

Abstract
Background:
Lung cancer is the leading cause of cancer-related mortality worldwide. Detection of promoter hypermethylation of tumor suppressor genes in exfoliated cells from the lung provides an assessment of field cancerization that in turn predicts lung cancer. The identification of genetic determinants for this validated cancer biomarker should provide novel insights into mechanisms underlying epigenetic reprogramming during lung carcinogenesis.
Methods:
A genome-wide association study using generalized estimating equations and logistic regression models was conducted in two geographically independent smoker cohorts to identify loci affecting the propensity for cancer-related gene methylation that was assessed by a 12-gene panel interrogated in sputum. All statistical tests were two-sided.
Results:
Two single nucleotide polymorphisms (SNPs) at 15q12 (rs73371737 and rs7179575) that drove gene methylation were discovered and replicated with rs73371737 reaching genome-wide significance (P = 3.3×10–8). A haplotype carrying risk alleles from the two 15q12 SNPs conferred 57% increased risk for gene methylation (P = 2.5×10–9). Rs73371737 reduced GABRB3 expression in lung cells and increased risk for smoking-induced chronic mucous hypersecretion. Furthermore, subjects with variant homozygote of rs73371737 had a two-fold increase in risk for lung cancer (P = .0043). Pathway analysis identified DNA double-strand break repair by homologous recombination (DSBR-HR) as a major pathway affecting susceptibility for gene methylation that was validated by measuring chromatid breaks in lymphocytes challenged by bleomycin.
Conclusions:
A functional 15q12 variant was identified as a risk factor for gene methylation and lung cancer. The associations could be mediated by GABAergic signaling that drives the smoking-induced mucous cell metaplasia. Our findings also substantiate DSBR-HR as a critical pathway driving epigenetic gene silencing.
Lung cancer is the leading cause of cancer-related mortality in both sexes worldwide, mainly because of lack of established early screening strategies (1). The development of this disease over 30 to 40 years in smokers involves field cancerization, characterized as the acquisition of genetic and epigenetic changes in oncogenes and tumor suppressor genes throughout the lung epithelium (2). The epigenetic silencing of tumor suppressor genes by promoter hypermethylation has been recognized as a major and causal event for lung cancer initiation and progression (2). Moreover, the detection of gene methylation in exfoliated cells from the lungs of smokers provides an assessment of the extent of field cancerization and is a validated biomarker for predicting lung cancer risk (2–4).
The precise mechanism by which cigarette carcinogens disrupt the capacity of lung cells to maintain the epigenetic code during DNA replication and repair is largely unknown. Thus, the identification of genetic determinants contributing to the propensity for acquiring gene methylation in lung epithelium of smokers should provide new insights into the mechanisms underlying epigenetic reprogramming during lung carcinogenesis. Importantly, these genetic loci may also contribute to the genetic component affecting the risk for lung cancer that includes risk loci identified in several lung cancer genome-wide association studies (GWAS [5]). Emerging evidence suggested that genome-wide landscaping of the sequence-dependent allele-specific methylation for nonimprinted genes may help pinpoint the functional regulatory polymorphisms that may influence the disease susceptibility (6). Most recently, Shi et al. (7) conducted a genome-wide assessment for methylation quantitative trait loci (meQTL) and found 34 304 cis-meQTLs, mostly localized to CpG sites outside of genes, promoters, and CpG islands, and 585 trans-meQTLs largely overrepresented in promoter CpG islands. A strong enrichment of these meQTL single nucleotide polymorphisms (SNPs) for DNase hypersensitive sites and sequences bound by CCCTC-binding factor (CTCF) or modified histones was also identified in cell lines, although the effect of methylation of these CpG sites on gene transcription regulation warrants future investigation (7). The etiology of tumor development likely requires the acquisition of the silencing of hundreds of critical genes by promoter methylation, with most gene silencing occurring independent of allele-specific methylation and/or meQTL (Leng et al., unpublished data). Previously, we conducted a candidate gene–based study that implicated genetic variation in DNA replication and apoptosis pathways in modifying the propensity for gene methylation in the aerodigestive tract of smokers (8). The current study extends this work by conducting a two-stage GWAS (9) using smokers from two geographically independent cohorts to identify low-penetrance alleles affecting the propensity for acquiring gene methylation in the lungs.
Methods
Study Subjects
Two longitudinal smoker cohorts were used for the GWAS discovery (stage 1, the Lovelace Smokers cohort [LSC]) and replication (stage 2, the Pittsburgh Lung Screening Study [PLuSS]). The LSC has been actively enrolling smokers from the Albuquerque, NM metropolitan area since 2001 (8,10,11). The PLuSS Cohort was established in 2002 to support translational studies of the Pittsburgh Lung Cancer Specialized Programs of Research Excellence (12). A total of 1200 and 718 white (self-reported) smokers from LSC and PLuSS, respectively, were included in this study (Table 1). A detailed description for subject enrollment and collection of information and biological specimens is provided in the Supplementary Methods (available online). Chronic mucous hypersecretion (CMH) phenotype was defined by self-reported cough productive of phlegm for at least three months per year for at least two consecutive years (ie, the standard definition of chronic bronchitis) (13). All participants signed a consent form, and all protocols were approved by the institutional review board at each participating institution.
Table 1.
Characteristics of study populations in LSC and PLuSS*
| Characteristic | LSC (stage 1) | PLuSS (stage 2) |
|---|---|---|
| n | 1163 | 718 |
| Age, y, mean ± SD | 57.0±9.5 | 64.6±5.1 |
| Sex, male, % | 23.6 | 32.6 |
| Current smokers, % | 54.7 | 59.1 |
| Pack-years, mean ± SD | 41.6±21.4 | 55.1±20.7 |
| 10 – 30, % | 35.1 | 9.3 |
| 30 – 45, % | 32.0 | 22.6 |
| 45 – 166, % | 32.9 | 68.1 |
| Methylation prevalence, % | ||
| P16 | 20.7 | 15.6 |
| MGMT | 25.5 | 23.0 |
| DAPK | 14.5 | 13.0 |
| RASSF1A | 1.0 | 0 |
| GATA4 | 34.8 | 32.7 |
| GATA5 | 14.5 | 10.0 |
| PAX5α | 14.5 | 10.7 |
| PAX5β | 7.7 | 5.3 |
| SULF2 | 33.2 | 26.9 |
| PCDH20 | 35.1 | 24.7 |
| DAL1 | 6.8 | 5.7 |
| JPH3 | 23.0 | 7.0 |
| Pulmonary disorders, % | ||
| Chronic obstructive pulmonary disease | 32.7 | 44.6 |
| Chronic mucous hyper-secretion | 30.9 | 26.0 |
* LSC = the Lovelace Smokers Cohort; PLuSS = the Pittsburgh Lung Screening Study cohort; SD = standard deviation.
Gene Promoter Methylation in Sputum
The propensity for gene methylation was defined by the prevalence for methylation of 12 genes determined in cytologically adequate sputum samples (Table 1). Sputum adequacy, defined as the presence of deep lung macrophages or Curschmann’s spiral (14), was assessed by a pathologist. These 12 genes were selected based on our previous studies establishing their association with risk for lung cancer and specificity to methylation in lung epithelial cells (2–4). The performance of the 12-gene panel for predicting risk for lung cancer is comparable with that seen for the seven-gene or 11-gene panels developed in our previous studies (4). Given the low percentage (<3%) of lung epithelial cells in sputum samples that also highly varied between individuals, a two-stage nested methylation-specific polymerase chain reaction (PCR) was used to detect methylated alleles (3,4). Our assay can reproducibly detect one methylated allele in a background of 10 000 unmethylated alleles (3).
GWAS Genotyping and Quality Assurance
The HumanOmni2.5-4v1-H BeadChip (Illumina, San Diego, CA) was used to genotype 2 450 000 SNPs in 1200 white smokers from the LSC. We removed 37 patients because of low call rate (< 95%, n = 7), low heterozygosity (n = 1), low white ancestry (< 85%, n = 2) (Supplementary Figure 1, available online), and high relatedness with other samples (n = 27). Furthermore, SNPs were excluded if they had a call rate of less than 90%, a minor allele frequency (MAF) of less than 0.008, or a P value of less than 10–8 for the Hardy-Weinberg equilibrium test or were on the Y or pseudo-autosomal region of X. The MAF cutoff is a technical one to identify at least 20 heterozygotes for accurate genotype clustering required by GenomeStudio. After quality assessment, 1163 subjects with 1 599 980 SNPs remained in the genetic association analysis.
SNP Selection for Replication
Twelve tag SNPs that were associated with risk for gene methylation with P values of less than or equal to 1×10–5 in the GWAS discovery and MAFs of greater than or equal to 0.1 were selected for replication (Table 2). These SNPs were genotyped using TaqMan genotyping assay (Life Technologies, Carlsbad, CA) in the PLuSS set.
Table 2.
Summary of associations between the 12 SNPs and risk for gene methylation in the LSC and PLuSS studies
| SNP | Chr | Position* | Allele† | MAF‡ | Gene/RNA | LSC (stage 1)§ | PLuSS (stage 2)|| | Combined¶ | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | ||||||
| rs7604698 | 2 | 180078742 | C/A | 0.24 | ZNF385B | 1.83×10–6 | 1.25 (1.14 to 1.38) | .32 | 1.09 (0.92 to 1.29) | — | — |
| rs4459923 | 3 | 151685713 | G/A | 0.40 | TSC22D2 | 7.48×10–7 | 1.24 (1.14 to 1.35) | .27 | 1.09 (0.94 to 1.27) | — | — |
| rs4447788# | 3 | 151701304 | T/C | 0.30 | TSC22D2 | 2.03×10–6 | 1.24 (1.13 to 1.35) | .62 | 1.04 (0.89 to 1.22) | — | — |
| rs2398721 | 5 | 143568607 | C/T | 0.22 | KCTD16 | 4.45×10–6 | 1.28 (1.15 to 1.42) | .99 | 1.00 (0.85 to 1.18) | — | — |
| rs12414981 | 10 | 122732922 | A/G | 0.11 | MIR5694 | 4.85×10–6 | 0.72 (0.63 to 0.83) | .32 | 1.12 (0.90 to 1.38) | — | — |
| rs4751287 | 10 | 132559608 | T/G | 0.49 | MIR378C | 4.30×10–6 | 1.21 (1.12 to 1.31) | .57 | 0.96 (0.84 to 1.11) | — | — |
| rs1580746 | 12 | 86538292 | G/A | 0.39 | MKRN9P | 2.14×10–6 | 1.24 (1.13 to 1.35) | .94 | 1.01 (0.86 to 1.17) | — | — |
| rs73371737 | 15 | 23335869 | C/A | 0.11 | UBE3A | 2.86×10–6 | 1.34 (1.19 to 1.51) | .0076 | 1.37 (1.09 to 1.72) | 3.3×10–8 | 1.35 (1.21 to 1.50) |
| rs7179575** | 15 | 24905945 | C/T | 0.36 | GABRG3 | 1.79×10–5 | 0.82 (0.75 to 0.90) | .058 | 0.87 (0.75 to 1.00) | 3.8×10–6 | 0.83 (0.77 to 0.90) |
| HapAC†† | 0.07 | 1.07×10–6 | 1.79 (1.26 to 2.54) | .0012 | 1.57 (1.35 to 1.82) | 2.5×10–9 | 1.57 (1.35 to 1.82) | ||||
| rs16946867 | 18 | 4460922 | G/A | 0.17 | DLGAP1 | 6.10×10–7 | 0.74 (0.66 to 0.83) | .76 | 0.97 (0.81 to 1.17) | — | — |
| rs767457 | 20 | 22133332 | T/C | 0.36 | CR627206 | 4.16×10–6 | 1.22 (1.12 to 1.33) | .59 | 0.96 (0.83 to 1.11) | — | — |
| rs62202801‡‡ | 20 | 22190904 | C/T | 0.35 | CR627206 | 3.00×10–6 | 1.24 (1.13 to 1.35) | .30 | 0.93 (0.80 to 1.07) | — | — |
* Based on genome build 36. Statistical tests were two-sided. Chr = chromosome; CI = confidence interval; LSC = the Lovelace Smokers Cohort; MAF = minor allele frequency; OR = odds ratio; PLuSS = the Pittsburgh Lung Screening Study cohort; SNP = single nucleotide polymorphism.
† Allele after ‘/’ is the test allele and minor allele.
‡ MAF is from the LSC set. MAF from the PLuSS set shows minimal difference compared with that seen in LSC.
§ Age, sex, current smoking status, and pack-years were included in the generalized estimating equation models for adjustment.
|| Age, sex, current smoking status, pack-years, and pulmonary function were included in the generalized estimating equation models for adjustment.
¶ Meta-analysis was conducted for two 15q12 SNPs and HapAC only.
# R2 is 0.52 with rs4459923.
** R2 is 0.000098 with rs73371737.
†† HapAC is the haplotype allele A-C for rs73371737 and rs7179575 on chromosome 15. The MAF for HapAC is population haplotype frequency estimated using PHASE. Probability for haplotype alleles other than AC was used as reference for comparison. The full results for haplotype-based analysis between rs73371737 and rs7179575 are in Supplementary Table 5 (available online).
‡‡ R2 is 0.69 with rs767457.
Imputation of Chromosome 15
Imputation of chromosome 15 in the LSC was conducted using BEAGLE (version 3.3.2) with phased haplotype data of European (EUR) populations (n for chromosomes = 758, EUR.chr15.phase1_release_v3.20101123) from 1000 Genomes project pilot 1 study as the reference panel. Masked analyses on 20% SNPs on chromosome 15 identified a high correlation of the observed vs expected allelic frequencies (Pearson correlation coefficient = 0.98). The estimated allelic dosages for SNPs (n = 165 451) with dosage R2 of greater than or equal to 0.3 and MAFs of greater than or equal to 0.05 in the LSC and EUR reference populations were included for assessing genetic association with risk for gene methylation.
Gene Expression Analysis
The genotype-expression correlation was conducted using primary human bronchial epithelial cells obtained by bronchoscopy (HBECs, n = 48) and distant normal lung tissues (n = 40). TaqMan real-time PCR was conducted to measure the expression of candidate genes in cDNA using the ΔCT method with β-actin as the endogenous control.
Meta-analysis of Four Lung Cancer Case-Control Studies
Four lung cancer case-control studies from New Mexico, Pittsburgh, MD Anderson Cancer Center (MDACC), and the National Cancer Institute (NCI) were used to evaluate the association between the two 15q12 SNPs and the risk for non–small cell lung cancer (NSCLC) (Supplementary Tables 1 and 2, available online) (10,15–17). The four studies had a detailed collection of smoking intensity. Because the two cohorts used in the two stages of the GWAS comprised moderate and heavy smokers, the association analysis was restricted to moderate and heavy smokers (pack years ≥ 10) of self-reported white ethnicity with a total sample size of 3737 case patients and 3974 control patients. The access to individual-level data was available for all studies except MDACC. Because of the exploratory nature of this analysis, associations were determined between rare homozygote and wild-type homozygote and between heterozygote and wild-type homozygote. The association results from the four studies were combined with a meta-analysis applying the inverse variance weighting method (18). Cochran’s Q statistic was used to test for heterogeneity, and the I2 statistic was used to quantify the proportion of the total variation caused by heterogeneity. Because there was no indication of heterogeneity between studies (P for Q > .10), the fixed-effect model was applied.
Pathway Analysis and Validation
A LD-based clumping approach in PLINK (version 1.06) was used to identify independent candidate genomic intervals that contained SNPs associated with risk for gene methylation. Genes (n = 389) (Supplementary Table 3, available online) located within these candidate intervals were identified and were applied in the pathway analysis using Ingenuity software. Detailed procedures are available in the Supplementary Methods (available online). To validate DNA double-strand break repair by homologous recombination (DSBR-HR) as a critical pathway affecting the risk for gene methylation, a composite risk score (Supplementary Table 4, available online) was generated based on the four DSBR-HR genes (GEN1, ABL1, MRE11A, and RAD51) that contained SNPs associated with the risk for gene methylation in the LSC subjects (n = 89), from which repair capacity had previously been determined (11). DSBR capacity was measured by assessing chromatid breaks in cultured lymphocytes challenged with bleomycin (Supplementary Methods, available online). Only subjects with extreme number of risk alleles (≤ 1 [n = 28] vs ≥ 4 [n=12]) were selected for comparison of the DSBR capacity because of the following four reasons (11,19): 1) LSC subjects with four or more risk alleles had the largest increased risk for gene methylation relative to the subjects with two or three risk alleles (not shown); 2) DNA repair capacity measured in lymphocytes can be affected by multiple factors including genetic component, age of the donor, and smoking history; 3) the functional redundancy of multiple genes within the DSBR-HR pathway and a myriad of post-translational modifications that alter catalytic activities and the specificity of protein interactions; and 4) 11 of the 12 subjects with four or more risk alleles carry at least one risk allele for each of the four SNPs, thus potentially reflecting the largest variation of DSBR capacity in DSBR-HR.
Statistical Analysis
In the GWAS stage 1 study, the genetic association was assessed in 1163 subjects using generalized estimating equations (GEE) (20) with a vector of the methylation status of 12 genes (1 for a methylated gene and 0 for an unmethylated gene) for each individual as the outcome. GEE modeling was conducted under the assumption that the methylation status for each individual gene in the outcome vector was binomially distributed, and a logit link function was used. In addition, the unstructured working correlation structure among the 12 genes was assumed and was incorporated in the GEE modeling for parameter estimation. SNPs were assessed under an additive genetic model. Odds ratios (ORs) and 95% confidence intervals (95% CIs) were calculated to quantify the magnitude of the association per allele. Several clinical variables, including age, sex, and smoking history (smoking status and pack-years) were selected a priori and included in the GEE models for covariable adjustment.
In the PLuSS replication, GEE was applied to assess the association between each individual SNP and risk for gene methylation. Because PLuSS has higher COPD prevalence than LSC, pulmonary function was included in the GEE models for covariable adjustment together with age, sex, and smoking history. Results from the two stages were combined by a meta-analysis using the inverse variance weighting method (18). Simulation studies determined a P value of less than or equal to 5×10–8 for defining an association with genome-wide significance (21).
Logistic regression models were used to evaluate the association between rs73371737 and risk for CMH or NSCLC. Generalized linear models (GLMs) were used to assess the associations between SNPs and gene expression (delta Ct relative to β-actin) in primary HBECs and normal lung tissues, between rs73371737 and pack-years (log transformed) in NSCLC cases from New Mexico and Pittsburgh, and between composite risk score in DSBR-HR pathway (≥ 4 risk alleles versus ≤ 1 risk allele) and cells with chromatid breaks per 100 cells. For analyses with gene expression or cells with chromatid breaks as the phenotype, least square means and standard error of each phenotype were calculated in GLMs. Covariables adjusted in the models are described in the Supplementary Methods (available online). All statistical tests were two-sided. Statistical analyses were conducted in SAS 9.2, R 2.14, and PLINK 1.06.
Results
Comparison of the observed and expected distribution of the association estimates calculated for the GWAS stage 1 study indicated no evidence of population stratification (inflation factors = 1.03) (Supplementary Figure 2, available online). This was further confirmed by inclusion of European ancestry in the genetic association analysis (not shown). The distribution of association P values was skewed from the null distribution with 294 SNPs having P values of less than or equal to 10–4 greater than the 160 expected by chance (Figure 1).
Figure 1.

Manhattan plot for genome-wide association in the Lovelace Smokers Cohort discovery set. The P values for genetic associations for 1 599 980 single nucleotide polymorphisms on autosomal and X chromosomes and mitochondrial genomes that passed the quality assessment are plotted. The label under the x-axis is the chromosome coding with the order of one to 23 and the mitochondria genome (M). Statistical tests were two-sided.
Replication of the 12 SNPs most significantly associated with risk for gene methylation in the LSC was conducted in the PLuSS set. Two SNPs (rs73371737 and rs7179575) that were 1.5Mb apart at the chromosome 15q12 locus were associated with risk for gene methylation with P values of less than .06 in the PLuSS (Table 2). Meta-analysis combining the results from the two stages found that rs73371737 reached the GWAS significance (combined P value = 3.3×10–8, Table 2). The combined odds ratio for rs73371737 in the two cohorts was 1.35 (1.21 - 1.50) per A allele. The association with gene methylation between the two 15q12 SNPs was completely independent (not shown), and this was consistent with no linkage disequilibrium (LD) between these two SNPs (D’ = 0.02, r2 = 0.000098) in whites. The haplotype-based analysis found that each copy of the AC allele (rs73371737 – rs7179575) conferred 57% increased risk for gene methylation with a P value of 2.5×10–9 (Table 2; Supplementary Table 5, available online).
Human chromosome 15q11-13 is a complex locus with a high recombination rate in European populations (Supplementary Figure 3, available online) (22) and contains two brain-specific imprinted genes (UBE3A and ATP10A) and a cluster of three type-A γ-aminobutyric acid (GABAA) receptor subunit genes (GABRB3, GABRA5, and GABRG3) that are not imprinted in any human tissues (23–25). Additional association analyses based on imputation results on chromosome 15 did not identify new variants having association with risk for gene methylation stronger than that seen for rs73371737 and rs7179575 (Figure 2).
Figure 2.
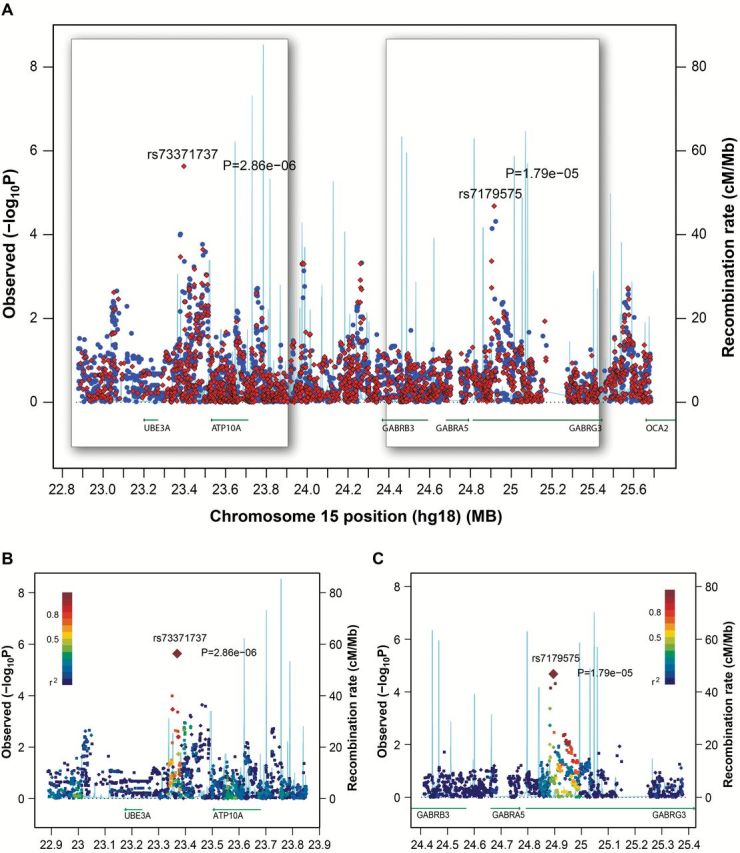
Imputation of chromosome 15q12. Imputation was conducted using phased genotype data of European (EUR) populations from the 1000 Genomes project. A) Genetic associations are plotted for 6557 variants (MAF ≥ 0.05) within the region of Chr15: 22.8 – 25.7Mb (NCBI build 36) that include 4810 imputed variants (dosage R2 is ≥ 0.3). Red diamonds are genotyped variants. Blue dots are imputed variants. The two 3D areas are detailed in panels (B) and (C) for rs73371737 and rs7179575. Diamonds are genotyped variants. Squares are imputed variants. Degree of linkage disequilibrium (LD) is schemed as the gradient of purple to blue color, with purple as perfect LD (R2 = 1) and blue as no LD (R2 = 0). Statistical tests were two-sided.
Among the five genes surrounding the two 15q12 SNPs, GABRB3, GABRA5, and GABRG3 code for three of nineteen subunits for the GABAA receptor, a heteromeric pentameric Cl-selective, ligand-gated ion channel (26). A functional GABAergic signaling pathway has been identified in airway epithelial cells and alveolar type II epithelial cells (27−29). Our recent study showed that smokers with CMH had a statistically significantly higher level of composite gene methylation index detected in sputum (13). These findings supported testing whether variants in 15q12 affect the expression of the three GABAA receptor subunits in the lung. Human HBECs (n = 48) showed expression levels of GABRB3 400- and 3000-fold higher than GABRA5 and GABRG3, respectively (Supplementary Table 6, available online). Thus, the genotype–gene expression association was analyzed for GABRB3 only. HBECs that carried the variant allele of rs73371737 had a statistically significant reduction in GABRB3 expression compared with wild-type homozygotes (P = .0025) (Figure 3A). This association was replicated in 40 normal lung tissues (P = 0.024) (Figure 3A). No statistically significant associations were identified between rs7179575 and GABRB3 expression in HBECs or normal lung tissues (Supplementary Table 7, available online). Furthermore, although expression levels of GABRB3 and ATP10A were moderately correlated (Pearson r = 0.30, P = .038) in human BECs, the associations of rs73371737 with ATP10A and UBE3A expression were not statistically significant (P > .88). Because of the association between rs73371737 and GABRB3 expression in the lungs, we further assessed whether rs73371737 affected the risk for CMH in former smokers. Genetic association analysis was restricted to white former smokers to minimize the reversible effect of smoking on CMH. A consistent association was identified between rs73371737 and risk for CMH in the LSC (772 former smokers, odds ratio [OR] = 1.73, 95% confidence interval [CI] = 1.18 to 2.52, P = .0046) and the PLuSS (311 former smokers, OR = 2.20, 95% CI = 1.04 to 4.62, P = .038), with a combined P value of 5.3×10–4 (Figure 3b; Supplementary Table 8, available online).
Figure 3.

Rs73371737 affects expression of GABRB3 gene, risk for chronic mucous hypersecretion (CMH), and non–small cell lung cancer (NSCLC), and pack-years in NSCLC cases. A) The variant A allele of rs73371737 is associated with reduced gene expression of GABRB3 in primary human bronchial epithelial cells (HBECs) (n = 48, P = .0025) and normal lungs (n = 40, P = .024). * indicates a statistically significant difference in GABRB3 gene expression between CA/AA and CC. Least square mean and standard error of delta Ct (GABRB3 – β-actin) is calculated using generalized linear models with adjustment for current smoking status because primary HBECs and normal lungs from current smokers have a 50% or greater reduction of GABRB3 expression than tissues from former smokers (P < .05). Higher delta Ct indicates lower relative gene expression. B) The variant A allele of rs73371737 is associated with increased risk (odds ratio [OR] = 1.82, P = 5.3×10–4) for CMH in white former smokers in Lovelace Smokers Cohort (n = 772) and PLuSS (n = 311). The size of the square indicates the relative weight in meta-analysis. Compared with wild-type homozygote (CC) of rs73371737, variant homozygote (AA) of rs73371737 is associated with increased risk (OR = 2.1, P = .0043) for NSCLC in 3737 case patients and 3974 control patients from four case-control studies (C2), while the association for heterozygote (CA) of rs73371737 with risk for NSCLC is not statistically significant (C1). Rs73371737 is imputed in National Cancer Institute (dosage R2 = 0.70) and MD Anderson Cancer Center (dosage R2 = 0.79) studies. D) Geometric means for pack-years in NSCLC cases from New Mexico and Pittsburgh by rs73371737 genotypes are shown. Subjects with AA genotype have statistically significantly lower pack-years than subjects with CC (P = .023) or CA (P = .043) genotypes. The asterisk indicates statistically significant differences in pack-years between AA and CC and between AA and CA. Statistical tests were two-sided. CI = confidence interval; CMH = chronic mucous hypersecretion; HBEC = human bronchial epithelial cell; LSC = the Lovelace Smokers Cohort; MDACC = MD Anderson Cancer Center; NCI = the National Cancer Institute; NM = New Mexico; NSCLC = non–small cell lung cancer; OR = odds ratio; PLuSS = the Pittsburgh Lung Screening Study cohort.
Because both gene methylation detected in sputum and CMH diagnosis predict lung cancer risk in prospective studies (3,4,30), SNPs associated with risk for gene methylation and/or CMH should affect the risk for lung cancer. We performed an exploratory analysis using four NSCLC case-control studies conducted in white populations (10,15–17). Of great interest, homozygotes for the rs73371737 A allele were consistently associated with increased risk for NSCLC across the four studies with a combined odds ratio of 2.14 (95% CI = 1.27 to 3.60, P = .0043) (Figure 3C2). However, heterozygotes of rs73371737 did not show increased risk for lung cancer (Figure 3C1). No statistically significant associations were observed for rs7179575 with risk for NSCLC (Supplementary Table 9, available online). Variant homozygotes of rs73371737 have statistically significantly lower pack-years than heterozygotes and wild-type homozygotes (P < .05) (Figure 3d; Supplementary Table 10, available online), providing additional support for higher cancer susceptibility associated with variant homozygotes. Although the epigenetic silencing of tumor suppressor genes by promoter hypermethylation is a major and causal event for lung cancer development, other mechanisms such as gene mutations that inactivate tumor suppressor genes and/or activate oncogenes are also involved in lung carcinogenesis (31). This may explain why a statistically significant association was only observed for rs73371737 variant homozygotes that presumably have the largest effect on gene expression and cancer risk.
The integration of multiple SNPs discovered through GWAS may provide additional insight regarding pathways that influence the susceptibility for acquiring gene methylation. The top 10 pathways identified by Ingenuity software are listed in Figure 4a and Supplementary Table 11 (available online), with DSBR-HR as the most statistically significant one. To validate DSBR as a critical pathway affecting the risk for gene methylation, DSBR capacity in peripheral lymphocytes was compared between LSC subjects with one or fewer 1 (n = 28) and four or more (n = 12) risk alleles from the four DSBR-HR genes (GEN1, ABL1, MRE11A, and RAD51) on our GWAS. Subjects with four or more risk alleles have statistically significantly reduced DSBR capacity compared with subjects with one or fewer risk allele (number of cells with chromatid breaks per 100 metaphases, 23.53±1.23 (least square mean and standard error) vs 18.94±0.72, P = .0063) (Figure 4B), validating DSBR as a major pathway affecting risk for gene methylation (11). This finding is consistent with a prospective study that identified DSBR capacity as a susceptibility biomarker for lung cancer (32). A risk score was also calculated for seven SNPs in CCR5 signaling in the macrophages pathway based on the associations with the risk for gene methylation. This pathway has strikingly different gene components relative to the DSBR-HR pathway. The association between the risk score and DSBR capacity was not statistically significant (P = .81, not shown), supporting the uniqueness of the DSBR-HR pathway in affecting risk for gene methylation by modulating DSBR capacity.
Figure 4.

DNA double-strand break repair by homologous recombination (DSBR-HR) as the major pathway affecting risk for gene methylation. A) The top 10 pathways (P for Fisher’s exact test < .002) that are enriched for single nucleotide polymorphisms (SNPs) associated with risk for gene methylation. Ratio represents the percentage of genes that contain SNPs associated with risk for gene methylation in that pathway and is indicated by yellow squares. A composite risk score is calculated based on SNPs associated with the risk for gene methylation (P < .0005) in four DSBR-HR genes (GEN1, ABL1, MRE11A, and RAD51). B) Smokers (n = 12) with higher composite score have statistically significantly reduced DSBR capacity in peripheral lymphocytes compared with smokers (n = 28) with lower score (P = .0063 using generalized linear model with adjustment for design variables). The asterisk indicates a statistically significant difference in number of cells with chromatid breaks per 100 metaphases between subjects with four or more risk alleles and one or fewer risk alleles. C) A conceptual model that integrates the mucous cell metaplasia, DSB repair, and gene methylation in the lungs of smokers is depicted. The overproduction of mucous associated with variants at 15q12 contributes to inflammation, possibly because of lack of clearance of particles from cigarette smoke and pathogens and activation of immune cells with release of multiple cytokines and eventually leads to increased DNA damage, especially DSBs as the most detrimental ones in the lungs of smokers. Thus, smokers with suboptimal DSBR capacity because of carrying variants in essential DSBR genes would be at a greater risk to acquire gene promoter hypermethylation that silences critical tumor suppressor genes, contributing to lung carcinogenesis. Statistical tests were two-sided.
Discussion
This is the first GWAS to identify genetic determinants for the propensity of acquiring gene promoter hypermethylation in lung epithelium from moderate and heavy smokers, a target population for lung cancer early screening. A functional variant at 15q12 was identified to be associated with the risk for gene methylation at genome-wide significance and was also shown to be a risk factor for lung cancer. The validation of DSBR through this GWAS as a major pathway affecting the susceptibility for gene methylation is likely impacted by the 15q12 variant through its effect on the expression of GABRB3 and associated mucus production that contributes to persistent inflammation, leading to increased DNA damage in the lungs of smokers (Figure 4C). Because epigenetic silencing of tumor suppressor genes by promoter methylation is a major and causal event for lung carcinogenesis, our studies suggest that the GABAergic signaling pathway could be a target for developing novel chemopreventive agents for evaluation in smokers with high risk for lung cancer.
Compelling evidence suggests that extensive DNA damage, manifested through DSBs, could be responsible for the acquisition of aberrant gene promoter methylation during lung carcinogenesis. A highly statistically significant association was observed between DSBR capacity measured in lymphocytes and the propensity for gene methylation detected in sputum from cancer-free smokers from the LSC (11). A subsequent study in the same cohort identified dietary factors including folate, leafy green vegetables, and multivitamin use as protective against the acquisition of gene methylation, possibly through the modulation of DNA repair and/or the reduction of DNA damage induced by tobacco-derived carcinogens because of their antioxidative effect (33). A strong mechanistic link between DSBs and induction of de novo methylation has also been established through in vitro studies. Mortusewicz et al. (34) found that DNMT1 is rapidly recruited to sites of DSBs in mammalian cells following laser microirradiation. Le Gac et al. (35) found that following treatment of cells with doxorubicin, which induces DSBs, DNMT1 is recruited by activated p53 and binds to functional Sp1 sites within promoters of the survivin, cdc2, and cdc25 genes. Moreover, the transcriptional repressor HDAC1 and the repressive chromatin mark H3K9me2 were also found at these promoters following DNA damage (35,36). Subsequent studies showed that following DSBs induced by doxorubicin, DNMT1 complexed with p53 was recruited to the survivin gene promoter followed by de novo methylation and gene silencing (37). Using an experimental model in which a defined DSB was induced in an exogenous promoter construct of the E-cadherin CpG island, O’Hagan et al. (38) identified that normal repair of a DSB can occasionally cause heritable silencing of a CpG island–containing promoter by recruitment of multiple proteins involved in silencing. Furthermore, the stress-related protein SIRT1 contributed to the spreading of the seeding of methylation within the promoter that further stabilized the gene silencing. Cuozzo et al. (39) provided even stronger support for a mechanistic link between DSBs, homologous recombination repair, and gene silencing by DNA methylation. In that study, a recombinant plasmid containing a 1-SCE1 restriction site within one copy of two inactivated tandem repeated green fluorescent protein (GFP) genes was introduced into Hela, or mouse embryonic stem cells. The restriction endonuclease 1-Sce1 was added to the cell to induce a DSB in the 5’ copy of the GFP gene. Rapid gene silencing associated with homologous recombination and DNA hypermethylation of the recombinant gene was detected, which could be blocked by treatment with the demethylating agent, 5-aza-deoxycytidine. Chromatin immunoprecipitation revealed that DNMT1 was bound specifically to the homologous-recombined GFP DNA. Together, these studies substantiate chronic DNA damage and reduced DNA repair capacity as important determinants for inducing gene methylation.
GABA is a major inhibitory neurotransmitter in the central nervous system (CNS) and generates fast inhibition in mature neurons via activation of GABAA receptors (26). GABAA receptors are expressed in nonneuronal cells in organs outside of the CNS, including lung, pancreas, and ovary (40). A functional GABAergic signaling pathway has been identified in the lung epithelial cells from which the adenocarcinomas and squamous cell carcinomas of the lung are derived (27–29). In addition, accumulating evidence suggests that GABAergic signaling is critical for mucous cell metaplasia in mouse and nonhuman primate models challenged with allergen, Il-13, or nicotine (27,28). The study population was comprised of former smokers in the LSC and PLuSS that have maintained abstinence from smoking for an average of 11.3 and 9.3 years, respectively. Thus, the CMH phenotype in former smokers should reflect the heritable change (eg, mucous cell metaplasia) in the lungs caused by the cumulative damage by cigarette smoke. Furthermore, the consistent association seen between rs73371737 and risk for CMH in former smokers from both cohorts is more likely because of its effect on the GABAergic signaling activity by modulating GABRB3 expression. This association between rs73371737, reduced GABRB3 expression, and higher risk for CMH may seem contradictory to the observation that GABAergic signaling drives mucous cell metaplasia in the lungs. However, the reduction of GABRB3 expression associated with rs73371737 could change the subunit composition of GABAA receptors and lead to elevated GABAergic signaling in the lung epithelial cells that drives mucous cell metaplasia (27–29). This premise is supported by the higher sensitivity to GABA for GABAA receptors shown in lung epithelial cells than neurons because of the abundant π subunit in lung epithelial cells (29).
The LD analysis on chromosome 15q12 using SNP and insertion/deletion polymorphism data from European populations in the 1000 Genomes Project did not identify polymorphisms in high LD (r2 ≥ 0.6) with rs73371737 (Figure 2). Genetic association analysis using imputation data on chromosome 15 also failed to identify polymorphisms having stronger association with the risk for gene methylation than rs73371737. These results support rs73371737 as a causal variant for affecting the risk for gene methylation. Rs73371737 is located 1Mb upstream of GABRB3. The consistent association between rs73371737 and GABRB3 expression in HBECs and normal lungs suggests that rs73371737 could regulate GABRB3 expression through an unknown in cis regulatory mechanism. Rs73371737 is located in an intergenic area between UBE3A to ATP10A, where genomic imprinting at 15q11-13 in brain begins attenuation. The genomic imprinting at 15q11-13 is then completely lost around the three GABAA receptor subunit genes (23–25). Thus, the existence of an imprinting/nonimprinting boundary (41) surrounding rs73371737 may be involved in the regulation of GABRB3, a hypothesis that requires future studies. Furthermore, analyses were also conducted to assess the association between rs73371737 and the methylation of CpGs annotated for UBE3A, ATP10A, GABRG3, GABRB3, and GABRA5 at chromosome 15q12 using The Cancer Genome Atlas lung cancer data with no statistically significant associations identified in either normal lungs or lung tumors (not shown). This indicates the high unlikelihood that the association observed in our GWAS is because of the effect of rs73371737 on CpG methylation of the five genes at chromosome 15q12 in the lungs.
Our study provides a proof-of-concept by which the propensity for gene methylation detected in sputum can be used as a functional readout to identify novel variants affecting lung cancer risk in smokers. This premise is further supported by showing that multiple SNPs in high LD with rs3117582 at 6p21.32–33, a known risk locus for lung cancer identified by multiple GWASs (5), were associated with increased risk for gene methylation in the LSC (ORs ≥ 1.13, P ≤ .05) (Supplementary Table 12 and Supplementary Figure 4, available online). Two genes (BAT3 and MSH5) were suggested to be responsible for the genetic association signals observed at this locus for lung cancer (5). BAT3 is implicated in the control of apoptosis and regulating heat shock protein. MSH5 is involved in DNA mismatch repair and meiotic recombination. Thus, the association of the 6p21.32–33 locus with the risk for lung cancer could in part be mediated by the effect of DNA repair and/or DNA damage–induced apoptosis on acquiring gene methylation in the lungs of smokers.
Chromosome 15q12 as a susceptibility locus for acquiring gene promoter hypermethylation in sputum, airway goblet cell metaplasia, and lung cancer was demonstrated in moderate and heavy smokers of white ethnicity. Whether this finding can be generalized to light and never smokers will require future research. Furthermore, it is intriguing that rs73371737 is more prevalent in African populations (MAF = 0.3), but not polymorphic in East Asian populations (Supplementary Table 13, available online), an observation that supports future assessment of the new 15q12 lung cancer risk locus in smokers from other ethnic groups.
Funding
This work was primarily supported by National Cancer Institute grant R01 CA097356 (SAB). The State of New Mexico, as a direct appropriation from the Tobacco Settlement Fund to SAB, through collaboration with University of New Mexico provided initial support to establish the Lovelace Smokers Cohort (LSC). Additional support was provided by National Cancer Institute P30 CA118100. The Pittsburgh Lung Screening Study cohort was established and supported through the National Cancer Institute Specialized Programs of Research Excellence in Lung Cancer grant P50 CA090440 to the University of Pittsburgh (JMS). Genotyping samples from Pittsburgh lung cancer case-control study was conducted using Cancer Biomarkers Facility at the University of Pittsburgh Cancer Institute, which was supported in part by National Institutes of Health award P30CA047904.
Supplementary Material
The study sponsors had no role in study design, the data collection, analysis, or interpretation, the writing of the manuscript, nor the decision to submit for publication.
We thank Dr. Nancy L. Crowley and Mr. Thomas J. Gagliano at Lovelace Respiratory Research Institute (LRRI) for GWAS data management and scientific editing of the figures, Ms. Christin M. Yingling and Amanda M. Snider and Mr. Christopher T. Dacugon at LRRI for DNA isolation, the staff from Lovelace Scientific Resources for recruiting and enrolling study subjects and collecting clinical samples and data, and the New Mexico residents who participate in the LSC.
References
- 1. Youlden DR, Cramb SM, Baade PD. The International Epidemiology of Lung Cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3(8):819–831. [DOI] [PubMed] [Google Scholar]
- 2. Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4(9):707–717. [DOI] [PubMed] [Google Scholar]
- 3. Belinsky SA, Liechty KC, Gentry FD, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66(6):3338–3344. [DOI] [PubMed] [Google Scholar]
- 4. Leng S, Do K, Yingling CM, et al. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res. 2012;18(12):3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Timofeeva MN, Hung RJ, Rafnar T, et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum Mol Genet. 2012;21(22):4980–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tycko B. Mapping allele-specific DNA methylation: a new tool for maximizing information from GWAS. Am J Hum Genet. 2010;86(2):109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi J, Marconett CN, Duan J, et al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nat Commun. 2014;5:3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leng S, Stidley CA, Liu Y, et al. Genetic Determinants for promoter hypermethylation in the lungs of smokers: a candidate gene-based study. Cancer Res. 2012;72(3):707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38(2):209–213. [DOI] [PubMed] [Google Scholar]
- 10. Leng S, Liu Y, Thomas CL, et al. Native American ancestry affects the risk for gene methylation in the lungs of Hispanic smokers from New Mexico. Am J Respir Crit Care Med. 2013;188(9):1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leng S, Stidley CA, Willink R, et al. Double-strand break damage and associated DNA repair genes predispose smokers to gene methylation. Cancer Res. 2008;68(8):3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilson DO, Weissfeld JL, Fuhrman CR, et al. The Pittsburgh Lung Screening Study (PLuSS): outcomes within 3 years of a first computed tomography scan. Am J Respir Crit Care Med. 2008;178(9):956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bruse S, Petersen H, Weissfeld J, et al. Increased methylation of lung cancer-associated genes in sputum DNA of former smokers with chronic mucous hypersecretion. Respir Res. 2014;15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saccomanno G, Archer VE, Auerbach O, Saunders RP, Brennan LM. Development of carcinoma of the lung as reflected in exfoliated cells. Cancer. 1974;33(1):256–270. [DOI] [PubMed] [Google Scholar]
- 15. Buch SC, Diergaarde B, Nukui T, et al. Genetic variability in DNA repair and cell cycle control pathway genes and risk of smoking-related lung cancer. Mol Carcinog. 2012;51(Suppl 1):E11–E20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landi MT, Chatterjee N, Yu K, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85(5):679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40(5):616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta-analysis. In: Egger M, Davey Smith G, Altman DG, eds. Systematic Reviews in Health Care: Meta-analysis in Context. London: BMJ Publishing Group; 2001:285–312. [Google Scholar]
- 19. Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63(2):349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42(1):121–130. [PubMed] [Google Scholar]
- 21. Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15(5):335–346. [DOI] [PubMed] [Google Scholar]
- 22. McCauley JL, Olson LM, Delahanty R, et al. A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am J Med Genet B Neuropsychiatr Genet. 2004;131B(1):51–59. [DOI] [PubMed] [Google Scholar]
- 23. Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17(1):12–13. [DOI] [PubMed] [Google Scholar]
- 24. Hogart A, Patzel KA, LaSalle JM. Gender influences monoallelic expression of ATP10A in human brain. Hum Genet. 2008;124(3):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hogart A, Natarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16(6):691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michels G, Moss SJ. GABAA receptors: properties and trafficking. Cris Rev Biochem Mol Biol. 2007;42(1):3–14. [DOI] [PubMed] [Google Scholar]
- 27. Fu XW, Wood K, Spindel ER. Prenatal nicotine exposure increases GABA signaling and mucin expression in airway epithelium. Am J Respir Cell Mol Biol. 2011;44(2):222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiang YY, Wang S, Liu M, et al. A GABAergic system in airway epithelium is essential for mucus overproduction in asthma. Nat Med. 2007;13(7):862–867. [DOI] [PubMed] [Google Scholar]
- 29. Xiang YY, Chen X, Li J, et al. Isoflurane regulates atypical type-A γ-aminobutyric acid receptors in alveolar type II epithelial cells. Anesthesiology. 2013;118(5):1065–1075. [DOI] [PubMed] [Google Scholar]
- 30. Brenner DR, Boffetta P, Duell EJ, et al. Previous lung diseases and lung cancer risk: a pooled analysis from the International Lung Cancer Consortium. Am J Epidemiol. 2012;176(7):573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014511(7511):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sigurdson AJ, Jones IM, Wei Q, et al. Prospective analysis of DNA damage and repair markers of lung cancer risk from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Carcinogenesis. 2011;32(1):69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stidley CA, Picchi MA, Leng S, et al. Multivitamins, folate, and green vegetables protect against gene promoter methylation in the aerodigestive tract of smokers. Cancer Res. 2010;70(2):568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A. 2005;102(25):8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Le Gac G, Estève PO, Ferec C, Pradhan S. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J Biol Chem. 2006;281(34):24161–24170. [DOI] [PubMed] [Google Scholar]
- 36. Esteve PO, Chin HG, Pradhan S. Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J Biol Chem. 2007; 282(4): 2615–2625. [DOI] [PubMed] [Google Scholar]
- 37. Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A. 2005;102(4):1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4(8):e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cuozzo C, Porcellini A, Angrisano T, et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3(7):e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gladkevich A, Korf J, Hakobyan VP, Melkonyan KV. The peripheral GABAergic system as a target in endocrine disorders. Auton Neurosci. 2006;124(1–2):1–8. [DOI] [PubMed] [Google Scholar]
- 41. Kawamura R, Tanabe H, Wada T, Saitoh S, Fukushima Y, Wakui K. Visualization of the spatial positioning of the SNRPN, UBE3A, and GABRB3 genes in the normal human nucleus by three-color 3D fluorescence in situ hybridization. Chromosome Res. 2012;20(6):659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.