

Abstract
Background:
Epigenetic alterations, such as histone methylation, modulate Myc signaling, a pathway central to oncogenesis. We investigated the role of the histone demethylase KDM4B in N-Myc–mediated neuroblastoma pathogenesis.
Methods:
Spearman correlation was performed to correlate MYCN and KDM4B expression. RNA interference, microarray analysis, gene set enrichment analysis, and real-time polymerase chain reaction were used to define the functions of KDM4B. Immunoprecipitation and immunofluorescence were used to assess protein-protein interactions between N-Myc and KDM4B. Chromatin immunoprecipitation was used to assess the binding of Myc targets. Constitutive and inducible lentiviral-mediated KDM4B knockdown with shRNA was used to assess the effects on tumor growth. Kaplan-Meier survival analysis was used to assess the prognostic value of KDM4B expression. All statistical tests were two-sided.
Results:
KDM4B and MYCN expression were found to be statistically significantly correlated in a variety of cancers, including neuroblastoma (R = 0.396, P < .001). Functional studies demonstrated that KDM4B regulates the Myc pathway. N-Myc was found to physically interact with and recruit KDM4B. KDM4B was found to regulate neuroblastoma cell proliferation and differentiation in vitro and xenograft growth in vivo (5 mice/group, two-tailed t test, P ≤ 0.001). Finally, together with MYCN amplification, KDM4B was found to stratify a subgroup of poor-prognosis patients (122 case patients, P < .001).
Conclusions:
Our findings provide insight into the epigenetic regulation of Myc via histone demethylation and proof-of-concept for inhibition of histone demethylases to target Myc signaling in cancers such as neuroblastoma.
The Myc family of transcription factors (c-Myc, N-Myc, and L-Myc) are central mediators of many different critical cellular processes (1–4). Additionally, alteration of Myc is one of the most common genetic abnormalities in human cancers, including neuroblastoma (5). Unfortunately, the Myc protein has proven to be difficult to target directly in anticancer strategies.
Myc activity is determined not only by its DNA binding sequences but also by local chromatin histone methylation status (6). Increased H3K4 methylation (active mark), but not H3K27 methylation (repressive mark), is characteristic of Myc-binding sites (6), which is consistent with recent studies that transcriptionally active epigenetic modifications mark genomic occupancy of Myc (7–9). An emerging theory is that Myc acts as a transcriptional amplifier, increasing transcription of genes that are already turned on, while genes not actively being transcribed are unaffected (8,9). However, two recent papers clearly demonstrated that Myc is also able to repress transcription (10,11). Nevertheless, Myc appears to be required for the induction and maintenance of normal histone methylation patterns associated with active chromatin in certain settings (12). Genetic disruption of MYCN in neural progenitors alters histone modifications that result in an increase in repressive H3K9me2/me3 marks and heterochromatinization, decreased DNA accessibility, and, ultimately, silencing of genes involved in Myc signaling (12), suggesting that Myc is required to maintain a euchromatin configuration by modifying histone methylation to facilitate its function. Similar results have been shown in cancer cells in which 12-hour inactivation of c-Myc resulted in global chromatin remodeling including elevated H3K9me3 (13). However, how H3K9me3/me2 is involved in mediating Myc function is not well understood. Additionally, the genetic alteration at glycine 34 (G34) of histone H3F3A, which is believed to affect the adjacent H3K36 methylation-related function, results in statistically significant N-Myc expression in pediatric glioblastoma (14), further supporting the biological connection between Myc activity and histone methylation.
The JmjC domain-containing histone demethylases, which are responsible for reversing most of the histone methyl marks in the human genome, play important roles in a number of physiologic processes such as stem cell maintenance, cell cycle regulation, and oncogenesis (15–18). Besides somatic mutations identified in the genes encoding histone demethylases such as UTX (19,20) and JARID1C (21), aberrant expression of histone demethylases has been observed in various cancers (16,18). KDM4B/JMJD2B and KDM4C/JMJD2C, which catalyze the demethylation of the repressive H3K9me3/me2 mark, are amplified in medulloblastoma (22), malignant peripheral nerve sheath tumor (23), and squamous cell carcinoma (24), suggesting a role in the pathophysiology of these tumors. However, the contribution of these histone demethylases to the activity of oncogenic drivers such as Myc is uncertain. Additionally, the opportunity to exploit this relationship as a therapeutic strategy has yet to be explored.
Methods
Affymetrix Microarray Analysis
RNA was extracted from SK-N-BE2 and NB-1691 cells 72 hours after transfection with two different siRNA oligos (siKDM4B#1, siKDM4B#2; siMYCN#1, siMYCN#2, sequence information is in the Supplementary Methods, available online). siRNA controls were purchased from Dharmacon (siKDM4B) and Origene (siMYCN), respectively. After quality control with Agilent RNA analyzer, RNA was subjected to hybridization using an Affymetrix HT HG-U133+ PM 16-Array Plate.
RNA and miRNA Extraction and Real-Time Polymerase Chain Reaction
RNA was extracted using RNeasy Mini Kit from Qiagen, while miRNA was extracted using miVana kit from Life Technologies. Real-time polymerase chain reaction (RT-PCR) was performed using Applied Biosystems’ 7500 Real-Time PCR system. The results were analyzed using delta delta Ct methods.
Data Mining
KDM4B gene expression data were downloaded from Oncomine (www.oncomine.com) or R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) and the R2 program was used to generate a Kaplan-Meier survival curve (http://r2.amc.nl). Correlation of KDM4B and MYCN was done using Spearman Correlation Analysis in the Prism program after downloading data from microarray datasets. We scanned for the optimal cutoff level of KDM4B expression and found a statistically significant difference in survival. As this is a multiple-testing approach, the P values were corrected by Bonferoni correction.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer’s protocol (Magna EZ-CHIP, Millipore). Details are described in the Supplementary Methods (available online). For primers of Myc binding sites and nonbinding sites, we referred to the Myc ChIP-sequencing data in human embryonic stem cells from the ENCODE project (http://genome.ucsc.edu/ENCODE/).
Mouse Experiments
All murine experiments were done in accordance with a protocol approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital. SK-N-BE2 and NB-1691 cells with shRNA knockdown of KDM4B were used to establish subcutaneous xenografts in four- to six-week-old male CB-17 severe combined immunodeficient mice (Taconic, Hudson, NY), with 5 mice for each group.
Statistical Analysis
GraphPad Prism software was used for data treatment and statistical analysis. All graph values represent means ± SD between two groups and were analyzed with the unpaired two-sided Student’s t test. A P value of less than .05 was considered statistically significant. The Kaplan-Meier method was used to estimate survival curves, and the log-rank test was used to test for differences between curves. All statistical tests were two-sided.
Microarray Deposition
The microarray data have been submitted in the GEO repository with accession numbers of GSE45969 and GSE45970.
Results
Assessment of the Functional Association of KDM4B With Myc in Neuroblastoma
Given the increasing appreciation for the role of epigenetic alterations in cancer and the ability of Myc to reprogram chromatin modification, we hypothesized that histone demethylases might be involved in the pathogenesis of neuroblastoma, an aggressive cancer of childhood in which dysregulated Myc signaling has a clear role in its pathogenesis. Spearman correlation analysis of many MYCN-correlated genes in our neuroblastoma microarray dataset showed that expression of 10 out of 17 human JmjC histone demethylases was associated with MYCN expression (Figure 1A), suggesting that the activity of N-Myc may be coordinated by histone demethylation. KDM4 family member KDM4B had the most statistically significant positive correlation with MYCN (R = 0.396, P < .001; Figure 1A) (R = 0.515, P < .001; Figure 1B), while KDM4A/JMJD2A and KDM4C/JMJD2C were negatively correlated with MYCN (R = -0.217, P = .04 and R = -0.286, P = .006, respectively) (Figure 1A). Separate analyses revealed that KDM4B expression was statistically significantly higher in MYCN-amplified tumors (P < .001, P = 0.01, respectively) (Figure 1, C and D) and that KDM4B expression was correlated with MYCN expression in various other cancers such as glioma, pancreatic adenocarcinoma, and sarcoma (Supplementary Figure 1A, available online).
Figure 1.

Assessment of the functional association of KDM4B with the Myc-signaling pathway. A) Spearman correlation analysis for histone demethylases and MYCN in neuroblastoma (Versteeg GSE16476). B) Spearman correlation analysis for KDM4B and MYCN in neuroblastoma (Delattre GSE12460). C-D) Expression of KDM4B in neuroblastoma from Versteeg dataset (GSE16476) (C) and Maris dataset (GSE3960) (D) were compared in MYCN-amplified and non-MYCN-amplified groups using a Student’s t test. MNA = MYCN amplification. E) Western blotting of KDM4B in SK-N-BE2 and NB-1691 cells confirms depletion of KDM4B following 72-hour siRNA transfection. F) Quantitative comparison of all chemical and genetic perturbation gene sets (n = 3403) from the MSigDB by gene set enrichment analysis (GSEA) for increased (left) and reduced (right) expression of global genes caused by KDM4B knockdown. Data are presented as a scatterplot of normalized P value/false discovery q value vs normalized enrichment score (NES) for each evaluated gene set. The dotted square gene sets indicate Myc pathway gene sets as listed in Supplementary Figure 1B (available online). G-H) Two examples of GSEA show that genes affected by depletion of KDM4B are regulated by the Myc pathway. I) MYCN gene signature derived from siRNA knockdown of MYCN in SK-N-BE2 and NB-1691 cells was included in gene sets for GSEA of KDM4B targets. J) Real-time polymerase chain reaction (PCR) validation of miR-19a and miR-20a decline after depletion of KDM4B with two different siRNA constructs (** P < .01, Student’s t test). K) Real-time PCR validation of other KDM4B targets (** P < .01, Student’s t test).
To assess KDM4B function, we analyzed the global gene expression profile after a 72-hour depletion of KDM4B in two MYCN-amplified neuroblastoma cell lines, SK-N-BE2 and NB-1691 (Figure 1E). Gene set enrichment analysis (GSEA) (25) revealed that genes regulated by KDM4B were statistically significantly associated with the Myc signaling pathway, among 3403 gene sets (P < .001) (Figure 1, F-H; Supplementary Figure 1B, available online). The genes downregulated by KDM4B depletion were enriched by Myc binding (Figure 1G) or upregulated by Myc overexpression or MYCN amplification (Figure 1H; Supplementary Figure 1B, available online). We also knocked down MYCN for global gene analysis, and the results showed that many Myc signatures were enriched (Supplementary Figure 1, C and D, available online). We also created a MYCN gene signature (top 100 genes downregulated by depletion of MYCN) (Supplementary Table 1, available online). GSEA showed that genes regulated by KDM4B were statistically significantly enriched with this MYCN signature (Rank #1, P < .001) (Figure 1I; Supplementary Figure 1B, available online). A substantial number of important cancer genes were regulated by KDM4B, such as MIR17HG (miR-19a and miR-20a)(also known as OncomiR-1); CDC25A, SOX2, and KITLG were also N-Myc targets (Figure 1, J and K; Supplementary Figure 1E, available online). Notably, at the 72-hour time point, depletion of KDM4B or N-Myc did not affect expression of the other protein (Figure 1E; Supplementary Figure 1F, available online), indicating that KDM4B primarily regulates N-Myc function via other mechanisms.
Nevertheless, KDM4B seems to specifically regulate a subgroup of N-Myc targets. For example, in SK-N-BE2 cells, 46% of genes downregulated by MYCN knockdown were also affected by KDM4B knockdown (Supplementary Figure 2A, available online). Gene ontology analysis showed that they were enriched for function in mitosis or cell cycle (Supplementary Figure 2B, available online). Those genes downregulated by MYCN but not by KDM4B knockdown such as RRP9 (essential for 18s rRNA processing during ribosome synthesis) and GEMIN5 (involved in RNA splicing) were enriched for RNA metabolism (Supplementary Figure 2, C and D, available online).
Assessment of the Physical Interaction of KDM4B and N-Myc
The finding that KDM4B depletion impaired N-Myc signaling without affecting N-Myc expression at a 72-hour time point prompted us to examine the mechanism whereby KDM4B regulated Myc activity. Histone demethylases such as LSD1 and KDM4B have been shown to interact with oncogenic transcription factors such as the estrogen receptor in breast cancer (26,27), presumably facilitating their transcriptional activities. To assess whether KDM4B was able to interact with N-Myc, we exogenously expressed KDM4B and N-Myc to perform reciprocal immunoprecipitation and showed that KDM4B and N-Myc did physically interact (Figure 2A). Endogenous KDM4B and N-Myc/Max were also found to be physically associated in SK-N-BE2 cells (Figure 2B). We further detected the physical interaction of N-Myc and KDM4B in primary human neuroblastoma xenografts (Figure 2C). Confocal immunofluorescence showed colocalization of KDM4B and N-Myc in the nucleus in over 90% of the SK-N-BE2 cells (Figure 2D; Supplementary Figures 2A and 3A, available online), while the colocalization signals were greatly diminished after knockdown of either KDM4B or MYCN (Supplementary Figures 2B, and 3B available online).
Figure 2.
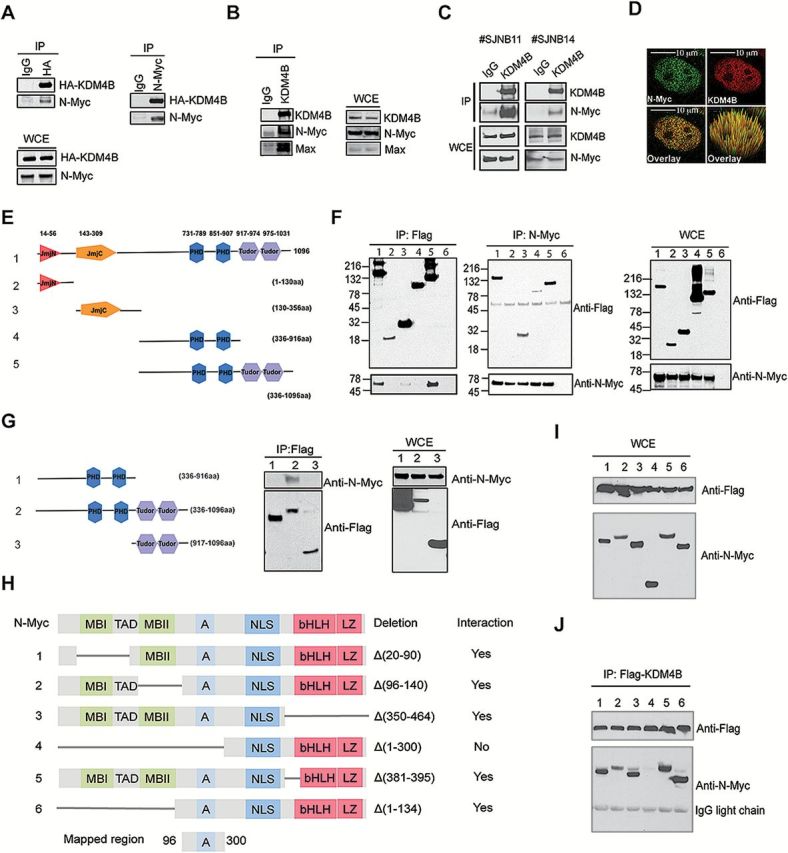
Assessment of the physical interaction of KDM4B and N-Myc. A) Reciprocal immunoprecipitation of HA-KDM4B and N-Myc overexpressed in 293T cells. 5% of total cell lysates was used as input. IP = immunoprecipitation; WCE = whole cell lysates. B) Immunoprecipitation of the endogenous KDM4B and N-Myc complex from SK-N-BE2 cells. 5% of total cell lysates was used as input. C) Immunoprecipitation of the endogenous KDM4B and N-Myc complex from human neuroblastoma xenografts. 5% of total cell lysates was used as input. D) Example of confocal immunofluorescence microscopy assessment of endogenous KDM4B and N-Myc in SK-N-BE2 cells (100X). More than 90% of the cells showed overlapping expression when 50 cells were evaluated. Scale bar = 10 µM. E) Flag-tagged constructs of different fragments of KDM4B. F) Reciprocal immunoprecipitation of flag-tagged KDM4B fragments and N-Myc overexpressed in 293T cells. Lane 6 is an IgG control. 5% of total cell lysates were used as input. G) Immunoprecipitation of flag-tagged KDM4B fragments as indicated and N-Myc overexpressed in 293T cells. 5% of total cell lysates were used as input. H) Deletion mutants of N-Myc. A = acidic central region; bHLH = basic helix-loop-helix; LZ = leucine zipper domain; MBI and MBII = MYC Box I and II; NLS = nuclear localization signal; TAD = transactivation domain. I) Input for immunoprecipitation in (J). J) Immunoprecipitation of flag-tagged KDM4B and MYCN deletion mutants as shown in (H) overexpressed in 293T cells.
KDM4B is composed of JmjN and JmjC domains, two PHD domains, and a tandem Tudor domain (Figure 2E). We used immunoprecipitation to map which domain(s) of KDM4B interact with N-Myc. We found that not only was the PHD/Tudor domain able to interact with N-Myc but also with the JmjC domain was as well, although the JmjC domain showed a much weaker interaction (Figure 2F). Interestingly, both PHD and the adjacent Tudor domains are required for the interaction of KDM4B and N-Myc, as they interacted only weakly when tested separately (Figure 2G). The interaction of N-Myc and the KDM4B PHD/Tudor domains was further validated using fluorescence resonance energy transfer (FRET) (Supplementary Figures 2C and 3C, available online). We next mapped which domain of N-Myc interacted with KDM4B by using deletion mutants (Figure 2H) (28). Immunoprecipitation assay revealed that KDM4B did not interact with the C-terminal basic helix-loop-helix leucine zipper (bHLH LZ) domain that dimerizes with MAX for DNA binding (Figure 2, I and J). The N-terminal transactivation domain (TAD) with MYC box I and II (MBI and MBII) was also not important for the interaction (Figure 2, I and J). Thus, the region between amino acid 96 and 300 that contains the acidic central region of N-Myc was mapped to interact with KDM4B (Figure 2, H-J). Taken together, these data demonstrate that KDM4B physically interacts with N-Myc, supporting the role of KDM4B in regulation of N-Myc signaling.
The Effect of KDM4B on Histone Methylation of N-Myc Target Genes
We next assessed whether KDM4B was able to bind N-Myc responsive genes in MYCN-amplified neuroblastoma cells using ChIP-PCR. KDM4B bound to the same region of the miR-17–92 gene cluster (MIR17HG/OncomiR-1), CDC25A, TRIP13, and VCAN as N-Myc (BS) (Figure 3A). Correspondingly, the H3K9me3/me2 levels were much lower than the nonbinding sites (NS) of N-Myc, KDM4B, and RNA polymerase II (Figure 3A). These data further indicate the interaction of KDM4B and N-Myc on the genomic locus. This finding is consistent with a recent study in murine embryonic stem cells, which showed that KDM4B interacts with Myc and that they co-occupy a subgroup of genes (29). Loss of KDM4B resulted in an increase of the H3K9me3/me2 repressive marks on the promoter of MIR17HG and a decrease in RNA polymerase II binding and acetyl H3K9 (Supplementary Figure 4A, available online), which are indicative of transcription reduction. These data demonstrate that KDM4B is able to modulate the histone methylation of Myc binding sites, thereby regulating gene expression.
Figure 3.
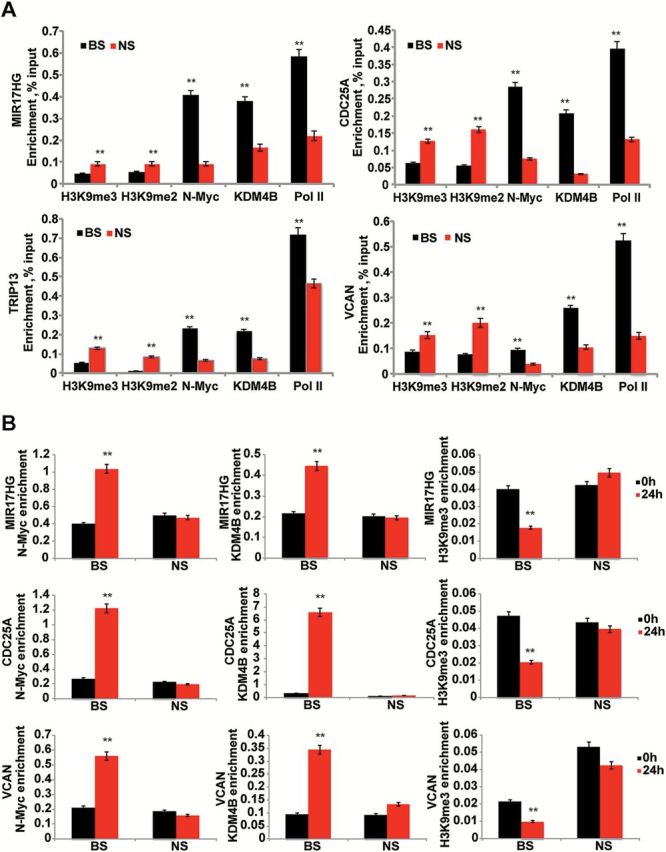
The effect of KDM4B on histone methylation of N-Myc target genes. A) Chromatin immunoprecipitation and real-time polyermase chain reaction (PCR) assess the binding of N-Myc, KDM4B, RNA polymerase II (Pol ll), H3K9me3/me2 on miR17HG, CDC25A, TRIP13, and SDC1 genes in SK-N-BE2 cells. BS = binding site; NS= nonspecific sites. B) Chromatin immunoprecipitation–PCR assessment of binding of N-Myc, KDM4B, and H3K9me3 on miR17HG and CDC25A after 24-hour N-Myc induction with 1 µg/mL of doxycycline in MYCN3 cells. The N-Myc and KDM4B enrichment represents percentage of input while the H3K9me3 enrichment represents percentage of histone H3.
We used an inducible N-Myc cell line, MYCN3, to assess whether N-Myc is able to recruit KDM4B to its binding sites, because KDM4B knockdown did not affect N-Myc binding (Supplementary Figure 4B, available online). N-Myc expression was increased after a 24-hour induction period with doxycycline (Supplementary Figure 4C, available online). The binding of N-Myc to its responsive genes, including MIR17HG, CDC25A, and VCAN occurred concomitantly with induction of N-Myc (Figure 3B). KDM4B binding to these genes increased at N-Myc binding sites but not the control nonbinding sites (Figure 3B), suggesting that induced N-Myc recruits KDM4B to the E box region. Concomitantly, H3K9me3 levels on Myc targets were decreased after normalizing to total histone H3 (Figure 3B). Because previous biochemical studies have indicated that KDM4 members are able to demethylate H3K36me3/me2 (30), we also assessed the H3K36me3 enrichment at these loci after N-Myc induction. The results showed that N-Myc induction increased H3K36me3 enrichment (Supplementary Figure 4D, available online), contrary to the expected action of KDM4B, suggesting a KDM4B-independent effect. Thus, our data indicate that the binding of N-Myc results in recruitment of KDM4B that subsequently remodels the regional chromatin through H3K9 methylation.
The Effect of KDM4B on Cell Proliferation, Differentiation, and Tumor Growth
To assess whether KDM4B is important for neuroblastoma cell growth, we used two different shRNAs for stable knockdown of KDM4B. Loss of KDM4B greatly reduced proliferation of MYCN-amplified (SK-N-BE2, NB-1691, IMR-32, NB-1643) and c-Myc overexpressed (SK-N-AS) cells (P < .001) (Figure 4, A-C, and data not shown). To exclude the possibility of off-target effects of shRNA knockdown, we introduced shRNA-resistant KDM4B back into cells, which statistically significantly rescued cell viability (P < .001), indicating that the observed effect is KDM4B specific (Figure 4D). Additionally, loss of KDM4B resulted in cell morphology changes such as neurite outgrowth, especially in SK-N-BE2 and NB-1643 cells (Figure 4E; Supplementary Figure 5A, available online), suggestive of induction of differentiation, consistent with upregulation of differentiation marker genes (Figure 4F; Supplementary Figure 5B, available online). We next studied whether KDM4B is required for tumor growth using xenograft models. Neuroblastoma cell lines were generated with stable knockdown of KDM4B using lentiviral-mediated shRNA. Depletion of KDM4B statistically significantly delayed tumor growth of these neuroblastoma xenografts (5 mice/group; P < .001) (Figure 4, G and H). To test whether KDM4B is important in tumor maintenance, we made inducible shRNA-KDM4B in SK-N-BE2 cells in which KDM4B was efficiently reduced after administration of doxycycline (Figure 4I). The results showed that growth of established tumors was statistically significantly suppressed by inducible knockdown of KDM4B (5 mice/group, P < .001) (Figure 4I), indicating that KDM4B is required for tumor maintenance.
Figure 4.

The effect of KDM4B on cell proliferation, differentiation, and tumor growth. A-C) Six days after lentiviral-mediated shRNA transduction and puromycin selection, cells were plated in triplicates for counting on consecutive four days (top). Western blotting shows the knockdown of KDM4B after six days of selection in puromycin. D) SK-N-BE2 cells were transduced with shRNA resistant KDM4B (rKDM4B). Cells were then transduced with shRNA against KDM4B for colony formation. E) Cell morphology change after KDM4B knockdown in SK-N-BE2 cells. Scale bar = 20 µm. F) Real-time polymerase chain reaction for assessment of differentiation marker genes after KDM4B knockdown. G-H) Growth of SK-N-BE2 and NB-1691 xenografts expressing shRNA GFP (shRNA-ctrl) or shRNA against KDM4B (shRNA-814) in mice (n = 5/group). Tumor volumes shown represent mean ± SD. Inlet shows in vitro knockdown efficiency of KDM4B. I) After puromycin selection of an inducible lentiviral shRNA-knockdown in SK-N-BE2 cells, 107 cells were subcutaneously implanted (n = 5/group). Four days later, when mice had palpable tumors, the mice were given 10mg/kg of doxycycline daily via gavage to induce shRNA knockdown. Tumor volumes represent mean ± SD. Inlet shows in vitro knockdown efficiency of KDM4B after doxycycline induction.
The Prognostic Value of KDM4B in Neuroblastoma
We further examined the clinical relevance of KDM4B expression in neuroblastoma. The gene transcriptome data from human neuroblastoma showed that KDM4B was highly expressed in neuroblastoma (NB) in comparison with well-differentiated ganglioneuroblastoma (GNB) and ganglioneuroma (GN) (122 cases, P < .001) (Figure 5A). KDM4B was also expressed at higher levels in stage 4 neuroblastoma than in stage 1 (35 cases, P = .04) (Figure 5B). Patients with recurrent neuroblastoma also had a higher expression of KDM4B (88 cases, P = .02) (Supplementary Figure 5C, available online). Importantly, high expression of KDM4B was associated with very poor prognosis in neuroblastoma (122 cases, P < .001) (Figure 5C). Even in patients with limited follow-up of five years (or deceased earlier), high KDM4B was still associated poor prognosis (122 cases, P < .001) (Supplementary Figure 5D, available online). However, when a Cox proportional hazard analysis on the separation calculated in the Kaplan scan was performed, the KDM4B high group lost their prognostic value (Supplementary Figure 5E, available online). Yet, when KDM4B was combined with INSS stages 1, 2, and 4s vs 3 and 4, a statistically significant result was still obtained (P < .001) (Supplementary Figure 5E, available online).
Figure 5.
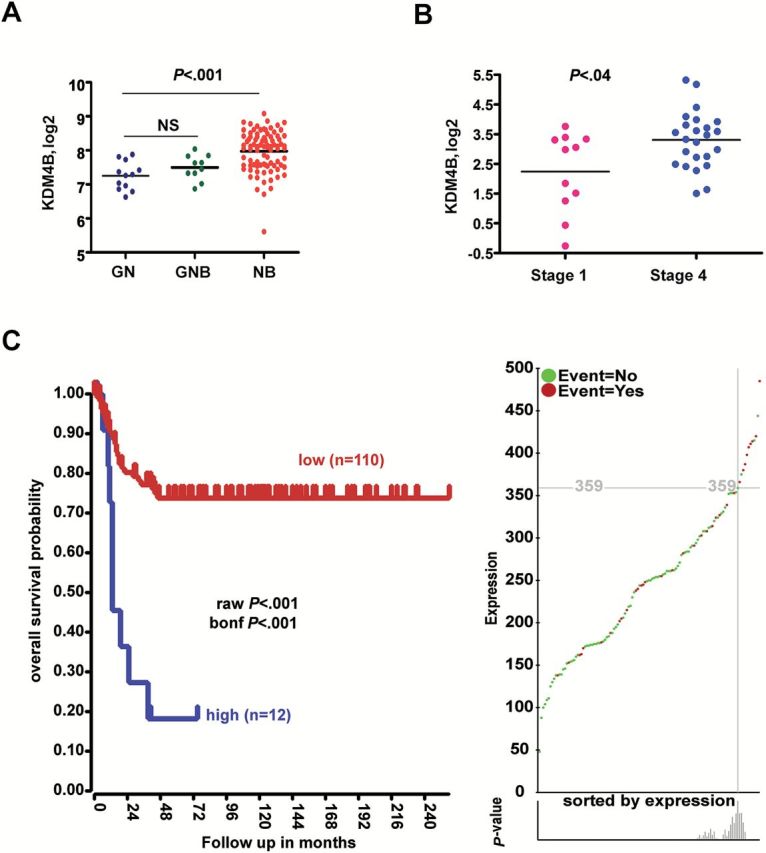
The prognostic value of KDM4B in neuroblastoma. A) KDM4B is more highly expressed in neuroblastoma (NB) compared with ganglioneuroblastoma (GNB) and ganglioneuroma (GN) (Versteeg dataset GSE16476). NS = not statistically significant. B) KDM4B is more highly expressed in patients with stage 4 neuroblastoma (Janoueix-Lerosey dataset, GSE12460). P values were computed by Student’s t test. All statistical tests were two-sided. C) Kaplan-Meier analysis shows that high KDM4B expression stratifies very poor prognosis in neuroblastoma patients from Versteeg dataset (GSE16476) (left). Gene expression and events (green and red) are shown on the right. The P value was corrected by Bonferoni correction.
Discussion
Epigenetic silencing of tumor suppressor genes by DNA methylation has a well-documented role in tumorigenesis (31). Accumulating evidence now suggests that deregulation of histone lysine methylation is also involved in cancer pathogenesis (17), because histone methylation regulates chromatin structure and gene transcription (32). As an important oncogenic transcriptional factor, Myc cooperates with epigenetic modulators for its full activity (33). Here we show that KDM4B, a H3K9me3/me2 demethylase, is highly expressed in MYCN-amplified neuroblastomas with poor outcome and is a novel protein-protein interaction partner of N-Myc. We have found that the PHD and Tudor domains of KDM4B cooperatively interact with N-Myc, which resembles the interaction of KDM4A and the androgen receptor (34), suggesting that the PHD and Tudor domains in KDM4B, like those in UHRF1, act together as a functional module in protein-protein interactions (35,36). Genetic ablation of KDM4B in MYCN-amplified neuroblastoma cells results in downregulation of important cancer genes such as MIR17HG, CDC25A, SOX2, KITLG, VCAN, and SDC1 that are also N-Myc targets. MIR17HG, the Oncomir-1/miR-17–92 cluster host gene, is a well-known Myc target and plays an important role in tumorigenesis including in neuroblastoma (2,37,38). CDC25A is an oncogenic phosphatase required both for progression through mitosis and for Myc-induced apoptosis (39,40). KITLG and SOX2 are pluripotent stem cell markers (41), and SOX2 has been shown to control tumor initiation and cancer stem cell functions (42). VCAN and SDC1 encode sulfate proteoglycan proteins that are involved in tumor angiogenesis (43,44). Thus, KDM4B plays an important role in regulation of genes that are involved in tumorigenesis and progression. Interestingly, the functions of N-Myc in the regulation of RNA metabolism seemed not to be affected by KDM4B, which is indicative of the specificity of KDM4B function. Inhibition of KDM4B primarily suppresses Myc function, inhibits neuroblastoma cell proliferation, induces cell differentiation, and delays tumor growth, indicating that targeting KDM4B may provide a new therapeutic approach for tumors in which Myc signaling is involved in oncogenesis.
Myc appears to recruit histone methylation modifiers to facilitate its function by maintaining DNA-encompassing target genes in an open configuration. For example, in a prior study, disruption of MYCN led to increased H3K9me2/me3 and decreased DNA accessibility (12), suggesting that N-Myc is able to maintain a euchromatin state via histone demethylases. For the first time, we provide evidence that DNA binding by N-Myc is associated with KDM4B, with which it physically interacts, thereby maintaining low levels of repressive H3K9me2/me3 marks from chromatin loci of Myc targets such as miR-17–92. Loss of KDM4B results in elevated H3K9me3/me2 marks at N-Myc binding loci, which is indicative of transcriptional repression. Myc is able to recruit multiple components including histone acetyltransferases and ATP-dependent chromatin remodeling complexes for its activity (45). KDM4B may collaborate with these epigenetic modifiers to enhance Myc function. Although it has been shown that KDM4 members are able to demethylate H3K36me3/me2, we failed to see reduction of H3K36me3 after N-Myc induction; instead, we observed reduced H3K9me3 but increased H3K36me3. These data indicate that N-Myc recruits KDM4B to remove H3K9me3, while elevated H3K36me3 might be KDM4B independent. H3K36me3 is important in G34 mutated glioma and is connected with N-Myc function (14). The elevated H3K36me3 suggests that N-Myc may recruit histone methyltransferases for its activity. Crystallographic structure studies show that the H3K9me3 and H3K36me3 substrates need to assume different conformations for catalysis by KDM4A (46); thus, it is possible that KDM4B can locate at H3K9 only when interacting with N-Myc. Another possibility is that KDM4 members may have a low affinity for H3K36me3 under physiological settings, as in vitro demethylation assays reveal that H3K9me3 is a more preferred modification state for KDM4A than H3K36me3 (47).
Although we have shown that KDM4B and N-Myc physically interact and affect N-Myc function, the global picture of the interaction of KDM4B and N-Myc on the chromatin level needs to be characterized by ChIP-seq or other approaches. The effect of KDM4B on H3K9me3 was modest and whether its catalytic function is really required for N-Myc activity needs to be further confirmed using specific KDM4B inhibitors.
In summary, our findings indicate that KDM4B plays an important role in regulation of N-Myc pathway in cancer. Pharmacological targeting of KDM4B might be an effective therapeutic approach in the treatment of neuroblastoma. Specific compounds against KDM4B are not available. However, development of KDM4B inhibitors would provide more insight into understanding basic and clinical questions in MYCN-mediated cancer.
Funding
This work was supported by the Assisi Foundation of Memphis, the US Public Health Service Childhood Solid Tumor Program Project Grant No. CA23099, the Cancer Center Support Grant No. 21766 from the National Cancer Institute, and by the American Lebanese Syrian Associated Charities (ALSAC).
Supplementary Material
The study funder had no role in design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; nor the decision to submit the manuscript for publication.
We are grateful to Professor Kristian Helin (Biotech Research and Innovation Centre, University of Copenhagen) for the gift of the HA-KDM4B plasmid and to the staff of the Cell and Tissue Imaging Center and Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital for technical assistance. We are grateful to the Vector Core Lab at St. Jude Children’s Research Hospital for making lentivirus vectors and to the Animal Imaging Center for assistance with animal experiments.
References
- 1. Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22(20):2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schulte JH, Horn S, Otto T, et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008;122(3):699–704. [DOI] [PubMed] [Google Scholar]
- 3. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nature Rev Cancer. 2008;8(12):976–990. [DOI] [PubMed] [Google Scholar]
- 4. Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genet Dev. 2010;20(1):91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29(9):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guccione E, Martinato F, Finocchiaro G, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8(7):764–770. [DOI] [PubMed] [Google Scholar]
- 7. Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143(2):313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin CY, Loven J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nie Z, Hu G, Wei G, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walz S, Lorenzin F, Morton J, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511(7510):483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sabo A, Kress TR, Pelizzola M, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511(7510):488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. Embo J. 2006;25(12):2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104(32):13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bjerke L, Mackay A, Nandhabalan M, et al. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov. 2013;3(5):512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. New Engl J Med. 2012;367(7):647–657. [DOI] [PubMed] [Google Scholar]
- 16. Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22(9):1115–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev. Genetics. 2007;8(11):829–833. [DOI] [PubMed] [Google Scholar]
- 18. Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13(5):297–311. [DOI] [PubMed] [Google Scholar]
- 19. van Haaften G, Dalgliesh GL, Davies H, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41(4):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pryor JG, Brown-Kipphut BA, Iqbal A, Scott GA. Microarray comparative genomic hybridization detection of copy number changes in desmoplastic melanoma and malignant peripheral nerve sheath tumor. Am J Dermatopathol. 2011;33(8):780–785. [DOI] [PubMed] [Google Scholar]
- 24. Yang ZQ, Imoto I, Fukuda Y, et al. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60(17):4735–4739. [PubMed] [Google Scholar]
- 25. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23(23):3251–3253. [DOI] [PubMed] [Google Scholar]
- 26. Kawazu M, Saso K, Tong KI, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6(3):e17830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia-Bassets I, Kwon YS, Telese F, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128(3):505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hatzi E, Murphy C, Zoephel A, et al. N-myc oncogene overexpression down-regulates leukemia inhibitory factor in neuroblastoma. Eur J Biochem. 2002;269(15):3732–3741. [DOI] [PubMed] [Google Scholar]
- 29. Das PP, Shao Z, Beyaz S, et al. Distinct and combinatorial functions of jmjd2b/kdm4b and jmjd2c/kdm4c in mouse embryonic stem cell identity. Mol Cell. 2014;53(1):32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fodor BD, Kubicek S, Yonezawa M, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20(12):1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 33. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359(3):742–746. [DOI] [PubMed] [Google Scholar]
- 35. Rothbart SB, Dickson BM, Ong MS, et al. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27(11):1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie S, Jakoncic J, Qian C. UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J Mol Biol. 2012;415(2):318–328. [DOI] [PubMed] [Google Scholar]
- 37. Mestdagh P, Bostrom AK, Impens F, et al. The miR-17–92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010;40(5):762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mendell JT. miRiad roles for the miR-17–92 cluster in development and disease. Cell. 2008;133(2):217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zornig M, Evan GI. Cell cycle: on target with Myc. Curr Biol. 1996;6(12):1553–1556. [DOI] [PubMed] [Google Scholar]
- 40. Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382(6591):511–517. [DOI] [PubMed] [Google Scholar]
- 41. Yeo JC, Ng HH. The transcriptional regulation of pluripotency. Cell Res. 2013;23(1):20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boumahdi S, Driessens G, Lapouge G, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511(7508):246–250. [DOI] [PubMed] [Google Scholar]
- 43. Yuan K, Hong TM, Chen JJ, Tsai WH, Lin MT. Syndecan-1 up-regulated by ephrinB2/EphB4 plays dual roles in inflammatory angiogenesis. Blood. 2004;104(4):1025–1033. [DOI] [PubMed] [Google Scholar]
- 44. Zheng PS, Wen J, Ang LC, et al. Versican/PG-M G3 domain promotes tumor growth and angiogenesis. Faseb J. 2004;18(6):754–756. [DOI] [PubMed] [Google Scholar]
- 45. He S, Liu Z, Oh DY, Thiele CJ. MYCN and the epigenome. Front Oncol. 2013;31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ng SS, Kavanagh KL, McDonough MA, et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448(7149):87–91. [DOI] [PubMed] [Google Scholar]
- 47. Klose RJ, Yamane K, Bae Y, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442(7100):312–316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.