV2 currents: ghrelin application inhibits calcium currents in HEK 293T cells expressing CaV2.1 or CaV2.2 and GHSR1a in a 1:1 molar ratio preincubated with SPA, despite the fact that constitutive GHSR1a activity is blocked (Fig. 2 A). In addition, we found that acute application of SPA was unable to remove CaV2 inhibition by constitutive GHSR1a activity (Fig. 2 B).
Figure 2.
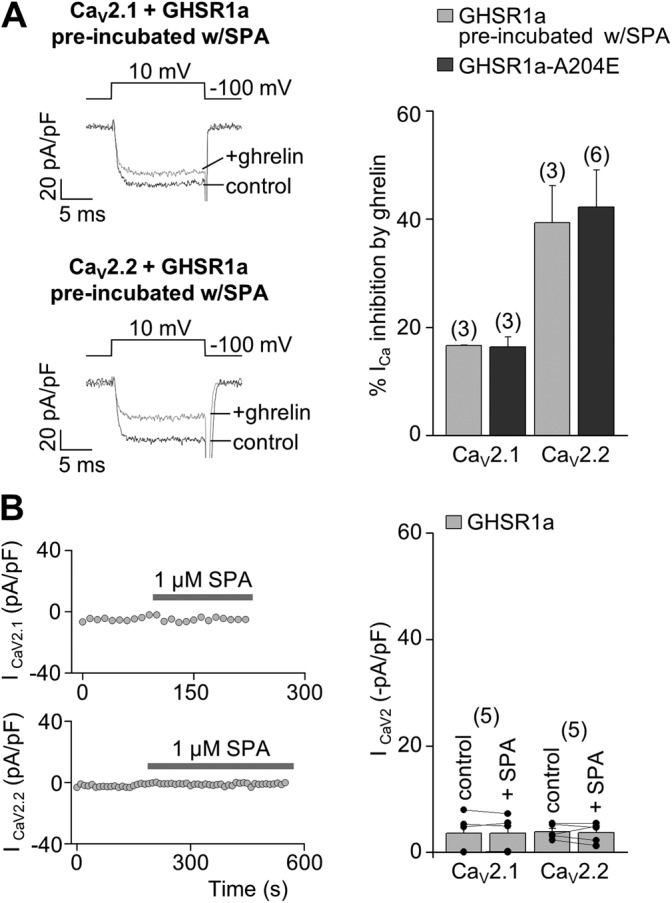
Constitutive GHSR1a activity inhibits by a long-term mechanism the CaV2 currents preserving CaV2 current inhibition by ghrelin-dependent GHSR1a activity. (A) Representative ICa traces (left) from HEK 293T cells expressing CaV2.1 (above) or CaV2.2 (below), CaVα2δ1, CaVβ3, and GHSR1a preincubated with 1 µM SPA, before (control) and after (+ghrelin) 500-nM ghrelin application, and averaged percentage of ICa inhibition by 500 nM ghrelin (right) from HEK 293T cells expressing CaV2.1 or CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a preincubated with 1 µM SPA or GHSR1a-A204E as a control condition. (B) Time courses of peak CaV2 currents (left) with the acute application of 1 µM SPA (gray bars) from HEK 293T cells expressing CaV2.1 (above) or CaV2.2 (below), CaVα2δ1, CaVβ3, and GHSR1a, and the averaged CaV2.1 or CaV2.2 currents before (control) and after (+SPA) acute application of 1 µM SPA. Two sample (A) and paired (B) Student’s t tests. Error bars represent mean ± SE.
Thus, our data show that constitutive GHSR1a activity inhibits CaV2.1 and CaV2.2 channels by a long-term mechanism that depends on GHSR1a expression levels. In contrast, ghrelin-dependent GHSR1a activity is more effective to inhibit CaV2.2 as compared with CaV2.1 currents, and this inhibition is fast and independent of GHSR1a expression levels.
Two different signaling cascades are involved in CaV2 inhibition by constitutive and ghrelin-dependent GHSR1a activities
Next, we tested the hypothesis that differential mechanisms mediate the inhibition of CaV2 channels by constitutive and ghrelin-dependent GHSR1a activities. We examined the signaling pathways engaged by GHSR1a that result in CaV2 inhibition. We first assessed the effectiveness of ghrelin-induced activation of GHSR1a-A204E on CaV2.2 channels expressed in HEK 293T cells. Neither the Gs protein inhibitor ChTx (500 ng/ml) nor the Gi/o protein inhibitor PTx (500 ng/ml) affected ghrelin-mediated inhibition of CaV2.2 currents (Fig. 3 A). We therefore used a mutant form of Gαq (Gq dominant-negative mutant [GqDN]) that acts as a dominant negative interfering with Gq-dependent signaling (Yu and Simon, 1998; Lauckner et al., 2005), and we found that GqDN occluded the inhibitory actions of ghrelin on CaV2.2 (Fig. 3 A).
Figure 3.

CaV2.2 inhibition by constitutive and ghrelin-dependent GHSR1a activity is signaled by Gi/o and Gq proteins, respectively. (A) Time course, representative traces, and averaged ICa inhibition by 500 nM ghrelin in HEK 293T cells transfected with CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a-A204E in control conditions or preincubated with 500 ng/ml ChTx or 500 ng/ml PTx, or cotransfected with a GqDN. (B) Representative traces and averaged ICa in HEK 293T cells expressing CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a or GHSR1a-A204E in control or preincubated with 500 ng/ml ChTx or 500 ng/ml PTx, or cotransfected with GqDN. (C) Representative traces and averaged ICa inhibition by 500 nM ghrelin in HEK 293T cells transfected with CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a preincubated with 500 ng/ml PTx. (D) Representative ICa in HEK 293T cells cotransfected with CaV2.2, CaVα2δ1, GHSR1a-A204E, and CaVβ3 or CaVβ2a without (control) or with (+ghrelin) 500 nM ghrelin without or with (+pp) an 80-mV prepulse application (left), and averaged percentage of ICa inhibition by 500 nM ghrelin in cells expressing CaV2.2, CaVα2δ1, and GHSR1a-A204E with the coexpression of CaVβ3 or CaVβ2a and MAS-GRK2-ct and prepulse application (+pp; right). (E) Representative ICa traces from cells cotransfected with CaV2.2, CaVα2δ1, CaVβ3, and GHSR1a or GHSR1a-A204E with (+pp) or without (Control) the application of an 80-mV prepulse (left) and averaged ICa from HEK 293T cells expressing CaV2.2, CaVα2δ1, GHSR1a, or GHSR1a-A204E, with the coexpression of CaVβ3 or CaVβ2a and MAS-GRK2ct and an 80-mV prepulse application (+pp; right). ANOVA with Dunnett’s (A and B) or Tukey’s post-test (D), and two-sample Student’s t test (E). *, P < 0.05. Error bars represent mean ± SE.
Then, we tested if the same G protein–signaling pathway also mediates the inhibitory actions of constitutive GHSR1a activity on CaV2.2 currents independently of ghrelin. We found that neither GqDN nor pretreatment with ChTx affects CaV2.2 current amplitude in cells expressing GHSR1a. In contrast, CaV2.2 currents in cells coexpressing GHSR1a pretreated with PTx were not different from the currents recorded in cells coexpressing GHSR1a-A204E (Fig. 3 B). This result suggests that GHSR1a inhibits CaV2.2 channels through Gi/o protein activation by an agonist-independent mechanism. Next, we incubated cells expressing the wild-type receptor GHSR1a with PTx, and then determine whether the agonist still modulates channel activity. We found that ghrelin inhibits the currents in this experimental condition, suggesting that the agonist-mediated and constitutive activity of GHSR1a modulates CaV2 channels by two independent pathways (Fig. 3 C).
GPCR-mediated inhibition of CaV2.2 is mediated for at least three different G proteins, and the downstream mechanisms can be voltage sensitive or voltage insensitive (Zamponi and Currie, 2013). The most common form of Gi/o-dependent inhibition of CaV2.2 channels involves direct binding of Gβγ to the channel, and it is relieved by strong depolarizing prepulses (voltage sensitive) (Ikeda, 1996; Raingo et al., 2007; Lipscombe et al., 2013). On the other hand, several Gα protein subtypes (Gq, Gi/o, and Gs) activate voltage-insensitive forms of CaV2 inhibition (Kammermeier and Ikeda, 1999; Kammermeier et al., 2000; Zamponi and Currie, 2013; Agosti et al., 2014). We found that inhibition of CaV2.2 channels by ghrelin-induced activation of GHSR1a-A204E is not relieved by 80-mV prepulses, consistent with a purely voltage-independent mechanism and a Gq-mediated pathway (Fig. 3 D). However, it is known that Gq-mediated inhibition of CaV2.2 channels turns into a voltage-sensitive inhibition by substituting the CaVβ3 for a membrane-bound form, CaVβ2a (Keum et al., 2014). Thus, we assayed ghrelin-mediated inhibition of CaV2.2 with CaVβ2a as auxiliary subunit. We found that ghrelin-mediated inhibition of CaV2.2 was not only reduced, as compared with the inhibition of CaV2.2 channels formed by CaVβ3, but also completely reversed by 80-mV prepulses (Fig. 3 C). Moreover, we coexpressed the MAS-GRK2-ct peptide to sequester Gβγ (Kammermeier and Ikeda, 1999; Raingo et al., 2007) and found that this maneuver fully occluded the inhibitory actions of ghrelin–GHSR1a-A204E on CaV2.2 channels coexpressed with either CaVβ2a or CaVβ3. Collectively, our results demonstrate that ghrelin-mediated GHSR1a activation inhibits CaV2.2 currents by a mechanism that involves Gq, Gβγ subunits of G proteins whose voltage dependency relies on the CaVβ subtype. On the other hand, we found that inhibition of CaV2.2 by constitutive GHSR1a activity is unaltered by strong prepulses to 80 mV, CaVβ subtype, or Gβγ sequestration (Fig. 3 E). Thus, our results suggest that agonist-dependent and agonist-independent forms of CaV2.2 inhibition by GHSR1a signal through different pathways.
CaV2 inhibition by constitutive GHSR1a activity is associated with a reduced channel density at the plasma membrane
Based on our results showing that the GHSR1a inverse agonist, SPA, requires long preincubation periods to occlude constitutive inhibition of CaV2 by GHSR1a, we decided to test if surface CaV2 density was affected by constitutive GHSR1a activity. First, we monitored the presence of CaV2 channels in the plasma membrane, using CaV2.1 and CaV2.2 channels tagged with GFP (CaV2.1-GFP and CaV2.2-GFP) that we have confirmed are functional in our experimental conditions (CaV2.1-GFP current, −47.0 ± 14.5 pA/pF, n = 9; CaV2.2-GFP current, −51.8 ± 12.3 pA/pF, n = 5). To identify the surface location of the GFP fluorescence signal, we used the red fluorescent membrane marker CellMask (Cogger et al., 2010). We found that CaV2.1-GFP– and CaV2.2-GFP–associated fluorescence signal was significantly lower in cells coexpressing GHSR1a, as compared with those coexpressing GHSR1a-A204E or those coexpressing GHSR1a and preincubated with SPA or PTx (Fig. 4 A). Next, we used Western blots to confirm that the CaV2.1 and CaV2.2 membrane protein level is decreased when cells coexpress GHSR1a. We used HEK 293T cells transfected with CaV2.1 or CaV2.2, as well as GHSR1a, GHSR1a-A204E, or the pcDNA3.1 empty vector. By using cadherin as a plasma membrane marker, and AKT and Hsp90 as cytoplasmic protein markers, we found that CaV2.1 and CaV2.2 protein quantity decreased in the plasma membrane protein fraction when cells coexpress GHSR1a, whereas GHSR1a-A204E coexpression failed to affect the amount of CaV2.1 and CaV2.2 plasma membrane protein (Fig. 4 B). Collectively, our data suggest that constitutive GHSR1a activity reduces surface expression of CaV2.1 and CaV2.2 channels by a Gi/o-dependent mechanism.
Figure 4.

GHSR1a decreases surface CaV2.1 and CaV2.2 density. (A) Photomicrographs and averaged percentages of green fluorescent plasma membrane signal of HEK 293T cells transfected with CaV2.1-GFP (left) and CaV2.2-GFP (right), its auxiliary subunits (Control) with GHSR1a or GHSR1a-A204E, and preincubated or not with 1 µM SPA or 500 ng/ml PTx. Green and red signals correspond to the eGFP tag on CaV2 and the membrane marker CellMask, respectively. Kruskal–Wallis with Dunn’s post-test; *, P < 0.01. (B) Western blot analysis displaying the CaV2.1-GFP and CaV2.2-GFP protein level in the plasma membrane (PM) or the cytoplasmic (Cyt) fraction of HEK 293T cells transfected with CaV2.1-GFP or CaV2.2-GFP and its auxiliary subunits (Control), and cotransfected with GHSR1a or GHSR1a-A204E (left), and averaged percentage of CaV2.1-GFP and CaV2.2-GFP PM protein level in each condition normalized against cadherin signal used as the plasma membrane loading control (right). Both AKT and Hsp90 as cytosolic markers. n = 2 and 3 for CaV2.1-GFP or CaV2.2-GFP, respectively. Error bars represent mean ± SE.
Constitutive and ghrelin-dependent GHSR1a activities inhibit native N- and P/Q-type currents
To test the effect of GHSR1a activities on calcium channels in native conditions, we used hypothalamic primary neuronal cultures of GHSR-eGFP reporter mice in which GHSR1a-expressing neurons are identifiable by green fluorescent signal (Mani et al., 2014). We first compared CaV currents recorded in GHSR1a-positive (GHSR1a+) and GHSR1a-negative (GHSR1a−) neurons. CaV currents were inhibited by 100 µM CdCl2 (percentage of inhibition: GHSR1a+, 99.91 ± 2.51%, n = 5; GHSR1a−, 99.28 ± 0.98%, n = 5; both NS vs. 100%; t test; P > 0.05). CaV currents recorded in GHSR1a+ neurons displayed the same voltage dependency as those recorded in GHSR1a− neurons, but they were significantly smaller (Fig. 5, A and B). Importantly, ghrelin application inhibited CaV currents in GHSR1a+ but not in GHSR1a− neurons (Fig. 5 A), and the effect was insensitive to 80-mV prepulse application, in agreement with our data in the expression system (percentage of IBa inhibition by ghrelin without 80-mV prepulse, 28.14 ± 6.16, and with 80-mV prepulse, 26.22 ± 7.27; NS; n = 4 neurons; P = 0.57; paired t test). We used ω-conotoxin-GVIA and ω-agatoxin-IVA to quantify the contribution of N- (CaV2.2) and P/Q-type (CaV2.1) channels, respectively, in this experimental system. We found that both current subtypes were significantly smaller in GHSR1a+ compared with GHSR1a− neurons in the presence or absence of ghrelin (Fig. 5 C). However, TTX-sensitive NaV currents in GHSR1a+ and GHSR1a− neurons were not different (t test; P > 0.05; Fig. 5 D). Thus, GHSR1a+ neurons have reduced N- and P/Q-type currents as compared with GHSR1a− neurons, and ghrelin inhibits those currents only in GHSR1a+ neurons.
Figure 5.

GHSR1a activity modulates native CaV2 currents in hypothalamic neurons from GHSR-eGFP reporter mice. (A) Representative IBa traces and averaged IBa before (control) and after (+ghrelin) 500-nM ghrelin application in hypothalamic GHSR1a− and GHSR1a+ neurons. (B) Averaged peak IBa–voltage (I-V) relationships (evoked from a holding of −80 mV), reversal (Vrev), and activation (V1/2) potential midpoints (calculated by Boltzmann linear function) obtained from GHSR1a− and GHSR1a+ neurons. (C) IBa time courses of application of 1 µM ω-conotoxin-GVIA (conoTx) and 0.2 µM ω-agatoxin-IVA (agaTx) with or without previous 500-nM ghrelin application from hypothalamic GHSR1a− (top) and GHSR1a+ neurons (middle and bottom; left). Averaged percentage of IBa sensitive to agaTx and conoTx from GHSR1a− and GHSR1a+ neurons, with (+ghrelin) or without 500-nM ghrelin application (right). (D) Representative and averaged INa from GHSR1a− and GHSR1a+ neurons. Paired (A) or two-sample (B and D) Student’s t test and ANOVA with Dunnett’s post-test (C). *, P < 0.05. Error bars represent mean ± SE.
To dissociate the effect of constitutive and ghrelin-dependent GHSR1a activities on native calcium channels, we transfected either GHSR1a-YFP or GHSR1a-A204E-YFP in hypothalamic rat-cultured neurons, which express minimal levels of endogenous GHSR1a under our experimental conditions (6 ± 2% of neurons bind F-ghrelin; n = 3 independent cultures). CaV currents recorded in neurons expressing GHSR1a-YFP were significantly smaller as compared with those recorded in either neurons expressing eGFP or GHSR1a-A204E-YFP neurons (Fig. 6 A). Ghrelin inhibited the same relative percentage of CaV currents in neurons expressing either GHSR1a-YFP or GHSR1a-A204E-YFP, whereas it failed to affect CaV currents in neurons expressing eGFP or nontransfected neurons (Fig. 6 B; normalized current traces). We next used ω-conotoxin-GVIA and ω-agatoxin-IVA to test CaV2 subtype contribution to the total CaV current in our experimental conditions. We found a smaller contribution of CaV2.1 and CaV2.2 in GHSR1a-YFP–expressing neurons as compared with the contribution found in GHSR1a-A204E-YFP–expressing neurons. Additionally, calcium currents in the presence of ghrelin showed a reduced contribution of CaV2 in both GHSR1a-YFP– and GHSR1a-A204E-YFP–expressing neurons (Fig. 6 C). Importantly, TTX-sensitive NaV currents were not affected among the different experimental conditions (Fig. 6 D). Thus, constitutive and ghrelin-dependent GHSR1a activities inhibit native N- and P/Q-type calcium currents in hypothalamic neurons.
Figure 6.

GHSR1a activity inhibits native CaV2 currents from rat hypothalamic neurons. (A) Representative and averaged IBa from nontransfected (nt) and GFP-, GHSR1a-YFP–, and GHSR1a-A204E-YFP–transfected neurons. (B) Normalized IBa traces before (control) and after (+ghrelin) 500-nM ghrelin application, and averaged percentage of IBa inhibition by ghrelin in each condition. (C) IBa time courses of application of 1 µM ω-conotoxin-GVIA (conoTx) and 0.2 µM ω-agatoxin-IVA (agaTx) with or without previous 500-nM ghrelin application from GFP-, GHSR1a-, and GHSR1a-A204E–transfected neurons (left). Averaged percentage of IBa sensitive to agaTx and conoTx from nontransfected (nt), GFP-, GHSR1a-, and GHSR1a-A204E–transfected neurons, with (+ghrelin) or without 500-nM ghrelin application (right). (D) Representative and averaged INa from nontransfected (nt) and GFP-, GHSR1a-, and GHSR1a-A204E–transfected neurons. ANOVA with Dunnett’s post-test (A–D). *, P < 0.05. Error bars represent mean ± SE.
Constitutive and ghrelin-dependent GHSR1a activities reduce GABA release from hypothalamic explants
In previous studies, we have suggested that GHSR1a activity decreases inhibitory tone on hypothalamic neurons (Cabral et al., 2012). Thus, we next evaluated if GHSR1a activity affects GABA release from explants of the ARC, a hypothalamic nucleus where GHSR1a is highly expressed (Zigman et al., 2006) and GABA release is essential for food intake regulation (Wu et al., 2009). To explore the role of constitutive GHSR1a activity, we used 48 h–fasted mice, in which hypothalamic GHSR1a mRNA levels were 1.5-fold higher compared with levels found in ad libitum–fed mice (Fig. 7 A, right). We studied the [3H]GABA release from explants of ARC, and we found a significant reduction of [3H]GABA release stimulated by high K+ in explants from fasted mice as compared with release detected in explants from ad libitum–fed mice (Fig. 7 A, left). Thus, our data show that fasting-induced increase of GHSR1a expression levels correlates with an inhibition of GABA release from ARC neurons.
Figure 7.
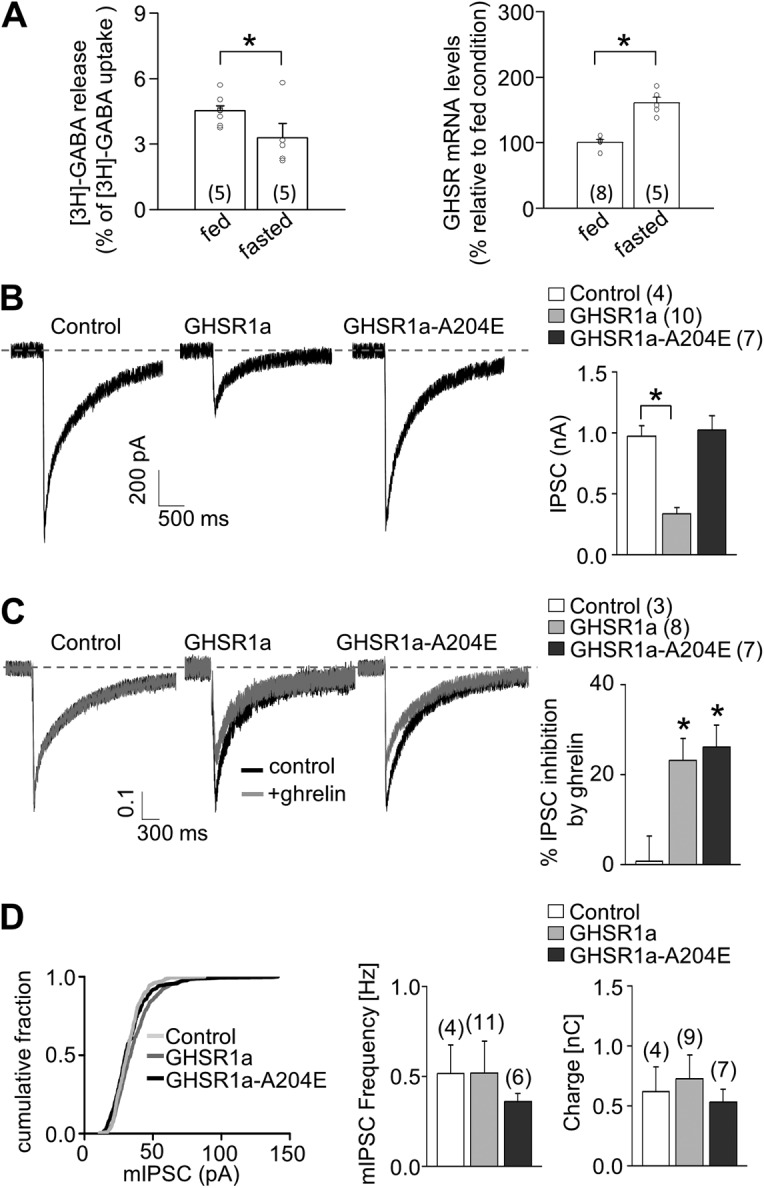
GHSR1a activity impacts GABA release. (A) [3H]GABA release (left) and GHSR1a mRNA levels (right) from ARC-enriched explants from ad libitum–fed or 48 h–fasted mice. (B) Representative traces and averaged IPSC size obtained from GHSR-null primary cultured neurons transduced or not with GHSR1a-YFP and GHSR1a-A204E-YFP. (C) Representative normalized traces with or without the application of 500 nM ghrelin, and average values of percentage of IPSC inhibition by ghrelin obtained from GHSR-null primary cultured neurons transduced or not with GHSR1a-YFP and GHSR1a-A204E-YFP. (D) Distribution of mIPSC size and averaged values for mIPSC frequencies and charge movement by 0.5-M sucrose solution application in GHSR-null primary cultured neurons transduced or not with GHSR1a-YFP and GHSR1a-A204E-YFP. Two-sample Student’s t test (A) and ANOVA with Dunnett’s post-test (B–D). *, P < 0.05. Error bars represent mean ± SE.
To test if constitutive GHSR1a activity–displayed inhibition of N- and P/Q-type currents has an impact on GABA release, we recorded IPSCs in hypothalamic neuronal cultures from GHSR-null mice transduced with either GHSR1a-YFP or GHSR1a-A204E-YFP lentiviral vectors, which allow a high percentage of GHSR1a-expressing neurons. In these experimental conditions, we found a reduced IPSC size triggered by electrical stimulation in GHSR1a-YFP–expressing cultures compared with both GHSR1a-A204E-YFP–expressing cultures and nontransduced cultures (Fig. 7 B). Moreover, we found that acute ghrelin application reduced by ∼20% the IPSC peak in cultures expressing either GHSR1a-YFP or GHSR1a-A204E-YFP (Fig. 7 C; normalized IPSC recordings), indicating that both GHSR1a variants are functional and that ghrelin-dependent GHSR1a activation can further inhibit IPSCs. Finally, we recorded GABAergic postsynaptic responses stimulated by hyperosmotic solution in the presence of TTX, a maneuver known to release neurotransmitter in a CaV-independent manner. We found no differences in these responses among nontransduced, GHSR1a-YFP–transduced, and GHSR1a-A204E-YFP–transduced cultures, suggesting that constitutive GHSR1a activity exclusively affects action potential–dependent and CaV2-mediated GABAergic neurotransmission. Importantly, the size of spontaneous IPSCs was not different among conditions (Fig. 7 D), indicating the lack of postsynaptic effects in the IPSC size reduction by GHSR1a activity.
DISCUSSION
We present evidence that GHSR1a, the GPCR with the highest constitutive activity, down-regulates CaV2.1 and CaV2.2 currents by a mechanism that is independent of its endogenous agonist, ghrelin, and involves Gi/o signaling and plasma membrane channel density reduction. We also show that ghrelin inhibits native and cloned CaV2.1 and CaV2.2 currents via activation of GHSR1a by a different signaling cascade that involves Gq protein activation. These two fundamentally different mechanisms could differentially contribute to regulate presynaptic Ca2+ entry and transmitter release from hypothalamic neurons.
Presynaptic CaV2 channel inhibition by GPCRs is an important mechanism mediating the effects of many transmitters and drugs that control neurotransmitter release (Catterall and Few, 2008). GHSR1a is expressed at axonal terminals, and a presynaptic role for this receptor has been suggested (Cowley et al., 2003; Cui et al., 2011; Yang et al., 2011; Ribeiro et al., 2014). In general, the ghrelin–GHSR1a system stimulates electrical and transcriptional activity in neurons (Cowley et al., 2003; Andrews et al., 2009; Shi et al., 2013). Also, it has been shown that GHSR1a signaling augments cytosolic Ca2+ levels in hypothalamic neurons (Howard et al., 1996; Chuang et al., 2011b; Yang et al., 2011). Here, we propose a novel mechanism that could potentially contribute to neuronal activation of hypothalamic neurons by controlling CaV2 channels located at the presynaptic terminals of inhibitory input neurons. Moreover, in agreement with previous data showing that GHSR1a can couple to several different pathways, we demonstrate that two fundamentally different mechanisms govern ghrelin-dependent and ghrelin-independent CaV2 inhibition by GHSR1a.
We show evidence that constitutive GHSR1a activity down-regulates CaV2 channel density by a Gi/o-dependent signaling pathway. GHSR1a is typically thought to involve Gq protein activation (Holst et al., 2003), although Gi/o involvement is also well documented (Bennett et al., 2009). Other GPCRs, such as the nociceptin receptor (opioid receptor–like 1 [ORL1]) and metabotropic glutamate receptor subtype 1, have been shown to inhibit CaV2 by agonist-binding independent mechanisms that require GPCR and channel protein direct interaction (Kitano et al., 2003; Beedle et al., 2004). ORL1 is a Gi/o-coupled receptor that inhibits CaV2.2 channels in dorsal root ganglion neurons, and it has been shown that basal and nociceptin-mediated ORL1 inhibition of CaV2.2 shared a Gβγ-mediated pathway, as both are avoided by Gβγ sequestering or depolarizing prepulse voltage application (Beedle et al., 2004). In contrast, here we show that CaV2 current inhibition by constitutive GHSR1a activity differs from ghrelin-evoked effect, as the former mode of inhibition is independent of Gβγ binding and unaffected by a depolarizing prepulse.
The long-lasting inhibition of CaV2 currents by GHSR1a constitutive activity implicates channel density reduction at the plasma membrane. Several GPCRs, including dopamine receptors and ORL1, use mechanisms involving channel internalization to impact CaV2 currents in the long term (Altier et al., 2006; Kisilevsky and Zamponi, 2008; Kisilevsky et al., 2008). In a clear demonstration of complexity in channel activity regulation by GPCRs, these receptors can inhibit the channels by voltage-dependent and voltage-independent modulation, as well as by cointernalization resulting from direct interaction between the activated receptor and the channel. We cannot discard the direct interaction between the GHSR1a and CaV2 channels in the mechanism involved in channel density reduction by GHSR1a constitutive activity. If present, however, this mechanism is likely insufficient to mediate channel density reduction, as protein Gi/o activation is required for the effect. The molecular mechanism by which GHSR1a-mediated Gi/o basal activation modifies channel density is unclear with our present data. We postulate that a reduced channel trafficking is a more suitable hypothesis over channel internalization, given the long-term effect of the GHSR1a inverse agonist (see Fig. 2 B) and the particular distribution of fluorescence signal from CaV2-GFP around the nucleus of transfected HEK 293T cells (see Fig. 4 A). More experiments are required to determine the intermediate molecular players of this pathway. Thus, there is increasing evidence that channel-trafficking regulation from and toward the plasma membrane could be universal mechanisms that GPCRs use to control voltage-gated calcium currents in neurons.
We found that ghrelin-dependent GHSR1a activity inhibits CaV2 channels by a rapid and reversible Gq-dependent signaling pathway. Gq activation generally inhibits CaV2 in a voltage-independent manner. Several signaling pathways have been proposed to mediate calcium channel inhibition, including PtdIns(4,5)P2 depletion from plasma membrane, increase of arachidonic acid production, and protein kinases activation (Wu et al., 2002; Liu and Rittenhouse, 2003; Gamper et al., 2004; Suh et al., 2010). The CaV2 inhibition induced by PtdIns(4,5)P2 depletion or arachidonic acid increment requires cytoplasmic subtypes of CaVβ (CaVβ3) and, as a consequence, both mechanisms fail to impair CaV2 currents if channels contain the membrane-anchored form of CaVβ, CaVβ2a (Heneghan et al., 2009; Suh et al., 2012). Accordingly, we found that the inhibition of CaV2.2 induced by ghrelin-evoked GHSR1a activation depends on the type of CaVβ subunit. In particular, the ghrelin-mediated CaV2.2 inhibition was significantly reduced and fully voltage dependent when CaVβ2a was present, whereas it was larger and voltage independent in the presence of CaVβ3. Additionally, we found that buffering Gβγ is enough to completely avoid the ghrelin-mediated inhibition of CaV2.2 independently of the CaVβ subtype. Previous reports have shown the same dependency of Gβγ for other GqPCR-mediated pathways (i.e., muscarinic type 1 and neurokinin type 1 receptors; Kammermeier et al., 2000). Thus, our data support the notion that CaVβ2a in the channel complex switches the voltage dependency of CaV2 inhibition by occluding Gq subunit–signaled effect and unmasks the inhibition by Gβγ direct binding, as recently demonstrated by Keum et al. (2014).
Peripheral administration of ghrelin potently increases food intake (Nakazato et al., 2001). Plasma ghrelin almost exclusively accesses the hypothalamic ARC, which expresses high levels of GHSR1a and is required for the orexigenic actions of peripheral ghrelin (Zigman et al., 2006; Schaeffer et al., 2013; Cabral et al., 2014). GABA release from ARC neurons is essential for food intake regulation (Wu et al., 2009), and GHSR1a is found at presynaptic terminals (Cowley et al., 2003; Yang et al., 2011). It has also been shown that GABA release by AgRP ARC neurons is required for ghrelin-induced food intake (Tong et al., 2008), and a ghrelin-induced reduction of GABA release has been proposed to mediate other hypothalamic actions of the hormone (Cabral et al., 2012). Thus, current results showing that ghrelin-induced GHSR1a activity inhibits native calcium channels, impairs CaV2-dependent GABA release from ARC explants, and modifies synaptic activity in hypothalamic neurons in culture offer a molecular mechanism that could mediate well-established effects of ghrelin. In contrast, the physiological role of constitutive GHSR1a activity has been hard to highlight in in vivo settings. Here, we confirmed that GHSR1a expression level is affected by the energy balance conditions, and found that this change correlates with GABA release from ARC explants, suggesting that GHSR1a constitutive activity may have functional implications (Holst et al., 2003; Kim et al., 2003; Pantel et al., 2006; Kineman and Luque, 2007; Liu et al., 2007). The fact that in vivo administration of the GHSR1a inverse agonist SPA reduces food intake and body weight in rats (Petersen et al., 2009), and that mice lacking GHSR1a exhibit a more severe phenotype as compared with ghrelin knockout mice, has also been interpreted as indications of a physiological role of the constitutive GHSR1a activity (Uchida et al., 2013). Notably, the A204E mutation has been associated with short stature and dominant transmission in human beings (Pantel et al., 2006). However, further studies are required to explore the physiological implications of the GHSR1a-related molecular events that occur in a ghrelin-independent fashion.
Current data open many intriguing questions including the physiological relevance of constitutive activity at synapses without ghrelin availability and mechanisms that regulate the agonist-independent actions of GHSR1a. Interestingly, GHSR1a is also present in the brain areas distant from circumventricular organs (Zigman et al., 2006) that are not accessed by plasma ghrelin (Cabral et al., 2013, 2014). It has been proposed that GHSR1a located in obviously inaccessible areas to plasma ghrelin could be modulated by centrally produced ghrelin (Cowley et al., 2003); however, more recent studies have clearly shown that ghrelin is not synthesized in the central nervous system (Sakata et al., 2009; Furness et al., 2011). Besides receptor expression levels, heterodimerization of GHSR1a with other GPCRs could also serve as an alternative mechanism to modulate specific functions of this receptor, independently of ghrelin binding (Jiang et al., 2006; Rediger et al., 2011; Kern et al., 2012; Schellekens et al., 2013). Thus, more efforts will be required to get insight on the complexity of the GHSR1a/actions in neurons.
Acknowledgments
We thank Dr. Jeffrey Zigman for providing GHSR-GFP mice.
This work was supported by grants from the National Agency of Scientific and Technological Promotion of Argentina (PICT2010-1954 and PICT2011-2142 to M. Perelló and PICT2011-1816 and PICT2013-1145 to J. Raingo) and National Institutes of Health (R03TW008925-01A1 to M. Perelló). E.J. López Soto, F. Agosti, A. Cabral, E.R. Mustafa, and V. Martínez Damonte were supported by National Scientific and Technical Research Council-Argentina and Comisión de Investigaciones Científicas fellowships.
The authors declare no competing financial interests.
Angus C. Nairn served as editor.
Footnotes
Abbreviations used in this paper:
- ARC
- arcuate nucleus
- ChTx
- cholera toxin
- F-ghrelin
- fluorescein-ghrelin
- GHSR1a
- growth hormone secretagogue receptor type 1a
- GPCR
- G protein–coupled receptor
- GqDN
- Gq dominant-negative mutant
- HEK
- human embryonic kidney
- IPSC
- inhibitory postsynaptic current
- mIPSC
- miniature IPSC
- ORL1
- opioid receptor–like 1
- PTx
- pertussis toxin
- SPA
- [d-Arg1,d-Phe5,d-Trp7,9,Leu11]–substance P
- TTX
- tetrodotoxin
References
- Agosti F., López Soto E.J., Cabral A., Castrogiovanni D., Schioth H.B., Perelló M., and Raingo J.. 2014. Melanocortin 4 receptor activation inhibits presynaptic N-type calcium channels in amygdaloid complex neurons. Eur. J. Neurosci. 40:2755–2765. 10.1111/ejn.12650 [DOI] [PubMed] [Google Scholar]
- Altier C., Khosravani H., Evans R.M., Hameed S., Peloquin J.B., Vartian B.A., Chen L., Beedle A.M., Ferguson S.S., Mezghrani A., et al. 2006. ORL1 receptor-mediated internalization of N-type calcium channels. Nat. Neurosci. 9:31–40. 10.1038/nn1605 [DOI] [PubMed] [Google Scholar]
- Andrews Z.B., Liu Z.W., Walllingford N., Erion D.M., Borok E., Friedman J.M., Tschöp M.H., Shanabrough M., Cline G., Shulman G.I., et al. 2008. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 454:846–851. 10.1038/nature07181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews Z.B., Erion D., Beiler R., Liu Z.W., Abizaid A., Zigman J., Elsworth J.D., Savitt J.M., DiMarchi R., Tschoep M., et al. 2009. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 29:14057–14065. 10.1523/JNEUROSCI.3890-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedle A.M., McRory J.E., Poirot O., Doering C.J., Altier C., Barrere C., Hamid J., Nargeot J., Bourinet E., and Zamponi G.W.. 2004. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat. Neurosci. 7:118–125. 10.1038/nn1180 [DOI] [PubMed] [Google Scholar]
- Bennett K.A., Langmead C.J., Wise A., and Milligan G.. 2009. Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super-agonists but not allosteric regulators for activation of the G protein Galpha(o1) by the Ghrelin receptor. Mol. Pharmacol. 76:802–811. 10.1124/mol.109.056101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral A., Suescun O., Zigman J.M., and Perello M.. 2012. Ghrelin indirectly activates hypophysiotropic CRF neurons in rodents. PLoS ONE. 7:e31462 10.1371/journal.pone.0031462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral A., Fernandez G., and Perello M.. 2013. Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience. 253:406–415. 10.1016/j.neuroscience.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral A., Valdivia S., Fernandez G., Reynaldo M., and Perello M.. 2014. Divergent neuronal circuitries underlying acute orexigenic effects of peripheral or central ghrelin: Critical role of brain accessibility. J. Neuroendocrinol. 26:542–554. 10.1111/jne.12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.Q., Piedras-Rentería E.S., Smith G.B., Chen G., Harata N.C., and Tsien R.W.. 2004. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron. 43:387–400. 10.1016/j.neuron.2004.07.014 [DOI] [PubMed] [Google Scholar]
- Catterall W.A., and Few A.P.. 2008. Calcium channel regulation and presynaptic plasticity. Neuron. 59:882–901. 10.1016/j.neuron.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Chuang J.C., Perello M., Sakata I., Osborne-Lawrence S., Savitt J.M., Lutter M., and Zigman J.M.. 2011a. Ghrelin mediates stress-induced food-reward behavior in mice. J. Clin. Invest. 121:2684–2692. 10.1172/JCI57660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang J.C., Sakata I., Kohno D., Perello M., Osborne-Lawrence S., Repa J.J., and Zigman J.M.. 2011b. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol. Endocrinol. 25:1600–1611. 10.1210/me.2011-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogger V.C., McNerney G.P., Nyunt T., DeLeve L.D., McCourt P., Smedsrød B., Le Couteur D.G., and Huser T.R.. 2010. Three-dimensional structured illumination microscopy of liver sinusoidal endothelial cell fenestrations. J. Struct. Biol. 171:382–388. 10.1016/j.jsb.2010.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley M.A., Smith R.G., Diano S., Tschöp M., Pronchuk N., Grove K.L., Strasburger C.J., Bidlingmaier M., Esterman M., Heiman M.L., et al. 2003. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 37:649–661. 10.1016/S0896-6273(03)00063-1 [DOI] [PubMed] [Google Scholar]
- Cui R.J., Li X., and Appleyard S.M.. 2011. Ghrelin inhibits visceral afferent activation of catecholamine neurons in the solitary tract nucleus. J. Neurosci. 31:3484–3492. 10.1523/JNEUROSCI.3187-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S., Farr S.A., Benoit S.C., McNay E.C., da Silva I., Horvath B., Gaskin F.S., Nonaka N., Jaeger L.B., Banks W.A., et al. 2006. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 9:381–388. 10.1038/nn1656 [DOI] [PubMed] [Google Scholar]
- Evron T., Peterson S.M., Urs N.M., Bai Y., Rochelle L.K., Caron M.G., and Barak L.S.. 2014. G Protein and β-arrestin signaling bias at the ghrelin receptor. J. Biol. Chem. 289:33442–33455. 10.1074/jbc.M114.581397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness J.B., Hunne B., Matsuda N., Yin L., Russo D., Kato I., Fujimiya M., Patterson M., McLeod J., Andrews Z.B., and Bron R.. 2011. Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience. 193:1–9. 10.1016/j.neuroscience.2011.07.063 [DOI] [PubMed] [Google Scholar]
- Gamper N., Reznikov V., Yamada Y., Yang J., and Shapiro M.S.. 2004. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J. Neurosci. 24:10980–10992. 10.1523/JNEUROSCI.3869-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandini M.A., Henríquez D.R., Grimaldo L., Sandoval A., Altier C., Zamponi G.W., Felix R., and González-Billault C.. 2014. CaV2.2 channel cell surface expression is regulated by the light chain 1 (LC1) of the microtubule-associated protein B (MAP1B) via UBE2L3-mediated ubiquitination and degradation. Pflugers Arch. 466:2113–2126. 10.1007/s00424-014-1476-4 [DOI] [PubMed] [Google Scholar]
- Heneghan J.F., Mitra-Ganguli T., Stanish L.F., Liu L., Zhao R., and Rittenhouse A.R.. 2009. The Ca2+ channel β subunit determines whether stimulation of Gq-coupled receptors enhances or inhibits N current. J. Gen. Physiol. 134:369–384. 10.1085/jgp.200910203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B., Cygankiewicz A., Jensen T.H., Ankersen M., and Schwartz T.W.. 2003. High constitutive signaling of the ghrelin receptor—identification of a potent inverse agonist. Mol. Endocrinol. 17:2201–2210. 10.1210/me.2003-0069 [DOI] [PubMed] [Google Scholar]
- Howard A.D., Feighner S.D., Cully D.F., Arena J.P., Liberator P.A., Rosenblum C.I., Hamelin M., Hreniuk D.L., Palyha O.C., Anderson J., et al. 1996. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 273:974–977. 10.1126/science.273.5277.974 [DOI] [PubMed] [Google Scholar]
- Ikeda S.R. 1996. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 380:255–258. 10.1038/380255a0 [DOI] [PubMed] [Google Scholar]
- Jiang H., Betancourt L., and Smith R.G.. 2006. Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol. Endocrinol. 20:1772–1785. 10.1210/me.2005-0084 [DOI] [PubMed] [Google Scholar]
- Jiang M., and Chen G.. 2006. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat. Protoc. 1:695–700. 10.1038/nprot.2006.86 [DOI] [PubMed] [Google Scholar]
- Kammermeier P.J., and Ikeda S.R.. 1999. Expression of RGS2 alters the coupling of metabotropic glutamate receptor 1a to M-type K+ and N-type Ca2+ channels. Neuron. 22:819–829. 10.1016/S0896-6273(00)80740-0 [DOI] [PubMed] [Google Scholar]
- Kammermeier P.J., Ruiz-Velasco V., and Ikeda S.R.. 2000. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Galpha q/11 and Gbeta gamma. J. Neurosci. 20:5623–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A., Albarran-Zeckler R., Walsh H.E., and Smith R.G.. 2012. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron. 73:317–332. 10.1016/j.neuron.2011.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum D., Baek C., Kim D.I., Kweon H.J., and Suh B.C.. 2014. Voltage-dependent regulation of CaV2.2 channels by Gq-coupled receptor is facilitated by membrane-localized β subunit. J. Gen. Physiol. 144:297–309. 10.1085/jgp.201411245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S., Yoon C.Y., Park K.H., Shin C.S., Park K.S., Kim S.Y., Cho B.Y., and Lee H.K.. 2003. Changes in ghrelin and ghrelin receptor expression according to feeding status. Neuroreport. 14:1317–1320. [DOI] [PubMed] [Google Scholar]
- Kineman R.D., and Luque R.M.. 2007. Evidence that ghrelin is as potent as growth hormone (GH)-releasing hormone (GHRH) in releasing GH from primary pituitary cell cultures of a nonhuman primate (Papio anubis), acting through intracellular signaling pathways distinct from GHRH. Endocrinology. 148:4440–4449. 10.1210/en.2007-0441 [DOI] [PubMed] [Google Scholar]
- Kisilevsky A.E., and Zamponi G.W.. 2008. D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels (Austin). 2:269–277. 10.4161/chan.2.4.6402 [DOI] [PubMed] [Google Scholar]
- Kisilevsky A.E., Mulligan S.J., Altier C., Iftinca M.C., Varela D., Tai C., Chen L., Hameed S., Hamid J., Macvicar B.A., and Zamponi G.W.. 2008. D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron. 58:557–570. 10.1016/j.neuron.2008.03.002 [DOI] [PubMed] [Google Scholar]
- Kitano J., Nishida M., Itsukaichi Y., Minami I., Ogawa M., Hirano T., Mori Y., and Nakanishi S.. 2003. Direct interaction and functional coupling between metabotropic glutamate receptor subtype 1 and voltage-sensitive Cav2.1 Ca2+ channel. J. Biol. Chem. 278:25101–25108. 10.1074/jbc.M303266200 [DOI] [PubMed] [Google Scholar]
- Kurrasch D.M., Huang J., Wilkie T.M., and Repa J.J.. 2004. Quantitative real-time polymerase chain reaction measurement of regulators of G-protein signaling mRNA levels in mouse tissues. Methods Enzymol. 389:3–15. 10.1016/S0076-6879(04)89001-3 [DOI] [PubMed] [Google Scholar]
- Lauckner J.E., Hille B., and Mackie K.. 2005. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA. 102:19144–19149. 10.1073/pnas.0509588102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D., Allen S.E., and Toro C.P.. 2013. Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. 36:598–609. 10.1016/j.tins.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Fortin J.P., Beinborn M., and Kopin A.S.. 2007. Four missense mutations in the ghrelin receptor result in distinct pharmacological abnormalities. J. Pharmacol. Exp. Ther. 322:1036–1043. 10.1124/jpet.107.123141 [DOI] [PubMed] [Google Scholar]
- Liu L., and Rittenhouse A.R.. 2003. Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc. Natl. Acad. Sci. USA. 100:295–300. 10.1073/pnas.0136826100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Soto E.J., and Raingo J.. 2012. A118G Mu Opioid Receptor polymorphism increases inhibitory effects on CaV2.2 channels. Neurosci. Lett. 523:190–194. 10.1016/j.neulet.2012.06.074 [DOI] [PubMed] [Google Scholar]
- Mani B.K., Walker A.K., Lopez Soto E.J., Raingo J., Lee C.E., Perelló M., Andrews Z.B., and Zigman J.M.. 2014. Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J. Comp. Neurol. 522:3644–3666. 10.1002/cne.23627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirr R., McFarland M.S., McTavish J., Luyt L.G., and Dhanvantari S.. 2011. Design and characterization of a fluorescent ghrelin analog for imaging the growth hormone secretagogue receptor 1a. Regul. Pept. 172:69–76. 10.1016/j.regpep.2011.08.011 [DOI] [PubMed] [Google Scholar]
- Nakazato M., Murakami N., Date Y., Kojima M., Matsuo H., Kangawa K., and Matsukura S.. 2001. A role for ghrelin in the central regulation of feeding. Nature. 409:194–198. 10.1038/35051587 [DOI] [PubMed] [Google Scholar]
- Pantel J., Legendre M., Cabrol S., Hilal L., Hajaji Y., Morisset S., Nivot S., Vie-Luton M.P., Grouselle D., de Kerdanet M., et al. 2006. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J. Clin. Invest. 116:760–768. 10.1172/JCI25303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G., and Franklin K.. 2001. The Mouse Brain in Stereotaxic Coordinates. Second edition Academic Press, Waltham, MA: 120 pp. [Google Scholar]
- Petersen P.S., Woldbye D.P., Madsen A.N., Egerod K.L., Jin C., Lang M., Rasmussen M., Beck-Sickinger A.G., and Holst B.. 2009. In vivo characterization of high basal signaling from the ghrelin receptor. Endocrinology. 150:4920–4930. 10.1210/en.2008-1638 [DOI] [PubMed] [Google Scholar]
- Raingo J., Castiglioni A.J., and Lipscombe D.. 2007. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat. Neurosci. 10:285–292. 10.1038/nn1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J., Khvotchev M., Liu P., Darios F., Li Y.C., Ramirez D.M., Adachi M., Lemieux P., Toth K., Davletov B., and Kavalali E.T.. 2012. VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat. Neurosci. 15:738–745. 10.1038/nn.3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rediger A., Piechowski C.L., Yi C.X., Tarnow P., Strotmann R., Grüters A., Krude H., Schöneberg T., Tschöp M.H., Kleinau G., and Biebermann H.. 2011. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J. Biol. Chem. 286:39623–39631. 10.1074/jbc.M111.287607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro L.F., Catarino T., Santos S.D., Benoist M., van Leeuwen J.F., Esteban J.A., and Carvalho A.L.. 2014. Ghrelin triggers the synaptic incorporation of AMPA receptors in the hippocampus. Proc. Natl. Acad. Sci. USA. 111:E149–E158. 10.1073/pnas.1313798111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata I., Nakano Y., Osborne-Lawrence S., Rovinsky S.A., Lee C.E., Perello M., Anderson J.G., Coppari R., Xiao G., Lowell B.B., et al. 2009. Characterization of a novel ghrelin cell reporter mouse. Regul. Pept. 155:91–98. 10.1016/j.regpep.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer M., Langlet F., Lafont C., Molino F., Hodson D.J., Roux T., Lamarque L., Verdié P., Bourrier E., Dehouck B., et al. 2013. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc. Natl. Acad. Sci. USA. 110:1512–1517. 10.1073/pnas.1212137110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellekens H., van Oeffelen W.E., Dinan T.G., and Cryan J.F.. 2013. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J. Biol. Chem. 288:181–191. 10.1074/jbc.M112.382473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L., Bian X., Qu Z., Ma Z., Zhou Y., Wang K., Jiang H., and Xie J.. 2013. Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat. Commun. 4:1435 10.1038/ncomms2439 [DOI] [PubMed] [Google Scholar]
- Suh B.C., Leal K., and Hille B.. 2010. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 67:224–238. 10.1016/j.neuron.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Kim D.I., Falkenburger B.H., and Hille B.. 2012. Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl. Acad. Sci. USA. 109:3161–3166. 10.1073/pnas.1121434109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q., Ye C.P., Jones J.E., Elmquist J.K., and Lowell B.B.. 2008. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 11:998–1000. 10.1038/nn.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A., Zigman J.M., and Perelló M.. 2013. Ghrelin and eating behavior: evidence and insights from genetically-modified mouse models. Front Neurosci. 7:121 10.3389/fnins.2013.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Bauer C.S., Zhen X.G., Xie C., and Yang J.. 2002. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 419:947–952. 10.1038/nature01118 [DOI] [PubMed] [Google Scholar]
- Wu Q., Boyle M.P., and Palmiter R.D.. 2009. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 137:1225–1234. 10.1016/j.cell.2009.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Atasoy D., Su H.H., and Sternson S.M.. 2011. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 146:992–1003. 10.1016/j.cell.2011.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., and Simon M.I.. 1998. Interaction of the xanthine nucleotide binding Goalpha mutant with G protein-coupled receptors. J. Biol. Chem. 273:30183–30188. 10.1074/jbc.273.46.30183 [DOI] [PubMed] [Google Scholar]
- Zamponi G.W., and Currie K.P.. 2013. Regulation of CaV2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta. 1828:1629–1643. 10.1016/j.bbamem.2012.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigman J.M., Jones J.E., Lee C.E., Saper C.B., and Elmquist J.K.. 2006. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494:528–548. 10.1002/cne.20823 [DOI] [PMC free article] [PubMed] [Google Scholar]