

Abstract
To investigate the expression profiles of long non-coding RNAs (lncRNAs) in atrial fibrillation (AF), atrial tissues from 3 AF patients and 3 non-AF patients that were collected for lncRNA expression microarray analyses to explore the role of lncRNA in the pathogenesis of AF. Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to identify the main functions of the differentially expressed genes and AF-related pathways. A total of 219 lncRNAs was found to be differentially expressed between AFs and controls. Among them, 156 were upregulated and 63 were downregulated. Eight out of 10 dysregulated lncRNAs such as uc001eqh.1 were validated by quantitative real-time PCR. GO categories, pathway analyses, and interaction network showed a consistent result that differentially expressed genes contribute to the pathogenesis of AF. In conclusion, the findings of our study provide a perspective on lncRNA in AF and the foundation for further study of the biological functions of lncRNAs in AF.
Keywords: Long non-coding RNA, atrial fibrillation, gene expression profile, pathway
Introduction
Atrial fibrillation (AF) is a highly prevalent disease with a significant genetic component [1,2], and is considered as the most common arrhythmia, which can cause or exacerbate heart failure and represents an important risk factor for ischemic stroke [3,4]. From a pathophysiological point of view, AF is characterized by atrial electrical remodeling, mainly mediated by ion-channel alterations [5-7] and structural remodeling (fibrosis and apoptosis), which favors arrhythmia recurrence and maintenance [8,9]. A noticeable feature of the electrical remodeling associated with AF is the abbreviation of the effective refractory period favoring reentry [10-12], primarily due to shortening of atrial action potential duration (APD). Over the last decade, much attention focused on microRNAs (miRNAs), a class of small non-coding RNAs that are involved in various biological and pathological processes. Growing studies have presented interesting connections between miRNAs and AF, and most of these miRNAs promote the electrical or structural remodeling in the atrium [13,14].
More recently, long non-coding RNAs (lncRNAs), generally defined as non-coding RNAs of more than 200 nt in length without known protein-coding function [15], have risen to prominence, with central roles in a diverse range of functions in cell biology [16,17]. In contrast to miRNAs, although an increasing number of lncRNAs have been characterized, the role of lncRNAs in AF has not been investigated. In this study, we preliminary explore the role of lncRNA in the pathogenesis of AF by identifying differentially expressed lncRNAs in AF patients using an lncRNA microarray and subsequently validating the microarray results using real-time quantitative reverse transcription PCR (qRT-PCR) for specific differentially expressed lncRNAs.
Materials and methods
Specimens
Atrial tissues were obtained from the left atrial appendage of 6 patients with rheumatic heart disease during mitral valve replacement, 3 with AF (AF group), 3 without AF (control group). Written informed consent was obtained from patients before surgery, and the study protocol was approved by the Ethics Committee of Taizhou People’s Hospital. All tissues were frozen in liquid nitrogen immediately after surgical resection.
Construction of the lncRNA microarray
Total RNAs were isolated from the six samples using Trizol (Invitrogen, USA) and were quantified using a NanoDrop spectrophotometer (NanoDrop, USA). Input of 100 ng of total RNA was used to generate Cyanine-3 labeled cRNA, according to the Agilent One-Color Microarray-Based Gene Expression Analysis Low for Input Quick Amp Labeling kit (v6.0). The hybridized arrays were washed, fixed and scanned using the Agilent DNA Microarray Scanner (Agilent, USA). Data were extracted using Agilent Feature Extraction software (version 11.0.1.1). Quantile normalization and subsequent data processing were performed using the GeneSpring GX v11.5.1 software package (Agilent, USA). Differentially expressed lncRNAs between the two groups were identified by Fold Change filtering. The threshold set for upregulated lncRNAs was more than five-fold and for downregulated lncRNAs more than three-fold. The lncRNAs discussed in this article were carefully collected from the most authoritative databases, such as RefSeq, UCSC Knowngenes, Ensembl and many related literature.
Validation by qRT-PCR
Total RNA extraction and cDNA transcription were conducted as above. For real-time qRT-PCR, we added 1 µl of cDNA to 12.5 µl of SYBR-Green Gene Expression Master Mix (Applied Biosystems, USA), 10.5 µl of DEPC-treated water and 0.5 µl of reverse and forward primers. cDNA was amplified for 40 cycles on the ABI 7500 Real-Time PCR system (Applied Biosystems, USA). The primers sequences used are listed in Table 1. GAPDH was used as a reference to obtain the relative expression of target lncRNAs which was determined with the comparative cycle threshold (CT) (2-ΔCT) method, in which ΔCT = CTtarget lncRNA-CTGAPDH.
Table 1.
Differentially expressed lncRNAs validated by qRT-PCR
Gene ontology (GO) and pathway analysis for the differentially expressed lncRNAs
Differentially expressed mRNAs screened by Volcano Plot filtering were further analyzed with GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) database to examine the functions of the genes and to find the pathways they participated.
Construction of lncRNA-transcription factor co-expression network
The lncRNA-transcription factor (TF) co-expression regulatory network was constructed by using the test of hypergeometric distribution. The TF correlated with lncRNAs was calculated based on the enrichment. Pearson correlation coefficients between all aberrant lncRNAs and TFs were calculated.
Statistical analysis
All statistical analyses were performed using the Student’s t-test with SPSS software version 17.0 (SPSS Inc, USA). A P value less than 0.05 was considered statistically significant, and all the statistical tests were two-sided. False discovery rate (FDR) was calculated to correct the P value.
Results
LncRNA profile
LncRNA microarrays are powerful tools for studying the biological function of lncRNAs. As showed in Figure 1, a total of 219 lncRNAs were identified to be differentially expressed with fold-change > 2. Among them, 156 and 63 lncRNAs were upregulated and downregulated in AF tissues compared with controls, respectively. According to the microarray data, we selected lncRNAs that were upregulated by more than five-fold and downregulated by more than three-fold. In addition, poorly conserved lncRNAs were excluded. The conservation of lncRNAs was determined using the online Basic Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Ultimately, 50 differentially expressed lncRNAs that were highly conserved were identified. Among them, 28 were upregulated and 22 were downregulated (Table S1). These results indicate that lncRNAs are involved in the pathogenesis of AF.
Figure 1.
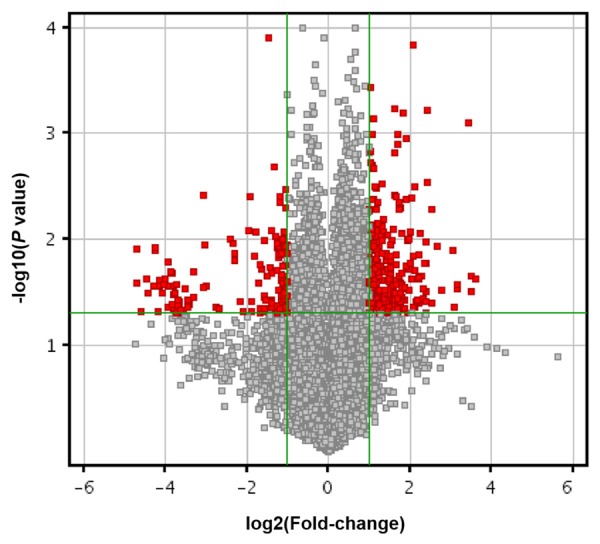
Volcano plot of lncRNAs between AF and controls. The vertical green lines delimit 2.0-fold up-and down-regulation. Red plots represent lncRNAs with > 2.0 fold-change and corrected P value < 0.05.
Gene ontology GO and pathway analysis
The GO project is a collaborative effort to construct and use ontologies to facilitate the biologically meaningful annotation of genes and their products in a wide variety of organisms. We performed GO analysis for mRNAs as shown in Figure 2. Differentially expressed transcripts in biological process were involved in differetiation, extracellular matrix organization, biosynthetic process, protein autophosphorylation, mitochondrion distribution, regulation of gene silencing by miRNA, peptidyl-tyrosine phosphorylation, ephyrin clustering, and T cell activation. Differentially expressed transcripts in cellular component were involved in focal adhesion, membrane, centrosome, intermediate filament cytoskeleton, dendrite, nucleus, stress fiber, and extracellular matrix. Differentially expressed transcripts in molecular function were involved in protein tyrosine kinase activity, calcium ion binding, DNA binding, protein binding, metal ion binding, androgen binding, Rab guanyl-nucleotide exchange factor activity, glycosaminoglycan binding, RNA polymerase III activity, and transforming growth factor beta binding.
Figure 2.
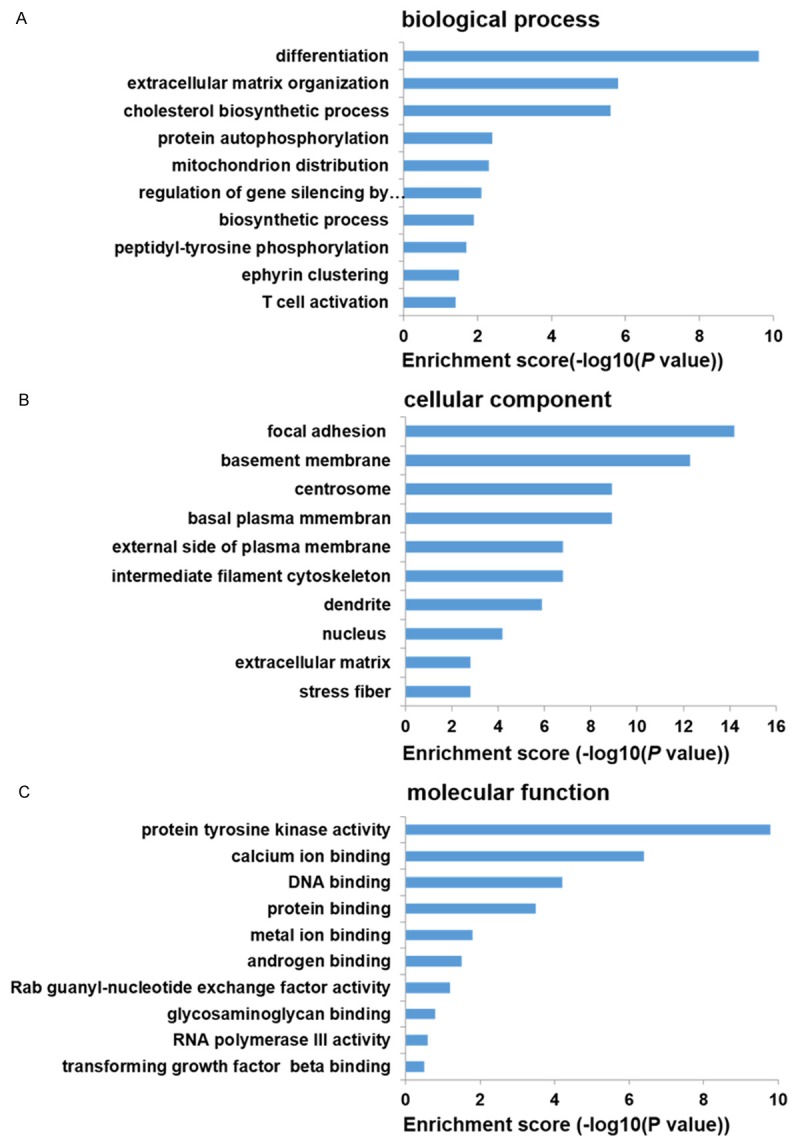
The top 10 results of GO analysis. A. biological process. B. cellular component. C. molecular function.
We further performed the KEGG pathway enrichment analysis for differentially expressed genes to identify pathways and gene networks represented among the sets of protein-coding mRNAs identified in the AF gene expression signature. The most significant pathways were involved in rennin-angiotensin system, calcium signaling pathway, NF-kappa B signaling pathway, ras signaling pathway, primary immunodeficiency, Wnt signaling pathway, axon guidance, olfactory transduction, fanconi anemia pathway, and neuroactive ligand-receptor interaction (Figure 3).
Figure 3.
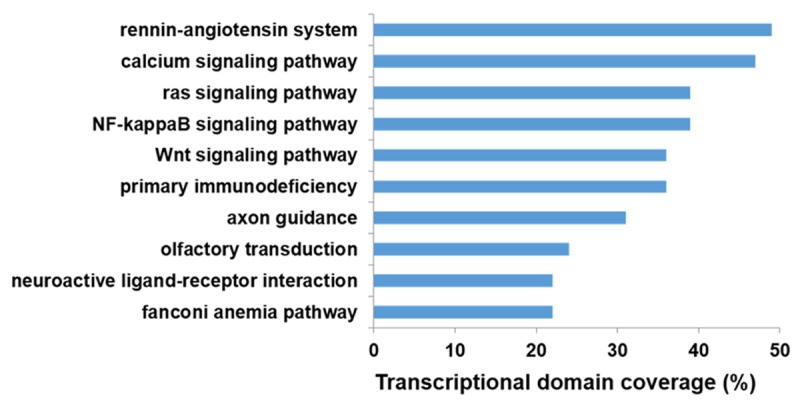
The top 10 results of KEGG pathway enrichment analysis.
Construction of lncRNA-TF co-expression network
By using cumulative hypergeometric test, each lncRNA could correlate with one to tens of TF and could get several pairs of lncRNA-TF and each pair of lncRNA-TF is the enrichment result of several genes. Using P-value < 0.001 and absolute value of correlation coefficient ≥ 0.99, each pair of lncRNA-TF was ranked according the P value. Then we selected the top 100 pairs of lncRNA-TF and constructed the binary network (Figure 4A), which descripted the relationship between lncRNAs and TFs and provided additional information for further studies. We further introduced target genes into the TF-lncRNA network to determine the TF-lncRNA-target genes ternary network based on the results of TF-lncRNA co-expression analyses. We selected the top 300 pairs of lncRNAs, TFs and target genes and constructed the ternary network (Figure 4B).
Figure 4.
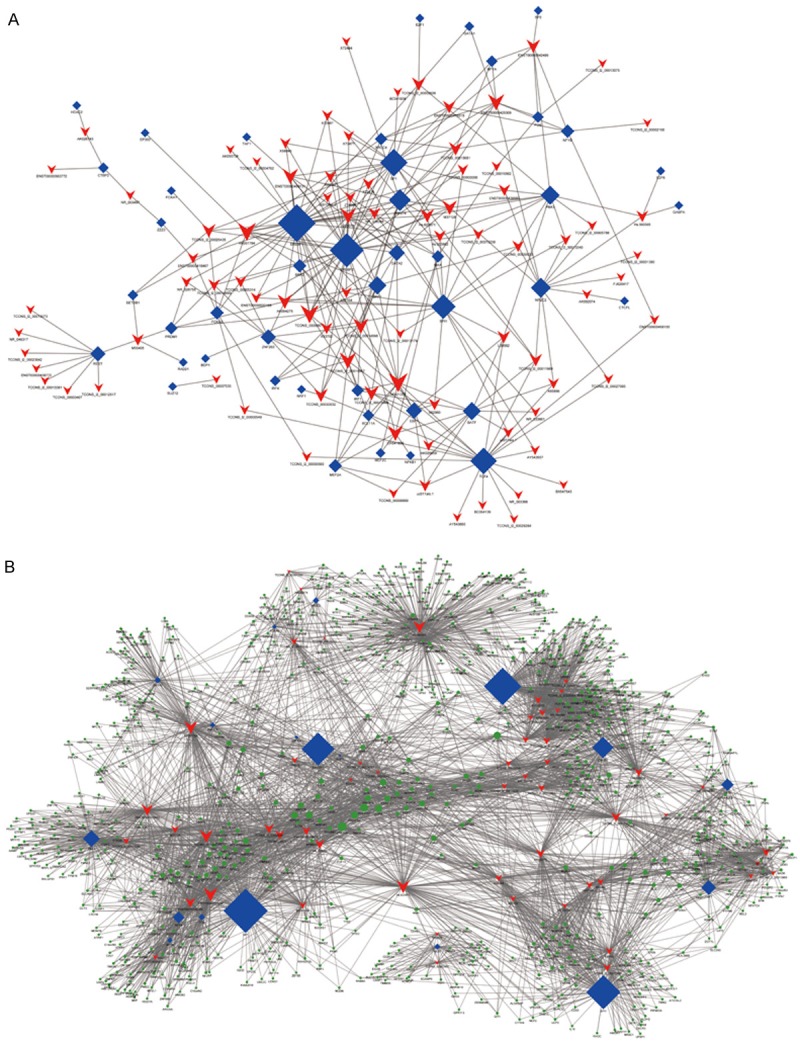
Co-expression networks of AF-associated genes and co-regulated lncRNAs. A. AF-lncRNA network. B. AF-lncRNA-target gene network. Blue diamonds represent as TFs, red plots represent as lncRNAs, and green plots represent as target genes.
Validation of differentially expressed lncRNAs
We randomly chose 5 upregulated lncRNAs (ENST00000575612, uc001eqh.1, BC064139, ENST00000425309, ENST00000500112) and 5 downregulated lncRNAs (TCONS_00006371, Z74666, X85157, X72487, and NR_033661) for qRT-PCR validation to confirm the microarray results. Four out of 5 upregulated lncRNAs (P = 0.024, 0.00035, 0,026 and 0.005 for ENST00000575612, uc001eqh.1, BC064139, and ENST00000425309, respectively) and 4 out of the five downregulated lncRNAs (P = 0.038, 0.0014, 0.00026 and 0.004 for TCONS_00006371, Z74666, X85157, and NR_033661, respectively) showed a significantly different expression (Table 1).
Discussion
AF is the most common sustained arrhythmia, especially in the elderly, and has a significant genetic component. Recently, several independent investigators have demonstrated a functional role for microRNAs in the pathophysiology of this cardiac arrhythmia [13,14]. In contrast to miRNAs, lncRNAs have not been fully investigated. Braveheart (Bvht), a heart-associated lncRNA in mouse, was identified as a critical regulator of cardiovascular commitment from nascent mesoderm, representing the first example of an lncRNA with potential implications in cardiovascular development [18]. The human lncRNA ANRIL has been associated with a locus implicated in cardiovascular disease [19]. In addition, human MIAT was identified as an lncRNA associated with increased risk of myocardial infarction [20]. Although an increasing number of lncRNAs have been characterized, the role of lncRNAs in AF remains largely unknown.
This study focused on determining the lncRNAs expression profile in atrial tissues from AF patients using microarray and preliminary explore the role of lncRNA in the pathogenesis of AF. We identified 50 differentially expressed lncRNAs. In order to confirm whether the differentially expressed lncRNAs profile is more informative and, potentially, a more faithful indicator of AF, we randomly selected five upregulated lncRNAs (ENST00000575612, uc001eqh.1, BC064139, ENST00000425309, ENST00000500112) and five downregulated lncRNAs (TCONS_00006371, Z74666, X85157, X72487, NR_033661) for qRT-PCR validation. qRT-PCR validated four of five upregulated and downregulated lncRNAs. Our results illustrated significant changes of lncRNAs expression in atrial tissues of AF patients and dysregulated lncRNAs may play regulatory roles in the mechanism of AF, which may further provide potential therapeutic targets for prophylaxis and treatment of AF.
The Gene Ontology project provides a controlled vocabulary to describe gene and gene product attributes in any organism. In our study, the main biological processes involving the differentially expressed lncRNAs included many closely connected to atrial remodeling, such as cell motility, protein autophosphorylation and extracellular matrix organization. However, perhaps the most important current challenge is that the knowledge embedded in pathways regarding how various genes interact with each other is not currently exploited. Microarray technology makes it possible to measure the expression levels of almost all the human genes and therefore facilitates the identification of genes and pathways that are related to disease initiation and development. Based on the findings of our study, many pathways are involved in AF, including atrial electrical remodeling and renin-angiotensin system. For instance, the calcium signaling pathway plays an important role in the electrical remodeling of AF and may induce the recurrence of AF. The renin-angiotensin system seems to be involved in the genesis of arrhythmia by the following two mechanisms: 1) the induction of atrial fibrosis and structural remodelling by mitogen-activated protein kinase (MAPK) expression and reduction of collagenase activity; 2) the induction of electrical remodelling by shortening of the atrial effective refractory period (AERP) and of the action potential duration [21].
It has been found that 80% of noncoding RNAs are lncRNAs, which are involved in gene expression and function regulations. Not like mRNA, the functions of most lncRNAs are still not determined. Some of the lncRNAs are proved to be regulated by key TFs and defined a unique collection of functional lncRNAs that are highly conserved and implicated in diverse biological process. In order to investigate the function of lncRNAs in AF, we calculated the TF correlated with lncRNAs based on the enrichment by using cumulative hypergeometric test and further combined differentially expressed lncRNAs with TF to construct a coexpression network. We found that many lncRNAs were significantly correlated with multiple protein-coding genes and may participate in the pathogenesis of AF.
Although we have identified some differentially expressed lncRNAs in AF, it is too early for us to confirm their relationship. Therefore, subgroup analysis of lncRNAs should be performed to explore this relationship in the future. In addition, most differentially expressed lncRNAs have no official Human Genome Nomenclature Committee symbol and their function is still unclear. Further functional studies are required to elucidate their roles in AF.
Acknowledgements
This study was supported by The Six Talents Peak Project of Jiangsu Province, China (grant No. 2013-WSN-020). We thank Wenqi Li for his assistance with statistical analysis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, Nolte IM, Segrè AV, Holm H, Handsaker RE, Westra HJ, Johnson T, Isaacs A, Yang J, Lundby A, Zhao JH, Kim YJ, Go MJ, Almgren P, Bochud M, Boucher G, Cornelis MC, Gudbjartsson D, Hadley D, van der Harst P, Hayward C, den Heijer M, Igl W, Jackson AU, Kutalik Z, Luan J, Kemp JP, Kristiansson K, Ladenvall C, Lorentzon M, Montasser ME, Njajou OT, O’Reilly PF, Padmanabhan S, St Pourcain B, Rankinen T, Salo P, Tanaka T, Timpson NJ, Vitart V, Waite L, Wheeler W, Zhang W, Draisma HH, Feitosa MF, Kerr KF, Lind PA, Mihailov E, Onland-Moret NC, Song C, Weedon MN, Xie W, Yengo L, Absher D, Albert CM, Alonso A, Arking DE, de Bakker PI, Balkau B, Barlassina C, Benaglio P, Bis JC, Bouatia-Naji N, Brage S, Chanock SJ, Chines PS, Chung M, Darbar D, Dina C, Dörr M, Elliott P, Felix SB, Fischer K, Fuchsberger C, de Geus EJ, Goyette P, Gudnason V, Harris TB, Hartikainen AL, Havulinna AS, Heckbert SR, Hicks AA, Hofman A, Holewijn S, Hoogstra-Berends F, Hottenga JJ, Jensen MK, Johansson A, Junttila J, Kääb S, Kanon B, Ketkar S, Khaw KT, Knowles JW, Kooner AS, Kors JA, Kumari M, Milani L, Laiho P, Lakatta EG, Langenberg C, Leusink M, Liu Y, Luben RN, Lunetta KL, Lynch SN, Markus MR, Marques-Vidal P, Mateo Leach I, McArdle WL, McCarroll SA, Medland SE, Miller KA, Montgomery GW, Morrison AC, Müller-Nurasyid M, Navarro P, Nelis M, O’Connell JR, O’Donnell CJ, Ong KK, Newman AB, Peters A, Polasek O, Pouta A, Pramstaller PP, Psaty BM, Rao DC, Ring SM, Rossin EJ, Rudan D, Sanna S, Scott RA, Sehmi JS, Sharp S, Shin JT, Singleton AB, Smith AV, Soranzo N, Spector TD, Stewart C, Stringham HM, Tarasov KV, Uitterlinden AG, Vandenput L, Hwang SJ, Whitfield JB, Wijmenga C, Wild SH, Willemsen G, Wilson JF, Witteman JC, Wong A, Wong Q, Jamshidi Y, Zitting P, Boer JM, Boomsma DI, Borecki IB, van Duijn CM, Ekelund U, Forouhi NG, Froguel P, Hingorani A, Ingelsson E, Kivimaki M, Kronmal RA, Kuh D, Lind L, Martin NG, Oostra BA, Pedersen NL, Quertermous T, Rotter JI, van der Schouw YT, Verschuren WM, Walker M, Albanes D, Arnar DO, Assimes TL, Bandinelli S, Boehnke M, de Boer RA, Bouchard C, Caulfield WL, Chambers JC, Curhan G, Cusi D, Eriksson J, Ferrucci L, van Gilst WH, Glorioso N, de Graaf J, Groop L, Gyllensten U, Hsueh WC, Hu FB, Huikuri HV, Hunter DJ, Iribarren C, Isomaa B, Jarvelin MR, Jula A, Kähönen M, Kiemeney LA, van der Klauw MM, Kooner JS, Kraft P, Iacoviello L, Lehtimäki T, Lokki ML, Mitchell BD, Navis G, Nieminen MS, Ohlsson C, Poulter NR, Qi L, Raitakari OT, Rimm EB, Rioux JD, Rizzi F, Rudan I, Salomaa V, Sever PS, Shields DC, Shuldiner AR, Sinisalo J, Stanton AV, Stolk RP, Strachan DP, Tardif JC, Thorsteinsdottir U, Tuomilehto J, van Veldhuisen DJ, Virtamo J, Viikari J, Vollenweider P, Waeber G, Widen E, Cho YS, Olsen JV, Visscher PM, Willer C, Franke L Global BPgen Consortium; CARDIoGRAM Consortium; Erdmann J, Thompson JR PR GWAS Consortium; Pfeufer A QRS GWAS Consortium; Sotoodehnia N QT-IGC Consortium; Newton-Cheh C CHARGE-AF Consortium. Ellinor PT, Stricker BH, Metspalu A, Perola M, Beckmann JS, Smith GD, Stefansson K, Wareham NJ, Munroe PB, Sibon OC, Milan DJ, Snieder H, Samani NJ, Loos RJ. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45:621–31. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santulli G, Iaccarino G, De Luca N, Trimarco B, Condorelli G. Atrial fibrillation and microRNAs. Front Physiol. 2014;5:15. doi: 10.3389/fphys.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas IC, Sorrentino MJ. Bleeding risk prediction models in atrial fibrillation. Curr Cardiol Rep. 2014;16:432. doi: 10.1007/s11886-013-0432-9. [DOI] [PubMed] [Google Scholar]
- 4.Conen D, Chae CU, Glynn RJ, Tedrow UB, Everett BM, Buring JE, Albert CM. Risk of death and cardiovascular events in initially healthy women with new-onset atrial fibrillation. JAMA. 2011;305:2080–2087. doi: 10.1001/jama.2011.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brundel BJ, Van Gelder IC, Henning RH, Tuinenburg AE, Wietses M, Grandjean JG, Wilde AA, Van Gilst WH, Crijns HJ. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: differential regulation of protein and mRNA levels for K+ channels. J Am Coll Cardiol. 2001;37:926–932. doi: 10.1016/s0735-1097(00)01195-5. [DOI] [PubMed] [Google Scholar]
- 6.Santulli G, D’Ascia SL, D’Ascia C. Development of atrial fibrillation in recipients of cardiac resynchronization therapy: the role of atrial reverse remodelling. Can J Cardiol. 2012;28:245, e17. doi: 10.1016/j.cjca.2011.11.001. author reply 245, e17-8. [DOI] [PubMed] [Google Scholar]
- 7.Xie W, Santulli G, Guo X, Gao M, Chen BX, Marks AR. Imaging atrial arrhythmic intracellular calcium in intact heart. J Mol Cell Cardiol. 2013;64:120–123. doi: 10.1016/j.yjmcc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell. 2011;42:84–95. doi: 10.1016/j.molcel.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santulli G, D’Ascia C. Atrial remodelling in echocardiographic super-responders to cardiac resynchronization therapy. Heart. 2012;98:517. doi: 10.1136/heartjnl-2012-301731. author reply 517. [DOI] [PubMed] [Google Scholar]
- 10.D’Ascia SL, Santulli G, Liguori V, Marino V, Arturo C, Chiariello M, D’Ascia C. Advanced algorithms can lead to electrocardiographic misinterpretations. Int J Cardiol. 2010;141:e34–36. doi: 10.1016/j.ijcard.2008.11.144. [DOI] [PubMed] [Google Scholar]
- 11.D’Ascia SL, D’Ascia C, Marino V, Lombardi A, Santulli R, Chiariello M, Santulli G. Cardiac resynchronisation therapy response predicts occurrence of atrial fibrillation in non-ischaemic dilated cardiomyopathy. Int J Clin Pract. 2011;65:1149–1155. doi: 10.1111/j.1742-1241.2011.02732.x. [DOI] [PubMed] [Google Scholar]
- 12.Kapur S, Macrae CA. The developmental basis of adult arrhythmia: atrial fibrillation as a paradigm. Front Physiol. 2013;4:221. doi: 10.3389/fphys.2013.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson K, Wakili R, Ordog B, Clauss S, Chen Y, Iwasaki Y, Voigt N, Qi XY, Sinner MF, Dobrev D, Kaab S, Nattel S. MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation. 2013;127:1466–75. 1475e1–28. doi: 10.1161/CIRCULATIONAHA.112.001207. [DOI] [PubMed] [Google Scholar]
- 14.McManus DD, Lin H, Tanriverdi K, Quercio M, Yin X, Larson MG, Ellinor PT, Levy D, Freedman JE, Benjamin EJ. Relations between circulating microRNAs and atrial fibrillation: data from the Framingham Offspring Study. Heart Rhythm. 2014;11:663–669. doi: 10.1016/j.hrthm.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saxena A, Carninci P. Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays. 2011;33:830–839. doi: 10.1002/bies.201100084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaikkonen MU, Lam MT, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res. 2011;90:430–440. doi: 10.1093/cvr/cvr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, Kawai T, Kusunoki H, Yamamoto H, Takeya Y, Yamamoto K, Onishi M, Sugimoto K, Katsuya T, Awata N, Ikebe K, Gondo Y, Oike Y, Ohishi M, Rakugi H. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis. 2012;220:449–455. doi: 10.1016/j.atherosclerosis.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 20.Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Saito S, Nakamura Y, Tanaka T. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 21.Novo G, Guttilla D, Fazio G, Cooper D, Novo S. The role of the renin-angiotensin system in atrial fibrillation and the therapeutic effects of ACE-Is and ARBS. Br J Clin Pharmacol. 2008;66:345–351. doi: 10.1111/j.1365-2125.2008.03234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.