

Abstract
Malignant ectomesenchymoma is a rare tumor that contains both ectodermal and mesenchymal elements. So far, only 7 patients with a manifestation in the cerebrum (with confirmed clinicopathological data) have been reported. A 4-year-old girl was present at our hospital with a 3-week history of intermittent sudden dizzy with no apparent cause. MRI showed an irregular enhanced lesion in the left frontal-parietal lobe and lateral ventricle with peripheral gadolinium-enhancement with a significant surrounding edema. Total removal of the tumor was performed. Histological examination of the resected tumor revealed a mixed astrocytoma and anaplastic ependymoma component with undifferentiated mesenchymal spindle cell component. Generally speaking, the main malignant part in most cases of malignant ectomesenchymoma (MEM) is the mesenchymal component. In the present case, the malignant component was both in the mesenchymal and ectodermal part. In particular, the mesenchymal part was mainly composed of spindle cells, and the ectodermal part primarily consisted of gliomatous component and anaplastic ependymoma component. The patient was then treated with chemotherapy and as regard to the prognosis, there was no evidence of tumor recurrence at the 5 months’ follow-up. The long term follow-up is still in progress.
Keywords: Malignant ectomesenchymoma, cerebrum, intracranial mass, pediatric brain tumor
Introduction
MEM is a rare tumor, which is believed to arise from a migratory neuronal crest cell [1,2]. In the literature, more than 50 cases of MEM and adequate clinicopathological information have been reported, in which only 7 were the cerebrum affected cases [3,8]. The exact etiology is unknown. In most cases of intracranial MEM, the neuroectodermal element is often ganglionic [1,2,4,5,7] and the mesenchymal component is often composed of rhabdomyosarcoma [1,4,5,7]. However, we are here reporting a very rare pediatric MEM with special neuroectodermal elements (gliomatous component and anaplastic ependymoma component) and mesenchymal spindle cell elements. Review of the literature shows that around 50 cases of malignant ectomesenchymoma have been reported till now. To the best of our knowledge, the case presented here is the only one with neuroectodermal anaplastic ependymoma element and mesenchymal spindle cell element in the case of intracranial MEM.
Clinical summary
A 4-year-old girl presented with a 3 weeks’ history of intermittent sudden dizzy with no apparent cause. The patient did not exhibit any motor weakness, apraxia or difficulty with coordination. Results of the sensory examination were normal. She has weak physique in the past. There was no history suggestive of trauma, seizures, other visual difficulties, or coordination abnormalities. There was no family history of similar lesions or of tumor predisposition syndromes.
MRI (Figure 1) showed an irregular enhanced lesion in the left frontal-parietal lobe and lateral ventricle with peripheral gadolinium-enhancement with a significant surrounding edema. Then the patient underwent left frontal-parietal craniotomy.
Figure 1.
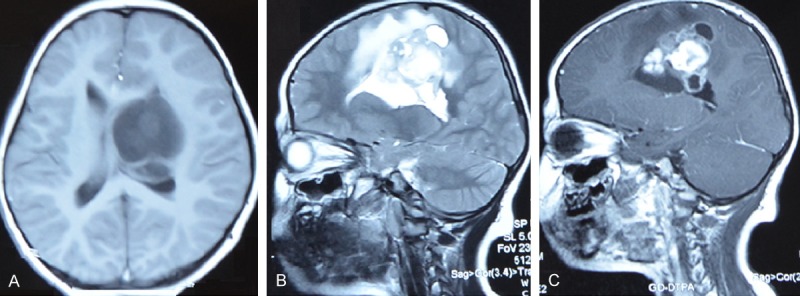
MR images disclosed a lesion in the left frontal-parietal lobe. A. MR images showed hypointense signal on T1-weighted MR images (axial). B. T2 revealed a large heterogeneous mass lesion in the right frontal lobe (sagittal). C. The central part of the lesion was observed on Gadolinium-enhanced T1 weighed MRI (sagittal).
During surgery, the tumor appeared polycystic and was well defined without obvious invasion of the surrounding brain parenchyma. The frozen section pathological analysis confirmed the presence of only ependymoma component (Figure 2A) and accordingly the frozen section diagnosis was ependymoma then. The tumor was resected piecemeal, finally resulting in gross total resection. The post-operative course was uneventful. She was suggested to receive post-operative chemotherapy. There was no evidence of tumor recurrence at the 5 months’ follow-up. The long term follow-up is still in progress.
Figure 2.
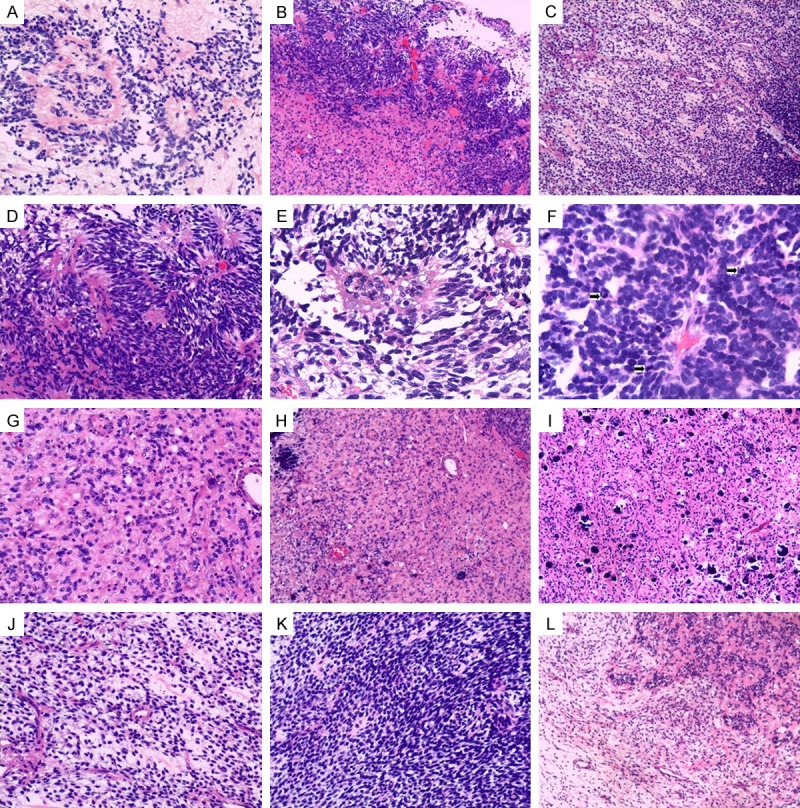
Histological features of MEM. (A) The ependymoma component in the frozen section. (B) Neuroectodermal, ependymoma and astrocytoma region. (C) Mesenchymal region of the tumor composed of spindle-shaped cells. (D-F) Neuroectodermal, ependymoma region of the tumor cells composed of perivascular pseudo-rosettes (D), vascular proliferation (E), and many mitoses (F). (G-I) Neuroectodermal, astrocytoma region of the tumor cells containing calcification (H, I) and some new small blood vessels (I), (J) Mesenchymal, scanty spindle-shaped cells with slightly myxoid background. (K) Mesenchymal, immature mesenchymal cells spread with schistose structure. (L) Occasionally, glial cells intermingled in this mesenchymal part.
Microscopically, the tumor consisted of two components, one neuroectodermal component (Figure 2B) and the other mesenchymal part (Figure 2C).
Pathological studies from the neuroectodermal component revealed mixed components of anaplastic ependymoma (Figure 2D) and astrocytoma (Figure 2G). The perivascular pseudo-rosettes (Figure 2D), vascular proliferation (Figure 2E), and many mitoses (Figure 2F) were seen in the anaplastic ependymoma. No mitoses or necrosis was found in astrocytoma portion. But calcification (Figure 2H, 2I) and new small blood vessels (Figure 2I) were present in astrocytoma portion. The MIB1 staining index in astrocytoma portion was 5%-8% and 10%-20% in the anaplastic ependymoma portion.
The latter part, mesenchymal component consisted of scanty spindle-shaped cells with slightly myxoid background (Figure 2J) (the MIB1 staining index was 3-5%), and the immature mesenchymal cells spread with schistose (Figure 2K) and nest-like structure with high proliferative activities ( the MIB1 staining index was up to 50%). Occasionally, glial cells were seen to intermingle in this mesenchymal part (Figure 2L).
Immunohistochemical staining was performed. In the astrocytoma cell portion, GFAP (glial fibrillary acidic protein) (Figure 3A), S-100 protein (Figure 3B), and vimentin were strongly positive. In the anaplastic ependymoma cell component, GFAP (Figure 3C), nestin (Figure 3D), MAP-2 (Figure 3E) vimentin and EMA (epithelial membrane antigen) (Figure 3F) showed clear positivity. The olig-2 and S-100 protein were partly positive in ependymoma portion. The astrocytic tumor cells portion and anaplastic ependymoma portion were devoid of reticulin fibers. The mesenchymal component cells were strongly positive for reticulin staining (Figure 3G) and positive immunostaining for GFAP (Figure 3H), nestin (Figure 3I), S-100 (Figure 3J) and Olig-2 (Figure 3K) were not detected. Also all of the specific mesenchymal markers (SMA, desmin, α-Sarcomeric) were not immunoreactive. INI-1 was positive in both components (Figure 3L).
Figure 3.

Immunohistochemical findings for GFAP (A). S-100 protein (B). In the astrocytoma cell portion. Immunohistochemical findings for GFAP (C), nestin (D), MAP-2(E), EMA (F) in the anaplastic ependymoma cell component. The mesenchymal component cells were strongly positive for reticulin staining (G), Immunostaining for GFAP (H), nestin (I), S-100 (J) and Olig-2 (K) were not detected. Also all of the specific mesenchymal markers (SMA, desmin, α-sarcomeric) were not immunoreactive. INI-1 was positive in both components (L).
We performed fluorescence in situ hybridization (FISH) for this tumor using the techniques previously described [9]. Briefly, 4-mm slides from the formalin-fixed paraffin-embedded tissue blocks were de-paraffinized before hybridization. Dual-color FISH was performed using LSI PTEN/CEP 10 dual color probe (Vysis/Abbott Molecular) for loss of PTEN. The EGFR gene copy number was determined by FISH using the LSI EGFR spectrum-orange/CEP 7 spectrum-green probe (Vysis/Abbott Molecular). Fluorescent signals were visualized and quantitated under fluorescence microscope. A minimum of 100 non-overlapping intact nuclei were assessed by hybridization. At least 30% in nuclei numbers is necessary for a signal to be scored as a deletion. Amplification of EGFR was defined as ratio of EGFR signal to CEP7 signal equal to or greater than two. For all of these probes, there was no evidence of 10 q deletion and EGFR gene amplification in this case.
From the above-mentioned immunohistochemical and genetic results, the neoplasm in this case was proved to contain parts of different characteristics, and was composed of the astrocytoma and anaplastic ependymoma portion (neuroectodermal component) and a non-neuronal mesenchymal component with no specific differentiation. So, finally the histological diagnosis was MEM.
Discussion
MEM is a very unusual tumor consisting of two elements, neuroectodermal element and mesenchymal element. Review of 7 previous cases of intracranial MEM found histological data in 7 of the cases [1-5,7,8]. Concerning the 8 cases of cerebral ectomesenchymomas reported up to now, the tumor consisted of (atypical) ganglion cells/ganglioneuroma and rhabdomyosarcomas.
The mesenchymal element most often consisted of rhabdomyosarcoma (57%) [1,4,5,7]. The neuroectodermal elements were most often ganglion cells (71%) [1-5]. In our case, the mesenchymal component was not rhabdomyosarcoma, but consisted of scanty spindle-shaped cells with low proliferative activities and the malignant immature mesenchymal cells with high proliferative activities. In contrast, the neuroectodermal elements consisted of a mixed astrocytoma and anaplastic ependymoma. The criteria for diagnosis of MEM were not defined well and somewhat ambiguous. The definition of ectomesenchymoma in a broad sense is that the tumor is composed of both neuroectodermal and one or more mesenchymal neoplastic elements [1,3,7]. These criteria seemed to fit in our case, so we might be able to adopt the diagnosis of MEM in the broad sense of this term.
It is worthwhile to notice that the frozen section histological diagnosis had possible pitfalls that pathologists can encounter, especially when there are only small frozen specimens available to examine. The typical biphasic aspects of the MEM can be difficult to be detected in small biopsies. In our case, the frozen section histological diagnosis was ependymoma, because the lesion was mainly represented by the perivascular pseudo-rosettes component in the frozen specimen. However, by examining more abundant tissue obtained from surgery, our final diagnose was MEM. This case told us to be cautious in the frozen section histological diagnosis of MEM because heterologous tumors cannot be identified when only small specimens were histologically examined. We believe that in the differential frozen section diagnosis of tumors in childhood and infancy, the ectomesenchymoma should always be kept in mind. The teratomas were also the major differential diagnosis that should be considered.
Limited therapeutic regime is available for this rare intracranial MEM. In the reported cases, three out of 4 patients died within 14 months of diagnosis, and none of these received complete multimodal therapy after clear diagnosis [7]. The best therapeutic approach for ectomesenchymoma seems to be a combination of surgery, radiotherapy, and chemotherapy. Previous studies have recommended sarcoma-based regimens for MEMs because rhabdomyosarcoma was the most common malignant component of MEM [1,4,7].
In our case, the mesenchymal component was not rhabdomyosarcoma, but consisted of the malignant immature mesenchymal cells with high proliferative activities. In the meantime, the anaplastic ependymoma component also had high proliferative activities. It was reported the children under 5 years of age with ependymoma can avoid radiotherapy and treated merely with prolonged adjuvant chemotherapy [10]. Our patient had complete resection and the age was 4 years old. Therefore, chemotherapy therapy (ifosfamide, cisplatin and etoposide) had been suggested immediately after the operation. No evidence of recurrence or metastasis was found after 5 months of surgery. A longer follow-up and multimodality therapy (aged over 5) could be of help to patients with this type of tumor.
Acknowledgements
This work was supported by the Capital health research and development of special (Grant No. 2011-1020-01), Beijing Municipal Natural Science Foundation (Grant No. 7122088) and the Joint Fund for basic and clinical research cooperation project of Capital Medical University (Grant No. 15JL89).
Disclosure of conflict of interest
None.
References
- 1.Freitas AB, Aguiar PH, Miura FK, Yasuda A, Soglia J, Soglia F, Aguiar CH, Vinko F, Silva NS. Malignant ectomesenchymoma. Case report and review of the literature. Pediatr Neurosurg. 1999;30:320–330. doi: 10.1159/000028818. [DOI] [PubMed] [Google Scholar]
- 2.Kleinschmidt-DeMasters BK, Lovell MA, Donson AM, Wilkinson CC, Madden JR, Addo-Yobo SO, Lillehei KO, Foreman NK. Molecular array analyses of 51 pediatric tumors shows overlap between malignant intracranial ectomesenchymoma and MPNST but not medulloblastoma or atypical teratoid rhabdoid tumor. Acta Neuropathol. 2007;113:695–703. doi: 10.1007/s00401-007-0210-0. [DOI] [PubMed] [Google Scholar]
- 3.Altenburger DL, Wagner AS, Eslin DE, Wagner AS, Eslin DE, Pearl GS, Pattisapu JV. A rare case of malignant pediatric ectomesenchymoma arising from the falx cerebri. J Neurosurg Pediatr. 2011;7:94–97. doi: 10.3171/2010.10.PEDS10261. [DOI] [PubMed] [Google Scholar]
- 4.Holimon JL, Rosenblum WI. “Gangliorhabdomyosarcoma”: a tumor of ectomesenchyme. Case report. J Neurosurg. 1971;34:417–422. doi: 10.3171/jns.1971.34.3.0417. [DOI] [PubMed] [Google Scholar]
- 5.Ingraham FD, Bailey OT. Cystic teratomas and teratoid tumors of the central nervous system in infancy and childhood. J Neurosurg. 1946;3:511–532. doi: 10.3171/jns.1946.3.6.0511. [DOI] [PubMed] [Google Scholar]
- 6.Paulus W, Slowik F, Jellinger K. Primary intracranial sarcomas: histopathological features of 19 cases. Histopathology. 1991;18:395–402. doi: 10.1111/j.1365-2559.1991.tb00869.x. [DOI] [PubMed] [Google Scholar]
- 7.Weiss E, Albrecht CF, Herms J, Behnke-Mursch J, Pekrun A, Brockmann K, Hess CF. Malignant ectomesenchymoma of the cerebrum. Case report and discussion of therapeutic options. Eur J Pediatr. 2005;164:345–349. doi: 10.1007/s00431-005-1646-7. [DOI] [PubMed] [Google Scholar]
- 8.Ito A, Kumabe T, Saito R, Sonoda Y, Watanabe M, Nakazato Y, Tominaga T. Malignant pediatric brain tumor of primitive small round cell proliferation with bland-looking mesenchymal spindle cell elements. Brain Tumor Pathol. 2013;30:109–116. doi: 10.1007/s10014-012-0106-0. [DOI] [PubMed] [Google Scholar]
- 9.Scartozzi M, Bearzi I, Mandolesi A, Pierantoni C, Loupakis F, Zaniboni A, Negri F, Quadri A, Zorzi F, Galizia E, Berardi R, Biscotti T, Labianca R, Masi G, Falcone A, Cascinu S. Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer. 2009;9:303. doi: 10.1186/1471-2407-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkatramani R, Ji L, Lasky J, Haley K, Judkins A, Zhou S, Sposto R, Olshefski R, Garvin J, Tekautz T, Kennedy G, Rassekh SR, Moore T, Gardner S, Allen J, Shore R, Moertel C, Atlas M, Dhall G, Finlay J. Outcome of infants and young children with newly diagnosed ependymoma treated on the “Head Start” III prospective clinical trial. J Neurooncol. 2013;113:285–91. doi: 10.1007/s11060-013-1111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]