

Abstract
The present study investigated the effect of valproic acid (VPA) on the inhibition of RET signaling and induction of apoptosis in human thyroid carcinoma cells. VPA inhibited the viability of ARO and WRO cells and also inhibited cyclin D1 and caused caspase-3 cleavage. VPA decreased the level of RET protein and blocked the activation of RET downstream targets including phosphorylated ERK, phosphorylated AKT, and p70S6K/pS6. VPA induced metabolic stress, activated AMP-activated protein kinase and increased autophagic flux. Pharmacological inhibition of autophagy (chloroquine) augmented VPA-inducible cytotoxicity, suggesting that autophagy was protective in VPA-treated cells. VPA has a wide spectrum of activity against human thyroid carcinoma cells, and its cytotoxicity can be augmented by inhibiting autophagy. Expression of VPA molecular targets in metastatic human thyroid carcinoma cells suggests that VPA has a potential to become a thyroid cancer therapeutic agent.
Keywords: Apoptosis, cytotoxicity, inhibited, oxidative stress, metastasis
Introduction
Histone deacetylase inhibitors (HDIs) comprise of a potent class of antineoplastic agents which enhance the apoptotic activity of DNA [1-3] and tubulin [4] targeting drugs. Usually HDIs promote the activity of the drugs which induce differentiation, growth arrest and cells apoptosis [5-7]. It is reported that imatinib-induced growth arrest and apoptosis in imatinib-sensitive and-resistant leukaemic cell lines is promoted by HDIs including SAHA [8], LAQ824 [9], MS-275 [10] and valproic acid [11]. The present study demonstrates the activity of valproic acid (VPA), to ER stress, autophagy, and oxidative stress in the MTC cells.
Thyroid cancer incidence has increased dramatically over the past few decades due to improvement in diagnostic accuracy and small papillary cancer detection [12]. It is believed that increase in the risk of various cancer types is associated with excess in body weight [13]. The value of height and body mass index has been shown to be related with increased thyroid cancer risk [14]. Among human cancers thyroid cancer which is a common endocrine malignant tumor accounts for 1% cancers. Anaplastic thyroid carcinoma (ATC) has a median survival period of 6 months subsequent to diagnosis and comprises a lethal disease [15-18]. ATC patients have extraglandular spread at the time of diagnosis and most of them develop distant metastases [19,20]. Although medullary thyroid carcinomas (MTC) only comprise 3-5% of thyroid malignancies, but they are responsible for 14% of all thyroid cancer deaths [21,22]. MTC is associated with mutations in the RET protooncogene and induce activation of the mitogenic MAPK kinase/ERK cascade and the antiapoptotic phosphoinositide 3-kinase (PI3K)/AKT pathway [23]. MTC patients with RET-positive are found to have increased probability of persistent disease and a lower survival rate [24]. Prognosis in patients with MTC depends most significantly on the stage of tumor progression at the time of diagnosis [24,25].
Materials and methods
Human thyroid tissue samples
The tissue samples from 28 patients with MTC were obtained from Uniformed Services University of the Health Sciences immediately following extraction and after paraffin-embedding stored at -80°C. Genetic analysis revealed mutations in the RET oncogene 14 cases (5 MTCs were positive for RET643 mutation and 8 MTCs were positive for RET918); 1 MTC harbored H2RAS mutation. The study was approved by the Institutional Review Boards at the Medstar Washington Hospital Center and the Human Use Committee at the Uniformed Services University of the Health Sciences.
Cell lines and culture
ARO (C634W RET mutated) and WRO (M918T RET mutated) human thyroid carcinoma cell lines were purchased from the Japanese Collection of Research Bioresources (JCRB, Shinjuku, Japan). The cells were maintained in minimum essential medium Eagle (MEM) (Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco-BRL) and 1% penicillin/streptomycin. Cultures were maintained at 37°C in humidified atmosphere of 5% CO2.
Cell viability assays
We used MTT assay to examine the viability of ARO and WRO cells. Briefly, the cells were plated at a density of 2.5 × 105 cells per well onto 96-well plates for 12 h. The media were replaced by the fresh media containing either saline (control) or various concentrations of VPA and incubated. After 48 h, 100 μL MTT solutions were added to each well and incubation was continued for 4 h more. Dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals formed by MTT. The microplate reader was used to measure the absorbance at 540 nm. All the analyses were performed three times.
DNA extraction and detection of mutations
The formalin-fixed and paraffin-embedded tissues obtained from MTC samples were subjected to DNA extraction by using QIAamp DNA FFPE tissue kit (QIAGEN). The mutations in RET, H, N, and K RAS were examined by employing the qBiomarker somatic mutation PCR arrays (QIAGEN).
Protein extraction and western blot analysis
The phosphate-buffered saline (PBS) washed cells were lysed with TNN buffer containing 1% Triton X-100, 1% Nonidet P-40, as well as the following protease and phosphatase inhibitors: aprotinin (10 mg/ml), leupeptin (10 mg/ml) (ICN Biomedicals, Inc., Asse-Relegem, Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM), NaVO3 (500 mM) and Na4P2O7 (500 mg/ml) (Sigma-Aldrich). After SDS-PAGE separation the protein were transferred onto PVDF. The blots were immunostained with primary antibodies followed by the secondary antibodies conjugated to horseradish peroxidase and detection by enhanced chemiluminescence reagent (ELPS, Seoul, Korea). The primary antibodies includedphosphorylated (p)-AKT1/2/3 (Ser473), total AKT, pERK1/2, total ERK, p-AMP-activated protein kinase (AMPK), p-p70S6K, total p70S6K, pS6, pERK, p-eLF2α, CCAAT/enhancer-binding protein homologous protein, cyclin-dependent kinase (CDK)-4, cleaved caspase-3, and cleaved poly(ADP-ribose) polymerase (PARP) (Cell Signaling Technology); HSP90, E-cadherin, connexin-43, connexin-31, cyclin D1.
The ECL Plus system (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA) was used for the visualization of bands.
Immunostaining
The tissue-array device containing a 2 mm needle was used to prepare tissue microarrays of the samples obtained from 28 patients with MTC. For tissue microarray sections and cancer cell lines immunohistochemistry was performed anti-HSP90, anti-pERK, and anti-LC3B.
Statistical analysis
The data presented are the mean of ± SD, and ANOVA and an unpaired Student’s t-test was used for statistical analysis. The differences were considered statistically significant at P ≤ 0.05. SPSS software for Windows operating system (version 10.0; SPSS, Chicago, IL, USA) was employed for statistical calculations.
Results
VPA inhibits growth, induces apoptosis, and sensitizes MTC cells to anoikis
We used MTT assay to investigate the effect of VPA on the viability of ARO and WRO cells and observed a concentration and time dependent effect (Figure 1A, 1B). VPA exhibited a significant inhibitory effect on the viability of ARO and WRO cells at a concentration of 20 μM after 48 h. The results of MTT assay were confirmed by employing Vi-CELL cell viability analyzer (Beckman Coulter) based on calculation of cell number. VPA decreased the cyclin D1 expression and induced apoptosis in ARO and WRO cells as revealed by western blot analysis (Figure 1C). In WRO cells VPA cells VPA induced apoptosis at a concentration of 20 μM after 48 h along with caspase-3 and PARP cleavage.
Figure 1.
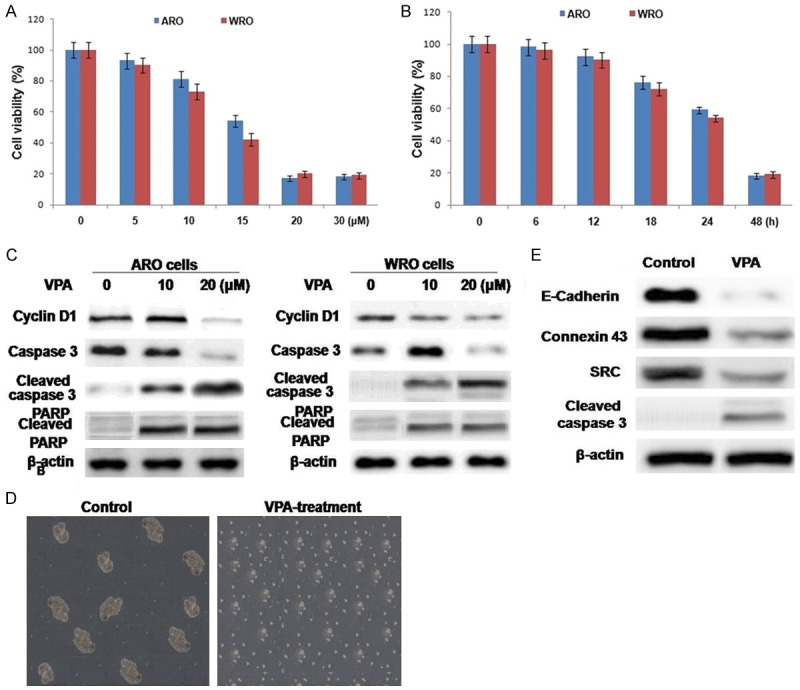
VPA inhibits viability and induces apoptosis in ARO and WRO cells. ARO and WRO cells were incubated with (A) various doses on VPA for the indicated (B) time. (C) Western blot analysis demonstrating the effects of VPA on molecules controlling cell cycle progression and markers of apoptosis. (D) The viability of ARO cells in non-adherent conditions was assessed by JC-1 staining. (E) Western blot analysis demonstrating inhibitory effects of VPA on molecules controlling cell adhesion in ARO cells.
ARO cells were cultured to induce the formation of multicellular spheroids in nonadherent conditions and then treated with VPA. After 48 h, cell viability was inhibited and the cells detached from the spheroid-like structure (Figure 1D). HSP90 client proteins including E-cadherin, SRC, and connexin-43 play a crucial role in the regulation of cell adhesion. The results from western blot analysis revealed reduction in the expression of HSP90 proteins on VPA treatment (Figure 1E). Thus VPA inhibits growth, induces apoptosis, and sensitizes to anoikis the ARO cell line, which harbors a C634W Ret mutation. At similar concentrations, VPA had diminished effects on WRO with the M918T Ret mutation.
VPA inhibits RET signaling in MTC-derived cells
The level of HSP90 expression on mRNA or protein levels was not significantly different in ARO and WRO cell lines and was not affected by VPA (Figure 2A). The protein level of known HSP90 clients (EGFR, SRC, and CDK4) in MTC cells was diminished after treatment with increasing concentration of VPA. VPA decreased the level of RET protein in ARO and WRO cells at concentrations of 5 and 15 μM, respectively (Figure 2B). Treatment with increasing concentrations of VPA (up to 20 μM) did not affect the level of RET mRNA either in ARO and WRO cells. Analysis of RET downstream signaling showed that loss of RET protein was associated with inhibition of pERK in both MTC cells (Figure 2B). The level of total ERK was not affected by VPA in ARO and WRO cells. Inhibition of pAKT was noted only in ARO cells, and the loss of pAKT correlated with a decreased protein level of total AKT. Because RET-dependent activation of mammalian target of rapamycin signaling was demonstrated in MTCs, we examined the phosphorylation status of the mammalian target of rapamycin downstream target, p70S6K. VPA decreased the level of phosphorylated p70S6K in MTC cells in a dose-dependent manner. Immunostaining with anti-HSP90, anti-RET, and anti-pERK demonstrated the inhibition of RET and pERK in VPA-treated ARO cells (Figure 2C).
Figure 2.
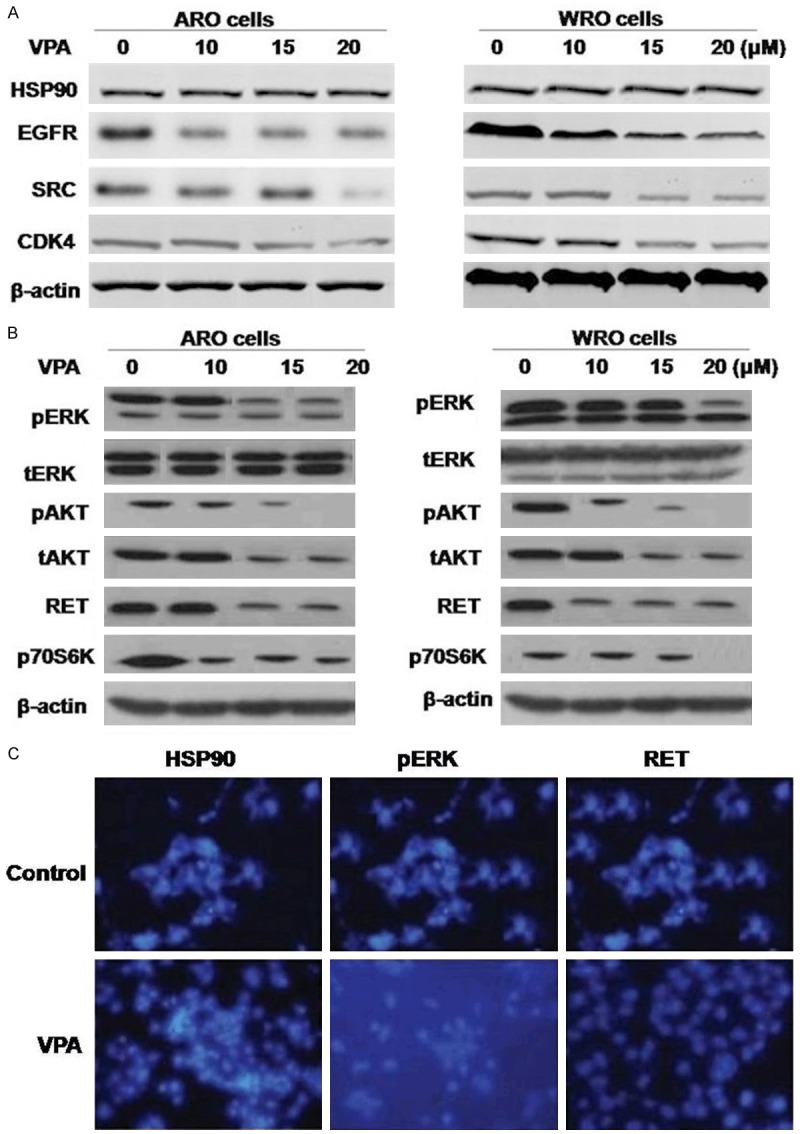
The effects of VPA on activation of RET signaling in thyroid cancer cells. A. ARO and WRO cells were treated with VPA at indicated doses for 48 hours and subjected to Western blot analysis with specific antibody as indicated. HSP90 protein levels remain unchanged; however, HSP90 client protein levels were diminished. B. In ARO and WRO cells inhibition of RET signaling was achieved. C. Immunostaining demonstrating loss of RET and pERK immunoreactivity in ARO cells that were treated with VPA.
VPA induces metabolic stress and autophagy in MTC-derived cells
The investigation of the effect of VPA on molecular markers of ER stress expression in ARO and WRO cells revealed no significant change on pERK and phosphorylated eukaryotic initiation factor 2α expression (Figure 3A). However, the expression of ribosomal protein S6 was inhibited in both the cell lines on VPA treatment after 48 h. VPA treatment also resulted in the AMPK phosphorylation. In addition, the expression of lipidated LC3B (LC3B-II) was induced and p62 was reduced on treatment with VPA in ARO cells (Figure 3B). For the purpose of autophagy confirmation by VPA, autophagy inhibitor chloroquine (CQ) was used. Exposure of the ARO and WRO cells to CQ for 48 h caused accumulation of LC3B-II protein (Figure 3C). Treatment with the combination of VPA and CQ exhibited synergistic effect on cleavage of caspase-3 in ARO and WRO cells (Figure 3C).
Figure 3.
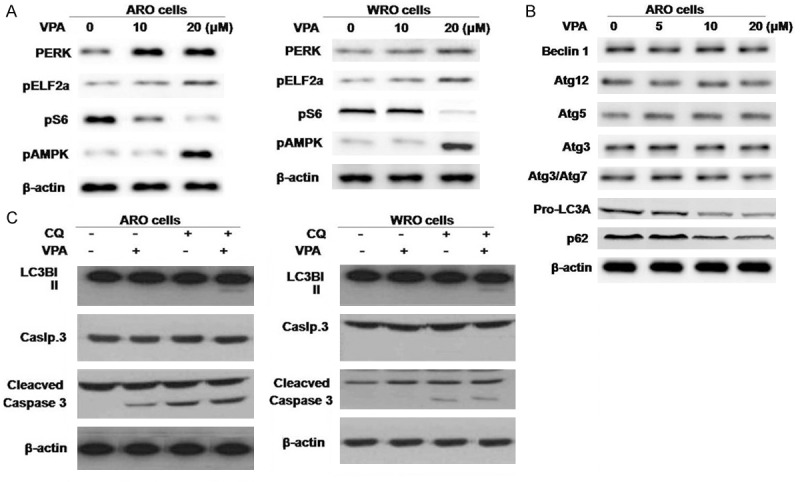
The effects of VPA on ER stress, metabolic stress, and autophagy. A. ARO and WRO cells were treated with VPA for 48 hours and subjected to Western blot analysis with antibody against molecular markers of ER stress and metabolic stress. B. Western blot demonstrating expression of autophagy markers in ARO cells treated with VPA for 48 hours. VPA induced expression of the lower migrating band of LC3B (LC3B-II) and increased the ratio of LC3B-II (16 kDa) vs. LC3BI (18 kDa). p62 expression was inhibited. C. MTC cells were treated with VPA 10 μM for 48 hours, with or without CQ.
Discussion
In this study we examined the effects of VPA on ARO and WRO cells at concentrations ranging from 5 to 30 μM. Our results demonstrated that VPA treatment induced both time- and dose-dependent degradation of RET kinase and subsequently inhibited RET downstream signaling. There was decrease in the expression of HSP90, EGFR, SRC and CDK4 which indicates that VPA exhibits inhibitory effect on HSP90. VPA treatment induced apoptosis in ARO and WRO cells after 48 h. The molecules with the ability to inhibit HSP90 in mitochondria of the carcinoma cells and induce apoptosis are known [25,26]. Our results also showed that VPA induces apoptosis in ARO and WRO cells by targeting the mitochondrial HSP90. It is reported that out of two components of RET, extracellular component has RET634 mutation and intracellular component has RET918 mutation. The sensitivity of RET643-positive ARO and WRO cells to VPA may be associated with its interaction with membrane-bound lipid rafts. Our study demonstrated that VPA targeted the adhesion kinase, Fyn, and Src kinases. The expression of these genes is believed to control the process of cell adhesion [27]. In carcinoma cells ER stress is induced on HSP90 signaling suppression and leads to accumulation of unfolded proteins [13-16]. VPA treated ARO and WRO cells induced inhibition of pS6 and activation of AMPK which indicates that VPA causes metabolic stress.
Metabolic stress is known to induce the process of autophagic flux. Our results revealed that VPA treatment induced autophagic vesicle associated form of LC3B (LC3B-II) in ARO cells after 48 h. It also caused sequestrosome 1 (p62) degradation and simultaneously induced LC3B-II suggesting autophagic flux activation in VPA-treated ARO cells. It is reported that autophagy can trigger cell death, but at the same time it can promote survival under conditions of metabolic stress. To investigate the role of autophagy in ARO and WRO cells, we used an inhibitor of autophagy, CQ. ARO and WRO cells showed apoptosis on treatment with VPA and CQ combination but exhibited no effect on cell viability when used separately. Thus suggesting that inhibition of autophagy with CQ enhanced the effects of VPA against cancer cells. Activation of autophagy may enhance or inhibit tyrosine kinase activity in various types of cancers. Our data suggest that induction of autophagy reduces the load of the toxic proteins in VPA treated ARO and WRO cells and therefore enhances cell survival. In conclusion, VPA can be a potent agent for the thyroid cancer therapeutic agent.
Disclosure of conflict of interest
None.
References
- 1.Catalano MG, Fortunati N, Pugliese M, Poli R, Bosco O, Mastrocola R, Aragno M, Boccuzzi G. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J Endocrinol. 2006;191:465–472. doi: 10.1677/joe.1.06970. [DOI] [PubMed] [Google Scholar]
- 2.Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem. 2004;92:223–237. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- 3.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 2005;65:3815–3822. doi: 10.1158/0008-5472.CAN-04-2478. [DOI] [PubMed] [Google Scholar]
- 4.Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G. Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr Relat Cancer. 2007;14:839–845. doi: 10.1677/ERC-07-0096. [DOI] [PubMed] [Google Scholar]
- 5.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–483. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs. 2002;12:1–13. doi: 10.1097/00001813-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Rosato RR, Grant S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol Ther. 2003;2:30–37. doi: 10.4161/cbt.190. [DOI] [PubMed] [Google Scholar]
- 8.Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101:3236–3239. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 9.Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–5135. [PubMed] [Google Scholar]
- 10.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1. Cancer Res. 2003;63:3637–3645. [PubMed] [Google Scholar]
- 11.Morotti A, Cilloni D, Messa F, Arruga F, Defilippi I, Carturan S, Catalano R, Rosso V, Chiarenza A, Pilatrino C, Guerrasio A, Taulli R, Bracco E, Pautasso M, Baraban D, Gottardi E, Saglio G. Valproate enhances imatinib-induced growth arrest and apoptosis in chronic myeloid leukemia cells. Cancer. 2006;106:1188–1196. doi: 10.1002/cncr.21725. [DOI] [PubMed] [Google Scholar]
- 12.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA. 2006;295:2164–2167. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 13.Calle EE, Thun MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- 14.Dal Maso L, Bosetti C, La Vecchia C, Franceschi S. Risk factors for thyroid cancer: an epidemiological review focused on nutritional factors. Cancer Causes Control. 2009;20:75–86. doi: 10.1007/s10552-008-9219-5. [DOI] [PubMed] [Google Scholar]
- 15.Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G. Emerging molecular therapies of advanced thyroid cancer. Mol Aspects Med. 2010;31:215–226. doi: 10.1016/j.mam.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Fassnacht M, Kreissl MC, Weismann D, Allolio B. New targets and therapeutic approaches for endocrine malignancies. Pharmacol Ther. 2009;123:117–141. doi: 10.1016/j.pharmthera.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Patel KN, Shaha AR. Poorly differentiated and anaplastic thyroid cancer. Cancer Control. 2006;13:119–128. doi: 10.1177/107327480601300206. [DOI] [PubMed] [Google Scholar]
- 18.Sakorafas GH, Sampanis D, Safioleas M. Cervical lymph node dissection in papillary thyroid cancer: current trends, persisting controversies, and unclarified uncertainties. Surg Oncol. 2010;19:e57–e70. doi: 10.1016/j.suronc.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Shimaoka K, Schoenfeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in patients with advanced thyroid carcinoma. Cancer. 1985;56:2155–2160. doi: 10.1002/1097-0142(19851101)56:9<2155::aid-cncr2820560903>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 20.Ahuja S, Ernst H. Chemotherapy of thyroid carcinoma. J Endocrinol Invest. 1987;10:303–310. doi: 10.1007/BF03348135. [DOI] [PubMed] [Google Scholar]
- 21.Hyer SL, Newbold K, Harmer C. Familial medullary thyroid cancer clinical aspects and prognosis. Eur J Surg Oncol. 2005;31:415–419. doi: 10.1016/j.ejso.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Cook M, Yu XM, Chen H. Notch in the development of thyroid C-cells and the treatment of medullary thyroid cancer. Am J Transl Res. 2010;2:119–125. [PMC free article] [PubMed] [Google Scholar]
- 23.Wells SA Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res. 2009;15:7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- 24.Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, Agate L, Vivaldi A, Faviana P, Basolo F, Miccoli P, Berti P, Pacini F, Pinchera A. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93:682–687. doi: 10.1210/jc.2007-1714. [DOI] [PubMed] [Google Scholar]
- 25.Flavin D. Medullary thyroid carcinoma relapse reversed with dichloroacetate: a case report. Oncol Lett. 2010;1:889–891. doi: 10.3892/ol_00000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;3:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 27.Liu G, Beggs H, Jurgensen C, Park HT, Tang H, Gorski J, Jones KR, Reichardt LF, Wu J, Rao Y. Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat Neurosci. 2004;7:1222–1232. doi: 10.1038/nn1331. [DOI] [PMC free article] [PubMed] [Google Scholar]