

Abstract
Growth factor receptors dysfunction has previously been correlated with glioma cell proliferation, ability to evade apoptosis, neo-angiogenesis and resistance to therapy. Antineoplastic molecules targeting growth factor receptors are in clinical handling, however the efficacy of these compounds has often been limited by the signaling redundancy. Here, we analyzed the effect of AG1433 (a PDGFR inhibitor), SU1498 (a VEGFR inhibitor) and BEZ235 (a PI3K/Akt/mTOR signaling pathways inhibitor) on glioblastoma cells in vitro. For this study, we used a low passage glioblastoma cell line (GB9B). Assessment of cell number over 72 h showed that the growth rate was 0.3024 and the doubling time of GB9B was 2.29 days. Similar cytotoxic effects were observed by using AG1433 and SU1498 treatment, while dual PI3K/Akt/mTOR inhibition by BEZ235 was more efficient in killing glioblastoma cells than individual PDGFR or VEGFR targeting. In SU1498 treated cells, caspase 3 activity was detected 3 hours after the treatment, while activation of caspase 8 and 9 was detected 48 hours later. AG1433 treatment induced caspase 3, 8 and 9, 3 hours after the treatment. BEZ235 treatment resulted in early caspase 3 and 8 activation, 3 hours after the treatment and an activation of caspase 9, 8 hours later.
Keywords: Glioblastoma, RTK, PDGFR, VEGFR
Introduction
Glioblastomas (GBs) are the most common and aggressive brain tumors. In spite of current approaches in their therapy including: surgical resection, radiotherapy and chemotherapy, the prognosis of these patients remains poor. The median survival rate is of 15 month [1]. These tumors may arise de novo or by malignant progression from astrocytomas. During this process, normal cells suffer some alterations of the expression and activity of growth factors (GF), their receptors or of the subsequent signaling pathways cascades. In order to fight these aberrant GB cells modifications, in the last years were developed a number of new therapies, including small-molecule receptor tyrosine kinases (RTK) inhibitors [2]. Because of their promising results obtained in preclinical experiments some of these drugs were also tested in clinical trials but the results were often limited [3,4]. Among the GF that are overexpressed in GB are platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) [5,6]. PDGF and its receptors (PDGFR’s) are already known to be involved in the differentiation, proliferation and apoptosis of GB cells. In the last years, several targeted therapies against PDGF or PDGFR’s have been tested. One inhibitor used in GB treatment is Imatinib (STI-571; Gleevec). The drug induced death in GB cells [7]. Although it was well tolerated, Imatinib proved limited response in clinical trials, either used alone or in combination with other therapies [8-11].
VEGF and its cognate receptors were shown to be the most important factors involved in the angiogenesis of GB [6]. Until now, targeted therapy against VEGFRs proved only modest results. Drugs like: vatalanib, cediranib, sorafenib, pazopanib, sunitinib or thalidomide were already used in clinical trials either alone or in combination with radiotherapy or other chemotherapeutic agents. In general, the effect of these inhibitors was rather limited [12-18].
PI3K/Akt/mTOR is frequently unregulated in GB. Because of its signaling components the pathway is involved in GB cells proliferation, differentiation, growth, survival and angiogenesis [19].
Therefore inhibition of various proteins involved in this pathway represents an attractive target for specialists. Several PI3K, Akt or mTOR small molecule inhibitors are tested in clinical trials but the results seem to be limited [20,21]. It has been suggested that a dual inhibition of PI3K/Akt/mTOR intracellular signaling may be a better alternative than the inactivation of a single growth factor receptor at the cell surface. Some of these dual inhibitors are now under investigation [22-24].
Among them is BEZ235, a potent PI3K/Akt/mTOR inhibitor [25], that was reported to induce autophagy in GB cells by G1 cell cycle arrest, and VEGF down-regulation [26]. Clinical studies are ongoing with promising preliminary results [24].
The treatment with small molecules that inhibit RTKs and their signal transduction usually induces apoptosis in several types of malignant diseases. In GB cells, both initiator (i.e. caspase 2, 8, 9, 10) and executor (i.e. caspase 3, 6, 7) caspases, are induced after RTKs inactivation [27-29].
In this work we investigated the antiproliferative effect of AG1433 (a PDGFR inhibitor), SU1498 (a VEGFR inhibitor) and of BEZ235 (a dual PI3K/Akt/mTOR inhibitor) in the human low passage GB9B cell line. We used a low passage cell culture because it is known to better preserves the original characteristics of the tumor. In order to determine the apoptotic effect of these small molecule inhibitors on GB9B cells, we decided to determine caspase 3, 8 and 9 activities after the treatment.
Materials and methods
Reagents
Culture medium used was DMEM/Nutrient Mixture F-12 Ham (Sigma Aldrich), Fetal Bovine Serum (FBS) (Gibco by Life TechnologiesTM), DMSO (dimethyl sulfoxide) purchased from Sigma Aldrich, Penicillin/Streptomycin antibiotics (Gibco), Trypsin (Gibco by Life TechnologiesTM), EDTA (Sigma Aldrich), Cell Growth Determination Kit MTT based [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] purchased from Sigma, Phosphate-buffered saline (PBS) purchased from Gibco, Cell Lysis Buffer (Invitrogen), ApoTarget Caspase-3 (CPP32) Colorimetric Protease Assay kit, ApoTarget Caspase-8 (FLICE) Colorimetric Protease Assay kit, ApoTarget Caspase-9 (Mch6/Apaf-3) Colorimetric Protease Assay kit purchased from Invitrogen, Life Tefnologies, USA. SU1498 a selective inhibitor of VEGFR2, purchased from Santa Cruz Biotechnologies, was diluted in DMSO to a stock solution of 100 mM and prepared to working solution by diluting in culture medium, AG1433 (Sigma Aldrich) is an inhibitor of PDGFR-β and a weak inhibitor of angiogenesis and VEGFR2, was prepared in DMSO (stock solution 100 mM) and diluted in culture medium for working solution and BEZ235 a dual inhibitor of PI3K/mTOR (Santa Cruz Biotechnologies) prepared as 100 nM stock solution in DMSO and diluted in culture medium as working solution were used for this study.
Cell culture and drug treatment
GB9B was obtained from Bagdasar Arseni Hospital in Bucharest. GB9B was obtained from a tumor sample of a patient diagnosed with glioblastoma. After a small fragment of the tumor was triturated and filtrated, the sample was centrifugated for 10 minutes and the cells resuspended and cultured using standard procedures. The samples were incubated at 37°C and 5% CO2 in a humidified incubator (CO2 Incubator Innova CO-incubated at 170) in 25 cm2 culture cell bottles. The cells were cultivated in DMEM/ Nutrient Mixture F-12 Ham supplemented with 10% FBS and 2 mM streptomycin/penicillin. The culture cells were transferred one time per week. The low passage glioblastoma control cells (untreated cells) and treated cells either with receptor inhibitors SU1498, AG1433 or with pathway inhibitor BEZ235 at final concentration of (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 µM, respectively nM for BEZ235) was used in the experiment. In the experiment the cells were incubated for, 48 or 72 hours with different drugs. We incubated cells, as control cells, both with culture medium and with 0.01% of DMSO.
Cell doubling time
After seeding a concentration of 2 × 104 of GB9B glioblastoma cells into 6-well plates and cultured in DMEM/Nutrient Mixture F-12 Ham for 72 hours. Medium was replaced every day for the duration of the experiment. The cells were trypsinized and counted every day in a Bürker hemocytometer, using trypan blue to count living cells. Doubling time and growth rate was determined using a free algorithm available online. **http://www.doubling-time.com/compute.ph.
Cell proliferation assay and IC50
We determined the effect of treatment on GB9B using cell proliferation assay, (MTT assay). The assay is based upon the cleavage of the yellow tetrazolium salt MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] into purple formazan crystals by the metabolically active cells. For this experiment we seeded GB9B in 96 well plates (3000/well) and left to attach overnight. Cells were treated for 48 and 72 hours with selective receptors inhibitors SU1948 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 µM), AG1433 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 µM) and BEZ235 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 nM) in triplicate wells. After incubation time MTT assay (Sigma Aldrich) was performed, incubating the 96 well plates with 10 μl MTT for 4 hours at 37°C, after this incubation time we added 50 µl of solubilization reagent. Appropriate control groups with diluents only and blank control were included. The ratio of optical density of treated cells and control cells was measured at 570 nm (492/630 nm) expressing the percentage of surviving cells. To determine IC50 (the concentration that produces cell death in 50% of cells), we used the formula: IC50 = [(50-X)/(Y-X)] × (W-Z) + Z, where X expresses the percent inhibition that is less than 50%, Y expresses the percent inhibition that is equal or higher than 50%, Z is expressed by the concentration that provides ×% inhibition and W is the concentration that provides the concentration of inhibition for Y%. To observe the morphological changes in cell shape we used a 10 × magnification microscopy [30].
Isolation of the cytosolic fraction
After SU 1498, AG1433 or BEZ235 treatment, the cells were lysed in Cell Lysis Buffer (Tris buffered saline containing detergent) incubated on ice for 10 minutes and centrifuged at 10000 × g for 1 minute, afterwards the supernatants were collected. Bradford method was performed to determine the protein concentration and samples of the supernatants containing 50-80 μg of proteins were used for caspase colorimetric assay.
Cell apoptosis assay
Apoptosis was analyzed using ApoTarget Caspase-3 (CPP32) Colorimetric Protease Assay kit, ApoTarget Caspase-8 (FLICE) Colorimetric Protease Assay kit, ApoTarget Caspase-9 (Mch6/Apaf-3) Colorimetric Protease Assay kit using the manufacturer’s recommendation. Caspases are cysteine protease, which exist as inactive pro-forms. By inducing apoptosis they are cleaved to active form. For experimental purposes we seeded into 96-well plates and either treated or left as control (untreated cells). We treated the cells for 3, 8, 24 and 72 hours with AG1433 (10 μM), SU1498 (10 μM) and BEZ235 (10 nM) in triplicate wells. The cells were then trypsinated and the cytosolic fraction was isolated at a concentration of 3 × 106 per sample. After adding 50 µl of 2 × Reaction Buffer/DTT (10 mM dithiothreitol) to each sample and 5 µl of 4 mM IEDT-Pna (Ile-Glu-Thr-Asp/p-nitroanilide) substrate per sample we incubated at 37°C in a dark incubator for 2 hours. The samples were read at an absorbance of 405 nm in a 96-well microplate reader at a spectrophotometer Star Fax-2100 (AWARENESS TECHNOLOGY INC).
Statistical analyses
Data are expressed as mean ± SD (standard deviation) and statistical comparisons were determined using a Student’s t-test, where 0.05 level of probability was consider statistically significant. All experiments were performed in triplicate.
Results
Glioblastoma cell growth patterns, growth rate and doubling time
In this study, we used GB9B glioblastoma cells. This is a low passage GB cell line, which was established from a tumor sample of a patient diagnosed with glioblastoma as described in “Materials and methods” section. The cells were cultured in standard conditions and frozen after passage three. After thawing in standard conditions, adherent monolayer cells showed continuous growth and could recover. The cell growth patterns of GB9B are presented in Figure 1. We determined the growth rate (GR) and doubling time (DT) for these cells. GB9B proliferates with a GR of 0.3024 and a DT of 2.29 days (Figure 1).
Figure 1.
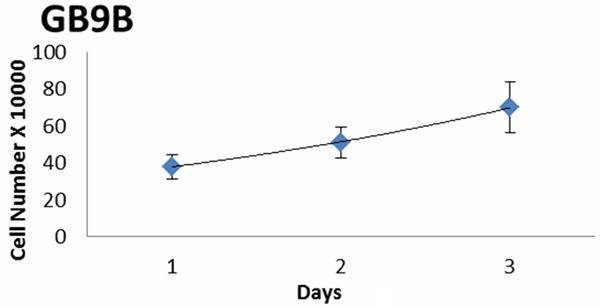
Cell doubling time and growth rate. Glioblastoma GB9B cells were seeded into 6-well plates at a concentration of 2 × 104 and cultured in DMEM/Nutrient Mixture F-12 Ham for 72 hours. We determined the growth rate (GR) and doubling time (DT) for these cells. GB9B proliferates with a GR of 0.3024 and a DT of 2.29 days.
The effect of AG1433 treatment on GB9B cells
It is already demonstrated the overexpression of PDGF ligands and receptors in human gliomas tissues [31-34] or GB cell lines [35]. Both the growth factor and its receptors are capable to mediate GB cells proliferation, differentiation and apoptosis [36]. Those observations led to the attempt to treat GB with small molecule PDGFR kinase inhibitors. In the present study, we analyzed the effect of AG1433 (a PDGFR small molecule inhibitor) on GB9B cells. To examine the effect of the drug on cell viability, exponential growing GB9B cells were exposed to increasing doses of AG1433 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 µM) for three days and the cytotoxic effect of the inhibitor was evaluated by MTT assay as described under “Material and methods” (Figure 2A). The studied cells answered in a dose-time dependent manner to the treatment 0.1 µM of AG1433 reduced cell survival by 3% at 48 hours and 5.3% at 72 hours after the treatment. The highest concentrations of AG1433 (100 µM) significantly decreased cell viability to 50.3 % at 48 hours and to 56.5% at 72 hours (Figure 2A). In order to determine AG1433 cytotoxic potency we calculated the IC50 value which was 51.63126 µM. Cell shape studied under a phase contrast microscope 10 × magnification were indicative for cell membrane alteration, note the rounded cell shape, detachment from substrate and cell fragments induced in cells treated with AG1433 at 72 hours (Figure 2B).
Figure 2.
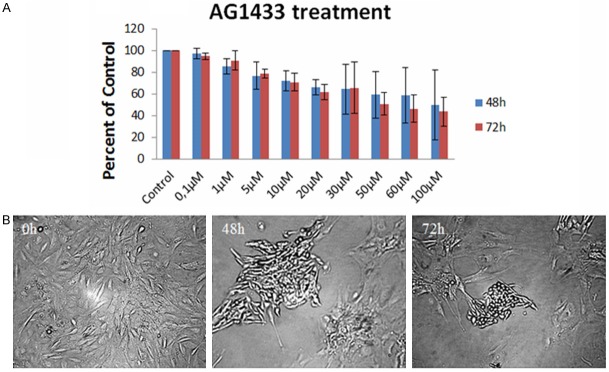
The effect of AG1433 on GB9B glioblastoma cells. GB9B cells were exposed to increasing doses of AG1433 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 µM) for three days. The cytotoxic effect of the inhibitor was evaluated by MTT assay. (A) Results are expressed as percentage of control. Data are mean and standard error of three separate experiments. Microscopy pictures (B) were taken at initial culture day, 48 h and 72 h after the treatment with 10 µM AG1433 (Magnification 10 ×).
The effect of SU1498 treatment on GB9B cells
Angiogenesis and tumorigenesis are two important characteristics of glioblastomas. VEGF and its cognate receptors are involved in the fight against them [37]. The growth factor is also involved in the survival and proliferation of GB [38]. Here we examined the antiproliferative effect of SU1498 (a VEGFR inhibitor) on GB9B cells. The cytotoxicity of the small molecule inhibitor was evaluated by MTT cell proliferation assay after treating the cells with increasing doses of SU1498 similar to AG1433 treatment. We observed that GB9B cells responded in a dose-time dependent manner to the treatment (Figure 3A). While 0.1 µM of SU1498 reduced cell survival only by 5.7% at 48 hours, the prolonged treatment to 72 hours induced 10% cytotoxicity the inhibition was of almost. Highest concentrations of SU1498 (100 µM) significantly decreased cell viability to 52.4% at 48 h and to 62.6% at 72 hours after the treatment (Figure 3A). The IC50 value for SU1498 was 73.5195 µM. Assessment of phase contrast microscopy on cells treated with SU1498 expressed alteration in shape (rounded cell), detachment from substrate and cell fragments after 72 hours treatment (10 × magnification) (Figure 3B).
Figure 3.
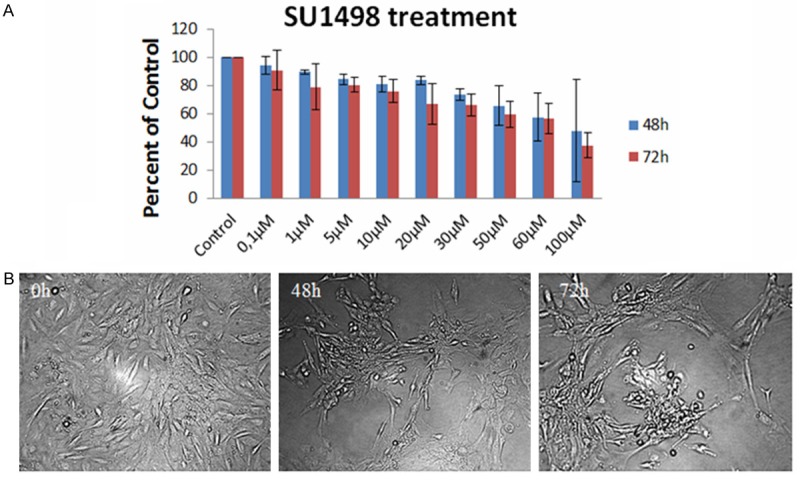
The effect of SU1498 on GB9B glioblastoma cells. GB9B cells were exposed to increasing doses of SU1498 (0, 1, 1, 5, 10, 20, 30, 50, 60 and 100 µM) for three days. The cytotoxic effect of the inhibitor was evaluated by MTT assay. (A) Results are expressed as percentage of control. Data are mean and standard error of three separate experiments. Microscopy pictures (B) were taken at initial culture day, 48 h and 72 h after the treatment with 10 µM SU1498 (Magnification 10 ×).
The effect of BEZ235 treatment on GB9B cells
The alteration of PI3K/Akt/mTOR pathway in human glioblastomas is already known [39]. The activation of this pathway promotes cells growth, proliferation and suppression of apoptosis along with the blockage of autophagy [40-43]. In our experiments we examined the cytotoxicity of BEZ 235 on GB9B cells. Its antiproliferative effect was evaluated by MTT cell proliferation assay, after treating the cells with increasing doses of the inhibitor (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 nM). The doses of BEZ235 used in our experiments were much lower when compared with AG1433 and SU1498. GB9B cells responded in a dose-time dependent manner to the treatment (Figure 4A). 0.1 nM reduced GB cells viability with 17.5% at 48 hours and with 15.3% at 72 hours. The best cytotoxic effect of the small molecule inhibitor was obtained at the concentration of 100 nM. This concentration determined a reduction of cell viability by 55.2% at 48 hours and by 67.5% at 72 hours. We also determined the concentration of BEZ235 to induce 50% cell death (IC50) in GB9B cell line. The IC50 value is very important in evaluation of the drug cytotoxic potency. We found that IC50 value was 34.3 nM (Figure 4A). Assessment of phase contrast microscopy (10x magnification) on cells treated with BEZ235 expressed alteration in cell shape (rounded cell), detachment from cell substrate and cell fragments after 48 hours and 72 hours of treatment (Figure 4B). We concludes that the dual PI3K/Akt/mTOR inhibitor BEZ235, was more efficient in killing glioblastoma cells than individual PDGFR or VEGFR small molecule inhibitors (AG1433 and SU1498), used in our experiments.
Figure 4.
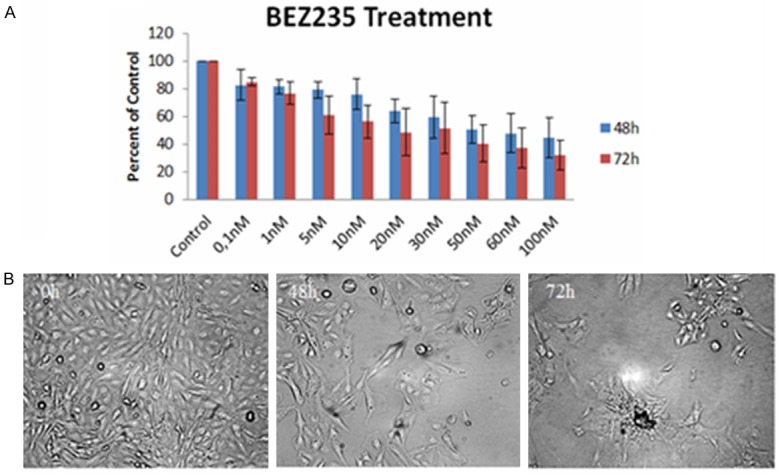
The effect of BEZ235 on GB9B glioblastoma cells. GB9B cells were exposed to increasing doses of BEZ235 (0.1, 1, 5, 10, 20, 30, 50, 60 and 100 nM) for three days. The cytotoxic effect of the inhibitor was evaluated by MTT assay. (A) Results are expressed as percentage of control. Data are mean and standard error of three separate experiments. Microscopy pictures (B) were taken at initial culture day, 48 h and 72 h after the treatment with 10 nM BEZ235 (Magnification 10 ×).
Caspase 3, 8 and 9 activity in GB9B cells after AG1433, SU1498 and BEZ235 treatment
It is already known that GB is characterized by resistance to apoptosis induced by therapy. One mechanism by which the GB cells evade apoptosis may be an impaired caspase function. Caspase 8 and caspase 9 are initiator caspases while caspase 3 is an executioner caspase. These proteases play significant role in GB apoptosis [43]. Therefore is important to know whether the inhibitors used for GB treatment are capable to activate those proteins. In our work we investigated the activity of caspase 3, caspase 8 and caspase 9 after treating GB9B cells with AG1433, SU1498 and BEZ235. Three hours after treating GB cells with AG1433, all three caspases were activated (Figure 5A). At 8 hours after the treatment only caspase 3 and caspase 8 remained activated (Figure 5B), while at 24 hours after the treatment only caspase 3 was still active (Figure 5C). At 48 hours after the AG1433 administration, caspase 3 and caspase 8 were still active (Figure 5D). After treating the GB9B cells with the VEGFR small molecule inhibitor SU1498, caspase 3 activated at 3 hours after administration and remained active at 8 hours and 48 hours after the treatment (Figure 5A, 5B, 5D). Caspase 9 and caspase 8 activated only at 48 hours after SU1498 administration (Figure 5D). When we treated GB9B cells with BEZ235, a dual PI3K/Akt/mTOR small molecule inhibitor, we observed at 3 hours after the treatment that caspase 3 and caspase 8 were activated (Figure 5A). At 8 hours and at 48 hours after the administration of the drug were activated all 3 caspases (Figure 5B and 5D) while at 24 hours after BEZ235 administration only caspase 3 and caspase 8 were active (Figure 5C).
Figure 5.
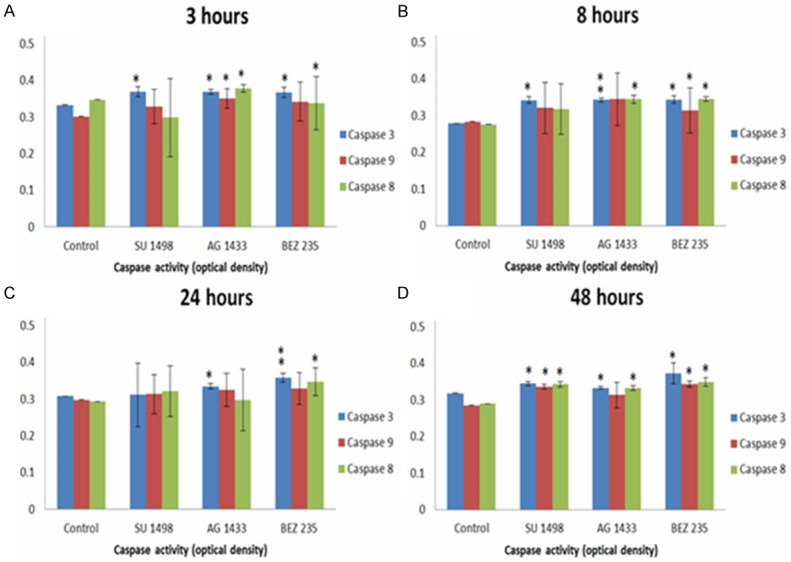
Assessment of the activity of caspase 3, caspase 8 and caspase 9 after treating GB9B cells with AG1433, SU1498 and BEZ235 at 3 hours (A), 8 hours (B), 24 hours (C) and 48 hours (D). The effect was assessed by apoptosis assay. *represents statistically significant values expressed by p-value < 0.05, **represents the highly statistically significant values expressed by p-value < 0.005.
Discussion
Growth factors (GF) and their receptors are proteins capable to stimulate cell proliferation and to regulate the growth of the cells in normal conditions. In neoplastic cells there, an alteration of the expression and activity of GFRs has been reported in the literature. In glioblastomas, a deadly form of brain tumor, the dysfunction of growth factors receptors stimulates cells proliferation, inhibits apoptosis and stimulates neo-angiogenesis.
PDGF family is involved in normal cells differentiation, development of embryos or normal response to tissue damage. The GF’s and their two receptors α and β are known to be overexpressed in glioblastomas [5]. As glioblastomas are highly vascularized, it was also suggested that VEGF and its receptors have an elevated expression in those tumors [44,45].
In the last years, several growth factor family inhibitors were developed for GB. These are targeted drug therapies design to inhibit the RTK’s and their intracellular pathways which are commonly aberrant in GB [46].
In our study we used a low passage GB cell line: GB9B, in order to determine the effect of some receptor tyrosine kinases inhibitors on GB cells survival. The use of cell lines is the first step which leads the research toward clinical trials. In fact, the most important preclinical studies which investigated the cytotoxicity of several targeted drugs used immortalized cell lines. However, the results of these studies often do not correspond to the in vivo observations. One of the causes might be that established tumor cell lines fail to reproduce the tumor heterogeneity. Another cause is that all long-time culture cancer cells accumulate a series of mutations. Unlike immortalized cell lines, low passage tumor cell cultures are capable to preserve original tumor phenotype and genotype [47,48]. The low passage GB9B proliferation rate was 0.3024 and the DT was of 2.29 days.
The targeted therapies against PDGFR have been used in several preclinical cancer studies with promising results. STI-571 (an unspecific PDGFR inhibitor) induced cell death in GB preclinical model [7,49] while in clinical trials the therapeutic effect of the drug was limited. STI-571 was used as a single-agent in phase I and II clinical studies. The results demonstrated that the drug didn’t have significant effects in glioblastoma patients [8,9]. In one study, STI-571 in combination with hydroxyurea showed positive results, however the clinical trial was stopped because of the paucity of results [10,11]. Other clinical studies that use STI-571 and hydroxyurea are still ongoing.
In our study GB cells responded in a dose and time dependent manner to the PDGFR inhibition by AG1433. However, the drug was unable to induce more than 57% cytotoxicity, even in a concentration 100 µM, which is reported to be very high. Thus, the cytotoxic effects of AG1433 on GB9B cells were modest.
Besides PDGFR, vascular endothelial factor receptors (VEGFR) were also associated with the development of brain tumors including GB. Malignant gliomas are characterized by a very active neo-angiogenesis. VEGF and VEGFR’s are the most important mediators of the angiogenic processes in those tumors. The expression of VEGFR was reported to be very low in normal brain cells and therefore targeted therapy against VEGFR was proposed to be a good choice for GB treatment [6]. In practice, targeted therapy against VEGFR proved to have only modest results. Vatalanib is a small molecule VEGF receptor tyrosine kinase that inhibits VEGFRs, as well as PDGFR-β and c-KIT. The drug is most selective for VEGFR-2. The inhibitor was used in combination with imatinib and hydroxyurea in a phase I pharmacokinetic study for recurrent GB treatment. Vatalanib was well tolerated and seemed to enhance the antiangiogenic activity [12]. Similar results were obtained in another phase I clinical trial in which the drug was used in combination with anti-epileptic drugs and radiotherapy and termozolomide for the treatment of new diagnosed GBM patients [13].
Another small molecule inhibitor that targets VEGFR’s is Cediranib a pan-VEGFR, PDGFR, and c-kit inhibitor. In a phase II clinical trial the substance proved to decrease tumor vessel size and permeability, blood volume and blood flow, consistent with vascular normalization. All these effects reversed after the interruption of drug administration [14]. A phase III randomized trial compared the efficacy of Cediranib as monotherapy with the administration of the inhibitor in combination with Lomustine, and with Lomustine alone in patients with recurrent GB. Unfortunately the study did not realize its primary end point to improve the progression-free survival. However Cediranib proved to have clinical efficacy on secondary end points like: time to deterioration in neurologic status and corticosteroid-sparing effects [50]. Sorafenib, another VEGFR2-3 small molecule inhibitor, was also used in a phase I/II clinical study in combination with temsirolimus for the treatment of recurrent GB or gliosarcoma. At the end of the study, only minimal activity in recurrent glioblastoma multiforme was seen at the maximum tolerated dosage of the 2 combined agents [15].
In 2013 bevacizumab in combination with sorafenib was used in a phase II study for in recurrent glioblastoma treatment. The combination did not improve outcome of patients with recurrent glioblastoma when compared with bevacizumab-treated controls [51]. The drug is still under investigation in other clinical studies for GB treatment. Pazopanib is another small molecule inhibitor that was tested in several clinical trials in association with lapatinib [16] where proved only limited results. The drug is under evaluation in ongoing clinical trials for GB. Sunitinib (SU11248), a tyrosine kinase inhibitor targeting VEGFR 1-2, PDGFR α-β, c-kit, bFGF, (CSF-1), FLT3 and RET did no showed activity as monotherapy in glioblastoma [17]. The drug is still investigated in other clinical trials. Thalidomide, an antiangiogenic agent was also tested in clinical trials for recurrent glioblastoma multiforme. The drug was well tolerated and may have some activity in the treatment of recurrent glioblastoma [18]. In another phase II clinical trial, which results were published in 2008, thalidomide in association with irinotecan showed promising activity against recurrent GBM [52]. Here we found that SU1498, a VEGFR inhibitor, had antiproliferative effect on GB9B cells in vitro. The cells responded in a dose-time dependent manner to the treatment. The SU1498 cytotoxic effect is comparable to AG1433 antiproliferative effect on GB9B cells. A very low drug concentration (0.1 μM) induced a significant cell death in GB9B cells.
Although AG1433 and SU1498 had good cytotoxic effect on the proliferation GB9B, their anti-tumor activity was not effective as expected. One explanation of this observation might be the coactivation of alternative tyrosine kinases which leads to the activation of other intracellular signaling cascades. Among them are phosphatidylinositol-3 kinases (PI3K) and protein kinase B (AKT)/mammalian target of rapamycin (mTOR), both very important in gliomagenesis by regulating cell growth and survival. It is already known that PI3K/AKT/mTOR pathway is overactivated in almost 70% of GBM [53-55]. Clinical trials with single PI3K inhibitors for GB are ongoing [56,57]. AKT inhibitors have also entered clinical trials but with minimal single-agent effect [58]. Meantime, clinical trials with mTOR inhibitor rapamycin and its analogs did not have a significant therapeutic effect on cancer diseases [59]. The dual inhibition of both pathways seems to be more effective in GB treatment. Among the dual inhibitors that are currently under investigation is BEZ235. The drug proved to be a potent dual inhibitor studied in preclinical models for various types of solid tumors including gliomas [26,60-63]. It is under investigation in clinical trials in patients with advanced solid tumors and the preliminary results are promising [24].
In our study we also investigated the importance of PI3K/AKT/mTOR pathway on GB9B cells survival. BEZ235 was used to assess the importance of this pathway in GB9B cells proliferation. We found that the inhibition of this pathway was more efficient in killing glioblastoma cells than the individual PDGFR or VEGFR small molecule inhibitors, used in our experiments.
Next, we wanted to determine whether the small molecule inhibitors used in our experiments are capable to activate caspases 3, 8 and 9 in GB9B cells. In fact, in the last years, were designed drugs that are capable to synthetically activate caspases. Among those are small molecule caspases activators [64]. Here we determined the capability of AG1433, SU1498 and BEZ235, all small molecule inhibitors, to activate caspases: 8, 9 and 3 in GB9B cell line. AG1433 activated all three caspases at 3 hours after administration. After 8 hours, only caspase 3 and caspase 8 were still activated. The same effect was observed at 48 hours after the treatment while at 24 hours only caspase 3 was activated. The treatment with the VEGFR inhibitor SU1498 activated only caspase 3 at 3 hours and 8 hours after drug administration while at 48 hours all three caspases were activated. The treatment of GB9B cells with the dual inhibitor BEZ235 determined at 3 hours and 24 hours after administration the activation of caspase 3 and caspase 8. At 8 hours and 24 hours after the drug administration all 3 caspases were activated.
Conclusion
The authors conclude: the present study showed that dual PI3K/Akt/mTOR inhibition by BEZ235 was more efficient in killing glioblastoma cells than individual PDGFR or VEGFR targeted by AG1433 and SU1498 respectively. Glioblastomas remains a very aggressive brain tumor, characterized by heterogeneity and very complex pathogenesis. Their treatment is still a challenge for oncologists and discovery of new targeted drugs is the hope for improving the survival of these patients.
Acknowledgements
The research is financed by: “Program of Excellence in multidisciplinary doctoral and postdoctoral research in chronic diseases”, Contract No. POSDRU/159/1.5/S/133377, co-financed project from the European Social Fund by Operational Sectoral Programme Human Resources Development 2007-2013 and PN-II-ID-PCE-2011-3-1041 UEFISCDI, Romania.
Disclosure of conflict of interest
None.
References
- 1.Darefsky AS, King JT Jr, Dubrow R. Adult glioblastoma multiforme survival in the temozolomide era: a population-based analysis of Surveillance, Epidemiology, and End Results registries. Cancer. 2012;118:2163–2172. doi: 10.1002/cncr.26494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carapancea M, Alexandru O, Fetea AS, Dragutescu L, Castro J, Georgescu A, Popa-Wagner A, Backlund ML, Lewensohn R, Dricu A. Growth factor receptors signaling in glioblastoma cells: therapeutic implications. J Neurooncol. 2009;92:137–147. doi: 10.1007/s11060-008-9753-8. [DOI] [PubMed] [Google Scholar]
- 3.Kilic T, Alberta JA, Zdunek PR, Acar M, Iannarelli P, O’Reilly T, Buchdunger E, Black PM, Stiles CD. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res. 2000;60:5143–5150. [PubMed] [Google Scholar]
- 4.Rich JN, Sathornsumetee S, Keir ST, Kieran MW, Laforme A, Kaipainen A, McLendon RE, Graner MW, Rasheed BK, Wang L, Reardon DA, Ryan AJ, Wheeler C, Dimery I, Bigner DD, Friedman HS. ZD6474, a novel tyrosine kinase inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor, inhibits tumor growth of multiple nervous system tumors. Clin Cancer Res. 2005;11:8145–8157. doi: 10.1158/1078-0432.CCR-05-0319. [DOI] [PubMed] [Google Scholar]
- 5.Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, Ali IU. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550–4553. [PubMed] [Google Scholar]
- 6.Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, Hoffman RM, Kerbel RS. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009;69:7243–7251. doi: 10.1158/0008-5472.CAN-09-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranza E, Mazzini G, Facoetti A, Nano R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J Neurooncol. 2010;96:349–357. doi: 10.1007/s11060-009-9975-4. [DOI] [PubMed] [Google Scholar]
- 8.Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, Frenay M, Rampling R, Stupp R, Kros JM, Heinrich MC, Gorlia T, Lacombe D, van den Bent MJ European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a european organisation for research and treatment of cancer brain tumor group study. J. Clin. Oncol. 2008;26:4659–4665. doi: 10.1200/JCO.2008.16.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razis E, Selviaridis P, Labropoulos S, Norris JL, Zhu MJ, Song DD, Kalebic T, Torrens M, Kalogera-Fountzila A, Karkavelas G, Karanastasi S, Fletcher JA, Fountzilas G. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. 2009;15:6258–6266. doi: 10.1158/1078-0432.CCR-08-1867. [DOI] [PubMed] [Google Scholar]
- 10.Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, Heinrich B, Mayer-Steinacker R, Karup-Hansen A, Fluge O, Nowak A, Mehdorn M, Schleyer E, Krex D, Olver IN, Steinbach JP, Hosius C, Sieder C, Sorenson G, Parker R, Nikolova Z. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. 2010;96:393–402. doi: 10.1007/s11060-009-9976-3. [DOI] [PubMed] [Google Scholar]
- 11.Dong Y, Jia L, Wang X, Tan X, Xu J, Deng Z, Jiang T, Rainov NG, Li B, Ren H. Selective inhibition of PDGFR by imatinib elicits the sustained activation of ERK and downstream receptor signaling in malignant glioma cells. Int J Oncol. 2011;38:555–569. doi: 10.3892/ijo.2010.861. [DOI] [PubMed] [Google Scholar]
- 12.Reardon DA, Egorin MJ, Desjardins A, Vredenburgh JJ, Beumer JH, Lagattuta TF, Gururangan S, Herndon JE 2nd, Salvado AJ, Friedman HS. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer. 2009;115:2188–2198. doi: 10.1002/cncr.24213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerstner ER, Eichler AF, Plotkin SR, Drappatz J, Doyle CL, Xu L, Duda DG, Wen PY, Jain RK, Batchelor TT. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. J Neurooncol. 2011;103:325–332. doi: 10.1007/s11060-010-0390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, Robins HI, Chang SM, Yung WK, Drappatz J, Mehta MP, Levin VA, Aldape K, Dancey JE, Wright JJ, Prados MD, Cloughesy TF, Gilbert MR, Wen PY. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012;14:1511–1518. doi: 10.1093/neuonc/nos264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J, McLendon RE, Suttle AB, Ma B, Curtis CM, Dar MM, de Bono J. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19:900–908. doi: 10.1158/1078-0432.CCR-12-1707. [DOI] [PubMed] [Google Scholar]
- 17.Balana C, Gil MJ, Perez P, Reynes G, Gallego O, Ribalta T, Capellades J, Gonzalez S, Verger E. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Target Oncol. 2014;9:321–329. doi: 10.1007/s11523-014-0305-1. [DOI] [PubMed] [Google Scholar]
- 18.Marx GM, Pavlakis N, McCowatt S, Boyle FM, Levi JA, Bell DR, Cook R, Biggs M, Little N, Wheeler HR. Phase II study of thalidomide in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2001;54:31–38. doi: 10.1023/a:1012554328801. [DOI] [PubMed] [Google Scholar]
- 19.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 20.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 21.Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J, Ronen SM, Kirkpatrick DL, Powis G, Yung WK. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol. 2010;12:559–569. doi: 10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner A, Bendell J, Dolly S, Morgan J, Ware J, Fredrickson J, Mazina K, Lauchle J, Burris H, De Bono J. A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors. ASCO Ann Meet Proc. 2011;29:3020. [Google Scholar]
- 23.Nghiemphu P, Omuro AM, Cloughesy T, Mellinghoff IK, Norden AD, Nguyen L, Rajangam K, Wen PY. A phase I safety and pharmacokinetic study of XL765 (SAR245409), a novel PI3K/TORC1/TORC2 inhibitor, in combination with temozolomide (TMZ) in patients (pts) with newly diagnosed malignant glioma. J Clin Oncol. 2010;28:3085. [Google Scholar]
- 24.Peyton J, Rodon Ahnert J, Burris H, Britten C, Chen L, Tabernero J, Duval V, Rouyrre N, Silva A, Quadt C. A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dispersion system (SDS) sachet, in patients with advanced solid tumors. ASCO Ann Meet Proc. 2011;29:3066. [Google Scholar]
- 25.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 26.Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C, Yung WK. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–2210. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 28.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 29.Yang X, Merchant MS, Romero ME, Tsokos M, Wexler LH, Kontny U, Mackall CL, Thiele CJ. Induction of caspase 8 by interferon gamma renders some neuroblastoma (NB) cells sensitive to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but reveals that a lack of membrane TR1/TR2 also contributes to TRAIL resistance in NB. Cancer Res. 2003;63:1122–1129. [PubMed] [Google Scholar]
- 30.Mahata S, Maru S, Shukla S, Pandey A, Mugesh G, Das BC, Bharti AC. Anticancer property of Bryophyllum pinnata (Lam. ) Oken. leaf on human cervical cancer cells. BMC Complement Altern Med. 2012;12:15. doi: 10.1186/1472-6882-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nister M. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–3219. [PubMed] [Google Scholar]
- 32.Martinho O, Longatto-Filho A, Lambros MB, Martins A, Pinheiro C, Silva A, Pardal F, Amorim J, Mackay A, Milanezi F, Tamber N, Fenwick K, Ashworth A, Reis-Filho JS, Lopes JM, Reis RM. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br J Cancer. 2009;101:973–982. doi: 10.1038/sj.bjc.6605225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, Broniscer A, Wetmore C, Zhang J, Jones C, Ellison DW, Baker SJ. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73:6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nister M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin CH, Schlessinger J, Westermark B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48:3910–3918. [PubMed] [Google Scholar]
- 36.Fomchenko EI, Holland EC. Platelet-derived growth factor-mediated gliomagenesis and brain tumor recruitment. Neurosurg Clin N Am. 2007;18:39–58. doi: 10.1016/j.nec.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Mackenzie F, Ruhrberg C. Diverse roles for VEGF-A in the nervous system. Development. 2012;139:1371–1380. doi: 10.1242/dev.072348. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Z, Kong Y, Yang S, Li M, Wen J, Li L. Upregulation of Flk-1 by bFGF via the ERK pathway is essential for VEGF-mediated promotion of neural stem cell proliferation. Cell Res. 2007;17:73–79. doi: 10.1038/sj.cr.7310126. [DOI] [PubMed] [Google Scholar]
- 39.Sami A, Karsy M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumour Biol. 2013;34:1991–2002. doi: 10.1007/s13277-013-0800-5. [DOI] [PubMed] [Google Scholar]
- 40.Fan QW, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol. 2010;347:279–296. doi: 10.1007/82_2010_67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 42.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 43.Gil del Alcazar CR, Hardebeck MC, Mukherjee B, Tomimatsu N, Gao X, Yan J, Xie XJ, Bachoo R, Li L, Habib AA, Burma S. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin Cancer Res. 2014;20:1235–1248. doi: 10.1158/1078-0432.CCR-13-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parliament MB, Allalunis-Turner MJ, Franko AJ, Olive PL, Mandyam R, Santos C, Wolokoff B. Vascular endothelial growth factor expression is independent of hypoxia in human malignant glioma spheroids and tumours. Br J Cancer. 2000;82:635–641. doi: 10.1054/bjoc.1999.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samoto K, Ikezaki K, Ono M, Shono T, Kohno K, Kuwano M, Fukui M. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res. 1995;55:1189–1193. [PubMed] [Google Scholar]
- 46.Carapancea M, Cosaceanu D, Budiu R, Kwiecinska A, Tataranu L, Ciubotaru V, Alexandru O, Banita M, Pisoschi C, Backlund ML, Lewensohn R, Dricu A. Dual targeting of IGF-1R and PDGFR inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in JNK-1 expressing cells. J Neurooncol. 2007;85:245–254. doi: 10.1007/s11060-007-9417-0. [DOI] [PubMed] [Google Scholar]
- 47.Chang-Liu CM, Woloschak GE. Effect of passage number on cellular response to DNA-damaging agents: cell survival and gene expression. Cancer Lett. 1997;113:77–86. doi: 10.1016/s0304-3835(97)04599-0. [DOI] [PubMed] [Google Scholar]
- 48.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 49.Hagerstrand D, Hesselager G, Achterberg S, Wickenberg Bolin U, Kowanetz M, Kastemar M, Heldin CH, Isaksson A, Nister M, Ostman A. Characterization of an imatinib-sensitive subset of high-grade human glioma cultures. Oncogene. 2006;25:4913–4922. doi: 10.1038/sj.onc.1209497. [DOI] [PubMed] [Google Scholar]
- 50.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, Degroot J, Gattamaneni R, Cher L, Rosenthal M, Payer F, Jurgensmeier JM, Jain RK, Sorensen AG, Xu J, Liu Q, van den Bent M. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galanis E, Anderson SK, Lafky JM, Uhm JH, Giannini C, Kumar SK, Kimlinger TK, Northfelt DW, Flynn PJ, Jaeckle KA, Kaufmann TJ, Buckner JC. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clin Cancer Res. 2013;19:4816–4823. doi: 10.1158/1078-0432.CCR-13-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puduvalli VK, Giglio P, Groves MD, Hess KR, Gilbert MR, Mahankali S, Jackson EF, Levin VA, Conrad CA, Hsu SH, Colman H, de Groot JF, Ritterhouse MG, Ictech SE, Yung WK. Phase II trial of irinotecan and thalidomide in adults with recurrent glioblastoma multiforme. Neuro Oncol. 2008;10:216–222. doi: 10.1215/15228517-2007-060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma--animal models and therapeutic challenges. Brain Pathol. 2009;19:112–120. doi: 10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 55.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 56.Pecchi S, Burger M, Nagel T, Schnell C, Menezes D, Knapp M, Shoemaker K, Wiesmann M, Zaror I, Dorsch M. Biological characterization of NVP-BKM120, a novel inhibitor of phosphoinosotide 3-kinase in Phase I/II clinical trials. Cancer Res. 2010;70:4498–4498. [Google Scholar]
- 57.Grana B, Burris H, Ahnert J, Razak A, De Jonge M, Eskens F, Siu L, Ru Q, Homji N, Demanse D. Oral PI3 kinase inhibitor BKM120 monotherapy in patients (pts) with advanced solid tumors: An update on safety and efficacy. J Clin Oncol. 2011:29. [Google Scholar]
- 58.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 59.Banerji U, Aghajanian C, Raymond E, Kurzrock R, Blanco-Codesido M, Oelmann E, Grinsted L, Burke W, Kaye S, Naing A. First results from a phase I trial of AZD8055, a dual mTORC1 and mTORC2 inhibitor. ASCO Ann Meet Proc. 2011;29:3096. [Google Scholar]
- 60.Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM, Weiss WA. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 62.Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–43. doi: 10.1593/neo.111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Philchenkov A, Zavelevich M, Kroczak TJ, Los M. Caspases and cancer: mechanisms of inactivation and new treatment modalities. Exp Oncol. 2004;26:82–97. [PubMed] [Google Scholar]