

Abstract
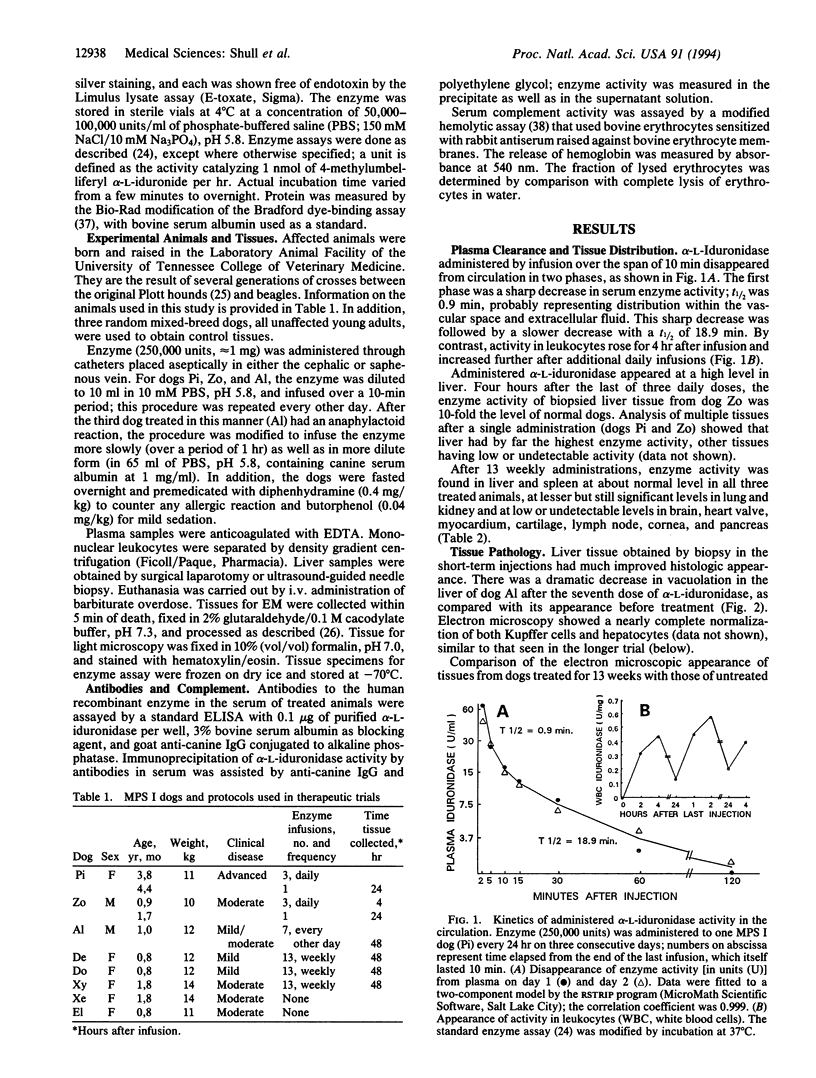
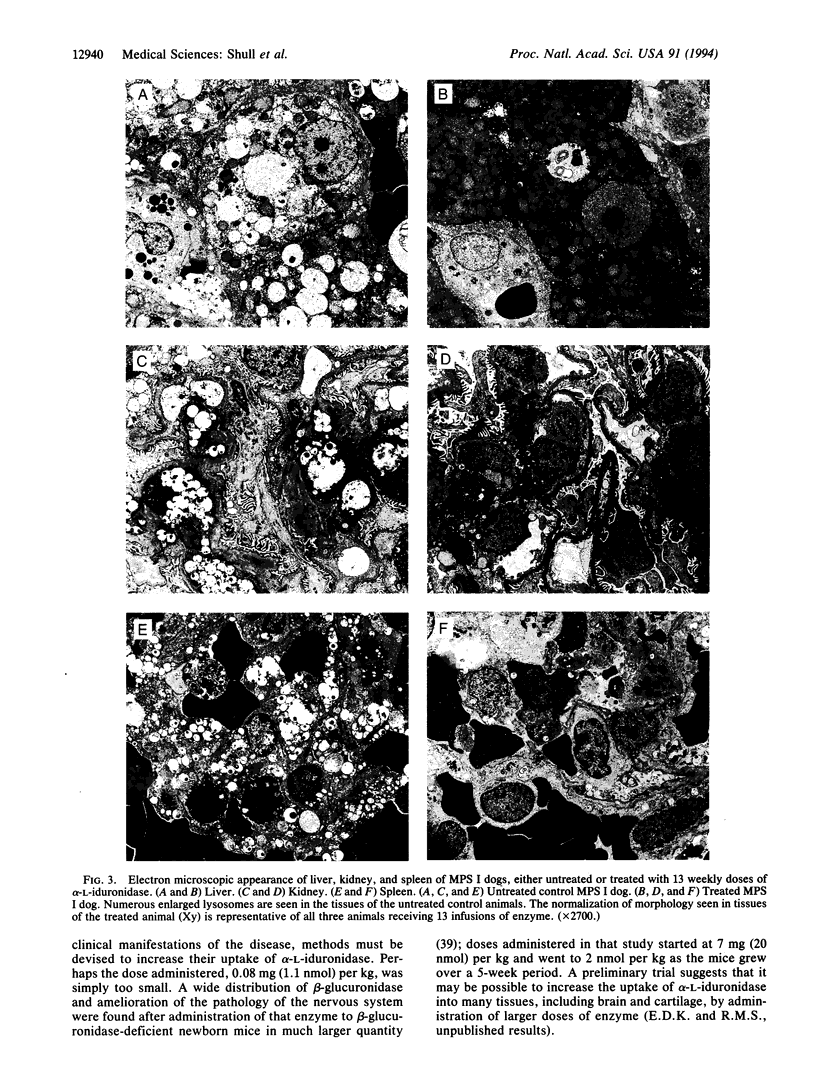
The Hurler syndrome (alpha-L-iduronidase deficiency disease) is a severe lysosomal storage disorder that is potentially amenable to enzyme-replacement therapy. Availability of a canine model of the disease and a sufficient supply of corrective enzyme have permitted a therapeutic trial lasting 3 mo. Recombinant human alpha-L-iduronidase, purified to apparent homogeneity from secretions of a stably transfected Chinese hamster ovary cell line, was administered i.v. to homozygous affected animals in doses of approximately 1 mg. The enzyme rapidly disappeared from the circulation in a biphasic manner, with t1/2 of 0.9 and 19 min, respectively, and was taken up primarily by the liver. Biopsy of the liver before and after a very short trial (seven doses administered over 12 days) showed remarkable resolution of lysosomal storage in both hepatocytes and Kupffer cells. After weekly administration of enzyme to three affected animals over a period of 3 mo, the level of enzyme was about normal in liver and spleen, lower but significant in kidney and lung, and barely detectable (0-5% of normal) in brain, heart valves, myocardium, cartilage, and cornea. Light and electron microscopic examination of numerous tissues showed normalization of lysosomal storage in liver, spleen, and kidney glomeruli, but there was no improvement in brain, heart valves, or cornea. Even though the treated dogs developed complement-activating antibodies against alpha-L-iduronidase, clinical symptoms could be prevented by slow infusion of enzyme and premedication.
Full text
PDF



Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Ashwell G., Harford J. Carbohydrate-specific receptors of the liver. Annu Rev Biochem. 1982;51:531–554. doi: 10.1146/annurev.bi.51.070182.002531. [DOI] [PubMed] [Google Scholar]
- Bach G., Friedman R., Weissmann B., Neufeld E. F. The defect in the Hurler and Scheie syndromes: deficiency of -L-iduronidase. Proc Natl Acad Sci U S A. 1972 Aug;69(8):2048–2051. doi: 10.1073/pnas.69.8.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach G., Moskowitz S. M., Tieu P. T., Matynia A., Neufeld E. F. Molecular analysis of Hurler syndrome in Druze and Muslim Arab patients in Israel: multiple allelic mutations of the IDUA gene in a small geographic area. Am J Hum Genet. 1993 Aug;53(2):330–338. [PMC free article] [PubMed] [Google Scholar]
- Barton N. W., Brady R. O., Dambrosia J. M., Di Bisceglie A. M., Doppelt S. H., Hill S. C., Mankin H. J., Murray G. J., Parker R. I., Argoff C. E. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991 May 23;324(21):1464–1470. doi: 10.1056/NEJM199105233242104. [DOI] [PubMed] [Google Scholar]
- Barton R. W., Neufeld E. F. The Hurler corrective factor. Purification and some properties. J Biol Chem. 1971 Dec 25;246(24):7773–7779. [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976 May 7;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breider M. A., Shull R. M., Constantopoulos G. Long-term effects of bone marrow transplantation in dogs with mucopolysaccharidosis I. Am J Pathol. 1989 Mar;134(3):677–692. [PMC free article] [PubMed] [Google Scholar]
- Bunge S., Kleijer W. J., Steglich C., Beck M., Zuther C., Morris C. P., Schwinger E., Hopwood J. J., Scott H. S., Gal A. Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common alpha-L-iduronidase mutations (W402X and Q70X) among European patients. Hum Mol Genet. 1994 Jun;3(6):861–866. doi: 10.1093/hmg/3.6.861. [DOI] [PubMed] [Google Scholar]
- Clarke L. A., Nelson P. V., Warrington C. L., Morris C. P., Hopwood J. J., Scott H. S. Mutation analysis of 19 North American mucopolysaccharidosis type I patients: identification of two additional frequent mutations. Hum Mutat. 1994;3(3):275–282. doi: 10.1002/humu.1380030316. [DOI] [PubMed] [Google Scholar]
- Clarke L. A., Scott H. S. Two novel mutations causing mucopolysaccharidosis type I detected by single strand conformational analysis of the alpha-L-iduronidase gene. Hum Mol Genet. 1993 Aug;2(8):1311–1312. doi: 10.1093/hmg/2.8.1311. [DOI] [PubMed] [Google Scholar]
- Dahms N. M., Lobel P., Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem. 1989 Jul 25;264(21):12115–12118. [PubMed] [Google Scholar]
- HERS H. G. INBORN LYSOSOMAL DISEASES. Gastroenterology. 1965 May;48:625–633. [PubMed] [Google Scholar]
- Hopwood J. J., Vellodi A., Scott H. S., Morris C. P., Litjens T., Clements P. R., Brooks D. A., Cooper A., Wraith J. E. Long-term clinical progress in bone marrow transplanted mucopolysaccharidosis type I patients with a defined genotype. J Inherit Metab Dis. 1993;16(6):1024–1033. doi: 10.1007/BF00711520. [DOI] [PubMed] [Google Scholar]
- Kakkis E. D., Matynia A., Jonas A. J., Neufeld E. F. Overexpression of the human lysosomal enzyme alpha-L-iduronidase in Chinese hamster ovary cells. Protein Expr Purif. 1994 Jun;5(3):225–232. doi: 10.1006/prep.1994.1035. [DOI] [PubMed] [Google Scholar]
- Kaplan A., Achord D. T., Sly W. S. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc Natl Acad Sci U S A. 1977 May;74(5):2026–2030. doi: 10.1073/pnas.74.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon K. P., Tieu P. T., Neufeld E. F. Architecture of the canine IDUA gene and mutation underlying canine mucopolysaccharidosis I. Genomics. 1992 Nov;14(3):763–768. doi: 10.1016/s0888-7543(05)80182-x. [DOI] [PubMed] [Google Scholar]
- Moskowitz S. M., Tieu P. T., Neufeld E. F. A deletion/insertion mutation in the IDUA gene in a Libyan Jewish patient with Hurler syndrome (mucopolysaccharidosis IH). Hum Mutat. 1993;2(1):71–73. doi: 10.1002/humu.1380020113. [DOI] [PubMed] [Google Scholar]
- Moskowitz S. M., Tieu P. T., Neufeld E. F. Mutation in Scheie syndrome (MPS IS): a G-->A transition creates new splice site in intron 5 of one IDUA allele. Hum Mutat. 1993;2(2):141–144. doi: 10.1002/humu.1380020215. [DOI] [PubMed] [Google Scholar]
- Pastores G. M., Sibille A. R., Grabowski G. A. Enzyme therapy in Gaucher disease type 1: dosage efficacy and adverse effects in 33 patients treated for 6 to 24 months. Blood. 1993 Jul 15;82(2):408–416. [PubMed] [Google Scholar]
- Pontow S. E., Kery V., Stahl P. D. Mannose receptor. Int Rev Cytol. 1992;137B:221–244. doi: 10.1016/s0074-7696(08)62606-6. [DOI] [PubMed] [Google Scholar]
- Richards S. M., Olson T. A., McPherson J. M. Antibody response in patients with Gaucher disease after repeated infusion with macrophage-targeted glucocerebrosidase. Blood. 1993 Sep 1;82(5):1402–1409. [PubMed] [Google Scholar]
- Sando G. N., Neufeld E. F. Recognition and receptor-mediated uptake of a lysosomal enzyme, alpha-l-iduronidase, by cultured human fibroblasts. Cell. 1977 Nov;12(3):619–627. doi: 10.1016/0092-8674(77)90262-8. [DOI] [PubMed] [Google Scholar]
- Sands M. S., Barker J. E., Vogler C., Levy B., Gwynn B., Galvin N., Sly W. S., Birkenmeier E. Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest. 1993 Jun;68(6):676–686. [PubMed] [Google Scholar]
- Sands M. S., Vogler C., Kyle J. W., Grubb J. H., Levy B., Galvin N., Sly W. S., Birkenmeier E. H. Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest. 1994 Jun;93(6):2324–2331. doi: 10.1172/JCI117237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott H. S., Litjens T., Hopwood J. J., Morris C. P. A common mutation for mucopolysaccharidosis type I associated with a severe Hurler syndrome phenotype. Hum Mutat. 1992;1(2):103–108. doi: 10.1002/humu.1380010204. [DOI] [PubMed] [Google Scholar]
- Scott H. S., Litjens T., Nelson P. V., Brooks D. A., Hopwood J. J., Morris C. P. alpha-L-iduronidase mutations (Q70X and P533R) associate with a severe Hurler phenotype. Hum Mutat. 1992;1(4):333–339. doi: 10.1002/humu.1380010412. [DOI] [PubMed] [Google Scholar]
- Scott H. S., Litjens T., Nelson P. V., Thompson P. R., Brooks D. A., Hopwood J. J., Morris C. P. Identification of mutations in the alpha-L-iduronidase gene (IDUA) that cause Hurler and Scheie syndromes. Am J Hum Genet. 1993 Nov;53(5):973–986. [PMC free article] [PubMed] [Google Scholar]
- Shull R. M., Hastings N. E., Selcer R. R., Jones J. B., Smith J. R., Cullen W. C., Constantopoulos G. Bone marrow transplantation in canine mucopolysaccharidosis I. Effects within the central nervous system. J Clin Invest. 1987 Feb;79(2):435–443. doi: 10.1172/JCI112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull R. M., Helman R. G., Spellacy E., Constantopoulos G., Munger R. J., Neufeld E. F. Morphologic and biochemical studies of canine mucopolysaccharidosis I. Am J Pathol. 1984 Mar;114(3):487–495. [PMC free article] [PubMed] [Google Scholar]
- Shull R. M., Munger R. J., Spellacy E., Hall C. W., Constantopoulos G., Neufeld E. F. Canine alpha-L-iduronidase deficiency. A model of mucopolysaccharidosis I. Am J Pathol. 1982 Nov;109(2):244–248. [PMC free article] [PubMed] [Google Scholar]
- Spellacy E., Shull R. M., Constantopoulos G., Neufeld E. F. A canine model of human alpha-L-iduronidase deficiency. Proc Natl Acad Sci U S A. 1983 Oct;80(19):6091–6095. doi: 10.1073/pnas.80.19.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu P. T., Menon K., Neufeld E. F. A mutant stop codon (TAG) in the IDUA gene is used as an acceptor splice site in a patient with Hurler syndrome (MPS IH). Hum Mutat. 1994;3(3):333–336. doi: 10.1002/humu.1380030330. [DOI] [PubMed] [Google Scholar]
- Walkley S. U., Thrall M. A., Dobrenis K., Huang M., March P. A., Siegel D. A., Wurzelmann S. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proc Natl Acad Sci U S A. 1994 Apr 12;91(8):2970–2974. doi: 10.1073/pnas.91.8.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley C. B., Belani K. G., Chang P. N., Summers C. G., Blazar B. R., Tsai M. Y., Latchaw R. E., Ramsay N. K., Kersey J. H. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet. 1993 Apr 15;46(2):209–218. doi: 10.1002/ajmg.1320460222. [DOI] [PubMed] [Google Scholar]
- Whitley C. B., Ramsay N. K., Kersey J. H., Krivit W. Bone marrow transplantation for Hurler syndrome: assessment of metabolic correction. Birth Defects Orig Artic Ser. 1986;22(1):7–24. [PubMed] [Google Scholar]
- von Figura K., Hasilik A. Lysosomal enzymes and their receptors. Annu Rev Biochem. 1986;55:167–193. doi: 10.1146/annurev.bi.55.070186.001123. [DOI] [PubMed] [Google Scholar]