

Abstract
Recent studies have reported that noise exposure at relatively low intensities can cause temporary threshold shifts (TTS) in hearing. However, the mechanism underlying the TTS is still on debate. Here, we report that an acoustic stimulation (100 dB SPL, white noise) induced TTS in mice, with the maximal ABR threshold elevations seen on the 4th day after noise exposure. On the other hand, there were no significant morphological changes in the cochlea. Further, there were paralleled changes of pre-synaptic ribbons in both the number and postsynaptic density (PSDs) during this noise exposure. The numbers of presynaptic ribbon, postsynaptic density (PSDs), and colocalized puncta correlated with the shifts of ABR thresholds. Moreover, a complete recovery of ABR thresholds and synaptic puncta was seen on the 14th day after the noise stimulations. Thus, our study may indicate that noise exposure can cause a decline in cochlear ribbon synapses and result in consequent hearing loss. The reduction of synaptic puncta appears reversible and may contribute to hearing restoration in mice after noise exposure.
Keywords: Noise exposure, hearing impairment, ribbon synapse plasticity, inner hair cell
Introduction
Noise induced hearing loss is one of the most common occupational diseases, which is caused by overexposure of sound [1]. Until decades ago, researches focused on noise induced permanent threshold shif (PTS) found noise exposure can adversely affect all three regions of the cochlea, the organ of Corti, the lateral wall and the spiral ganglion neurons (SGN) [2]. However, recent studies report that noise exposure at relatively low levels or intensities cause temporary hearing threshold shifts (TTS), which indicates the acute injury to cochlea is reversible [2]. Jeffery T’s study found decrease in compound action potentials (CAP) in noise induced TTS in human. CAP stands for a summation of multiple single neuron action potentials. They conjectured TTS may still cause massive damage to the synapses between cochlear inner hair cells (IHCs) and type I spiral ganglion neurons (SGNs), followed by a slowly developing process of degenerative SGN death. As we all known, death of SGN is irreversible, so TTS caused by overstimulation of noise may be associated with synapses between IHCs and SGNs-synaptic ribbons. The afferent synapse between IHCs and SGNs are mainly of the typical ribbon type [3], which is capable of high-speed neurotransmitter release in response to graded changes of membrane potential and ongoing recycling of released neurotransmitters. Because of these properties, ribbon synapses are recognized to play a critical role in the temporal signal processing in the cochlea [3]. Shi et al proved a relevance between hearing impairment and synaptic plasticity by using a guinea pig model [2].
However, it is still unclear at this time whether reduced number of ribbon synapses is a major cause of TTS, or, if so, whether the recovery of ribbon synapse numbers is responsible for the restoration of hearing after sound exposure, Here, we report that acoustic stimulation (100 dB SPL white noise) induced TTS in mice, with maximal ABR threshold elevations found on the 4th day after noise exposure. We utilized several experimental approaches, such as mouse modeling, auditory detection, whole mounts examination and confocal microcopy, to try to answer these concerns.
Material and methods
Animals and grouping
The experiments were performed on adult (8 weeks old) C57 mice with normal hearing, which were obtained from the Chinese Academy of Medical Sciences Animal Center (Beijing, China). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee of the Chinese PLA General Hospital. All surgery was performed under 10% chloral hydrate anesthesia, and all possible efforts were made to minimize suffering.
A total number of 50 animal were recruited and randomly divided into 5 groups (n = 10 in each): one control group (without noise) and 4 experimental groups according to the time of end point test after noise exposure (0 or instant, 4, 7 and 14 days after noise). The average body weight was 22 ± 2 g. Absence of outer or middle ear pathologies was confirmed. The mice in experiment groups were exposed to white noise at 100 dB SPL for 2 hours. ABR thresholds were taken before exposure, immediately following exposure in 0 day group, or on the 4th day in 4 days group, 7th day in 7 days group and14th day in the 14 days group after noise exposure. The mice were sacrificed after auditory testing for morphological investigations.
Assessment of auditory function
Bilateral auditory brainstem response (ABR) audiograms were obtained before exposure, ABR thresholds were taken before exposure, immediately following exposure in 0 day group, or on the 4th day in 4 days group, 7th day in 7 days group and 14th day in the 14 days group after noise exposure. ABR testing was double blinded, using the Intelligent Hearing Systems (Miami, FL). The smart-EP v2.21 was used to generate specific acoustic stimuli and to amplify, measure, and display the evoked brainstem responses. Mice were sedated using 10% chloral hydrate (0.45 ml/kg, Sigma, USA). Recording electrodes were placed at the vertex and the mastoid, while the reference electrode was placed in the rump area. Specific auditory stimuli (broadband clicks and tone bursts of 4, 8, 16 and 32 kHz) were delivered through an insert earphone (Intelligent Hearing systems, USA) placed directly in the ear canal. Auditory thresholds, which were based on the visibility and reproducibility of wave III, were obtained for click and tone bursts at each frequency by varying the sound pressure level (SPL) at 5-dB steps up and down in order to identify the lowest level at which an ABR pattern could be recognized (at least two reproducible peaks) [4]. ABR functions were analyzed for amplitude of wave I peak, each wave consisting of a starting negative (n) deflection and the following positive (p) deflection. Amplitudes of ABR waves I defined as Ip-In (latency, 1.2-1.9 ms). An algorithm for an automated determination of ABR amplitudes was programmed in MATLAB (MathWorks) [5]. For ABR growth functions, ABR amplitudes for wave I were derived from individual ears’ responses for increasing stimulus levels 20 dB above threshold to 90 dB (Figure 1A).
Figure 1.
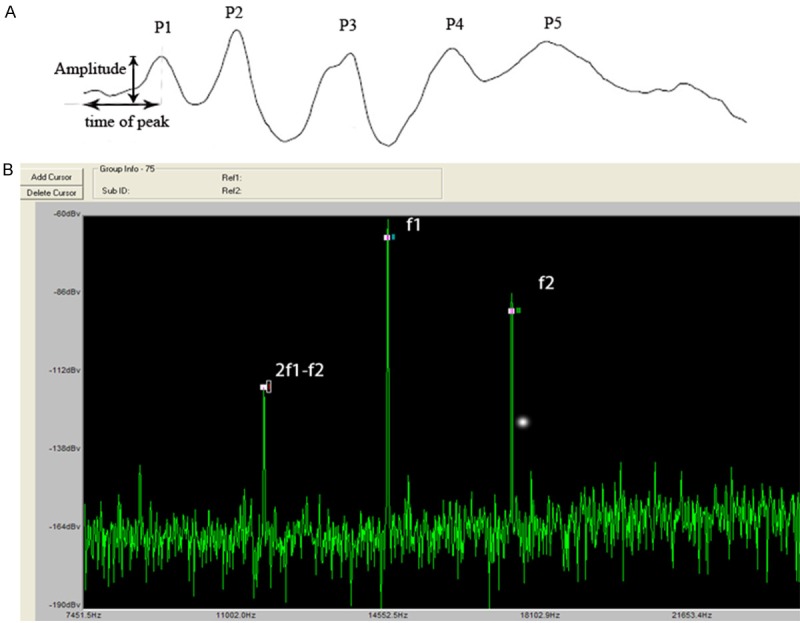
Illustrations of the methods to measure wave I of click and DPOAE. A. ABR wave I amplitude analysis includes three parts: time of peak (from beginning to Ip), SPL of click and amplitude of wave I (Ip-In). B. When we measured DPOAE, f1 and f2 connected to the earpiece. f2 was set at 4, 8, 16, and 32 kHz. The f2/f1 ratio was set at 1.2. The intensity of f2 = 55 dB, f1 = 65 dB. Amplitudes of 2f1-f2 were measured.
DPOAEs were measured using an ER-10B+ (Etymotic Research) microphone coupled with two EC1 speakers. Stimuli of two primary tones f1 and f2 (f2/f1 = 1.2) were presented with f2 = 55 dB, f1 = 65 dB increment and swept from 8 to 32 kHz in 1/2 octave steps. Stimuli were generated and attenuated digitally (200 kHz sampling). The ear canal sound pressure was preamplified and digitized. Amplitudes of 2f1-f2 were measured based on the baseline (Figure 1B).
Noise exposure
40 awake mice in separate stainless steel wire cages (approximately 15 × 5 × 5 cm) were exposed to white noise for 2 h to induce threshold shifts. To achieve the maximum TTS noise exposure, the noise level was adjusted to 100 dB SPL, measured at the center of each partition within the cage. The sound exposure chamber was fitted with a loudspeaker (YH25-19B, 25 W, 16 Ω, China) driven by a power producer (33220 A, China) fed from noise software. The noise sound files were created and equalized with audio editing software (Audition 3; Adobe System, Inc., San Jose, CA). Sound levels were calibrated with a sound level meter (model 1200; Quest Technologies, Oconomovoc, WI) at multiple locations within the sound chamber to ensure uniformity of the sound field. Sound levels were measured before and after exposure to ensure stability.
Cochlear tissue processing
At each evaluation time point (i.e. before noise exposure, immediately after exposure, and on post-exposure Days 4, 7 and 14), 5 mice were decapitated under deep anesthesia and the cochlea quickly removed from the skull and separated. The round and oval windows and the apex of the cochlea were opened for perfusion with 4% paraformaldehyde overnight. After fixation, the cochlea shell was decalcified with 10% EDTA for 4-6 hours, then the basal turn separated under a dissecting microscope in 0.01 mmol/L PBS solutions. The parietal gyrus of the basilar membrane was separated, and the vestibular membrane and the tectorial membrane were removed.
Immunohistology and confocal imaging
The separated basilar membrane was washed three times in 0.01 M PBS and pre-incubated for 30 min at room temperature in a blocking solution of 5% normal goat serum in 0.01 M PBS, followed by incubation with a combination of goat anti-mouse CtBP2 (E 16; Cterminal binding protein 2, C, end of combination of protein) antibody (1:200, SANTA CRUZ) and rabbit anti-mouse PSD95 antibody (1:200, SANTA CRUZ), at 4°C for overnight. The incubated specimens were washed in 0.01 M PBS three times and incubated in donkey anti-goat 488 and donkey anti-rabbit 568 (fluorescein isothiocyanate; SANTA CRUZ) (1:100) at room temperature for 60 min, and then washed three times again. A drop (approximately 40 μl) of DAPI (4, 6-diamidino-2-phenylindole; Santa Cruz) was added to the slide, on which the basement membrane was tiled under a dissecting microscope and covered with a coverslip. The specimens were imaged directly with fluorescent microscopy to examine the specificity of the primary antibody.
The laser scanning confocal microscope was a Olympus FV1000 configuration (Japan) with 180× oil immersion objective. The excitation wavelengths were 488 nm and 586 nm. Sequence scanning was performed on IHCs. The scanning interval for sequential scanning was set to 0.5 μm to ensure that each synapse was marked since the size of mature IHC ribbon synapses usually ranges from 150 nm to 200 nm.
Counting the number of cochlear ribbon synapse
Serial scanning generated 2D images, which were then used to mark IHC ribbon synapses. The image files were opened successively from top to bottom using the 3Ds Max Software. The images were magnified in “zoomed top view” to identify IHC ribbon synapses. The green fluorescence spot (indicating a pre synaptic ribbon) that appeared in each image was first marked by a sphere, whose size was then adjusted to match the area of green fluorescence. Before marking green fluorescence in the next image, the previous marked image was opened for comparison. In the next image, if the green fluorescence appeared at the same location as on the previous image, it was skipped to avoid redundant labeling. Additionally, only two adjacent images from serial scanning could overlap. After all the images were marked, the total number of marks was obtained using the layer manager of 3Ds Max [6]. After switching channels, the red fluorescence spots indicating PSD95 were counted using the same method described for the green fluorescence.
Light microscopy
At each evaluation time point, 4 cochleae were harvested for light microscopic examination. The mice were anesthetized and their cochleae were isolated and dissected. The cochleae were perfused through the round and oval windows with both 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), and incubated in the same fixative overnight at 4°C. The cochleae were then rinsed with 0.1 M (PBS) and incubated in 1% osmium tetroxide overnight, followed by immersion in 5% ethylenediaminetetraacetic acid (EDTA) for 2-4 days. The decalcified cochleae were then dehydrated in ethanol and propylene oxide and embedded in Araldite 502 resin (Electron Microscopy Sciences, Fort Washington, PA, USA). The cochleae were stained with toluidine blue and mounted in permount on microscope slides.
To count SGNs, we used the optical fractionators method to sample cell numbers in a defined fraction of the cochlear neural volume by optical dissection. Optical dissection was achieved using a 60x oil immersion lens to view along the Z-axis of sections having a thickness at least a few micrometers greater that the diameter of SGN cell bodies. The rationale for this requirement was that a few micrometers at the surface and bottom of the sections are needed to avoid the unevenness and artifacts at the surfaces of the sections. We cut a series of 50 μm cochlea sections for this approach. In order to count cells, a sampling grid was placed over the contour in a random fashion using Stereological Investigator software. Each box of the sampling grid contained an unbiased counting frame, representing a defined fraction of SGNs. The specific feature chosen for counting was the nucleolus of each cell.
Statistical analysis
Statistical analyses were performed using SPSS (IBM corporation, Somers, NY). All data were expressed as Mean ± SEM. Pre- and post-noise exposure ABR audiograms (specifically the SPL thresholds) at various evaluation time points were compared across frequencies and for individual frequencies, using a one-way ANOVA and Turkey’s post-hoc multiple comparison test. For multiple group comparisons, statistical significance of SGN counting was determined using Mann-Whitney U test. Two-way ANOVA followed by Student’s t-test with Bonferroni correction was used to compare hair cell numbers among all the groups. Comparison of amplitudes of wave I peaks in different groups included the amplitudes, the latency of peak and SPL of click. For multiple comparison of the number of pre- and post synapses, one-way ANOVA was used. To determine whether pre-synaptic ribbons number changes correlate with alterations in PSDs, student’s t test was used to test the correlation. A P value of < 0.05 was considered statistically significant.
Results
Temporary threshold shifts (TTS) and DPOAE after noise exposure
The averaged ABR threshold for click from the control and the groups tested at different times after the noise are 15.00 ± 6.79, 20.83 ± 9.06, 32.50 ± 6.33, 9.58 ± 9.22 and 15.00 ± 5.34 in the control group, 0 day group, 4 days group, 7 days group and 14 days group respectively, with peak threshold elevation occurring on Day 4 by nearly 17 dB compared with that before exposure (or the control) (Figure 2A, P < 0.01). ABR threshold improvement was seen on post-exposure Day 7, although no statistically significant difference was shown between post-exposure Day 4 and Day 7 (Figure 2A, P > 0.05). Apparent recovery of ABR threshold was identified on post-exposure Day 14, showing no statistically significant difference from the control (Figure 2A, P > 0.05).
Figure 2.
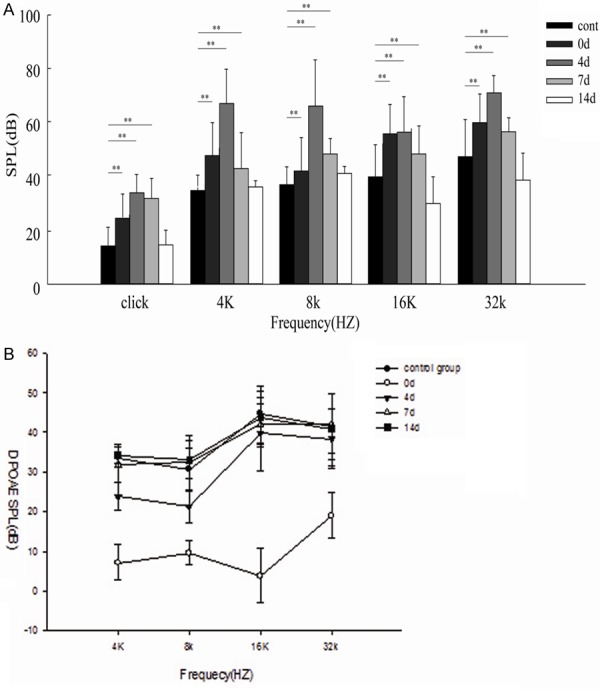
Temporary threshold shifts (TTS) and changes of DPOAEs caused by noise exposure. A. The ABR thresholds are elevated immediately following noise exposure, with maximum elevation of nearly 17 dB compared with before exposure on the 4th post-exposure day (P < 0.01). Thresholds on the 4th and 7th post-exposure days are not significantly different (P > 0.05). Thresholds are significantly elevated at 4, 8, 16 and 32 kHz immediately after exposure, but only damatically at 4 and 8 kHz on the 4th post-operative day ( P < 0.01). On the 7th post-exposure day, thresholds elevation show significant improvement across frequencies compared with those on the 4th post-exposure day (P < 0.05). On the 14th post-exposure day, thresholds at 4 and 8 kHz are not significantly different compared with before exposure (P > 0.05). B. In control group, DPOAEs in 4, 8, 16, 32KHZ were 33.5 ± 1.20, 30.7 ± 5.10, 44.6 ± 7.20, 41.5 ± 8.20. A severe reduction of DPOAEs at 0 day group in 4, 8, 16, 32KHZ was shown, 32.60 ± 4.40 , 31.2 ± 3.0, 41.5 ± 6.70, 42.3 ± 5.80, respectivly (p < 0.05, one-way ANOVA). However, DPOAEs of 16 and 32 KHZ returned to normal levels in 4 days group. They were 31.8 ± 4.60, 32.4 ± 6.80, 41.8 ± 5.40, 42.2 ± 7.5 at the 4 frquecies. On the 7th day after noise exposure, no loss of DPOAEs was detected at each frequency.
Additionally, the noise exposure caused significant ABR threshold elevation across all frequencies. On post-exposure Day 0 (instantly after exposure), statistically significant threshold changes were found at 4, 8, 16 and 32 kHz (Figure 2A), which increased dramatically at 4 and 8 kHz on post-exposure Day 4 (Figure 2A, P < 0.01), indicating increasing hearing impairment. On post-exposure Day 7, elevation of ABR thresholds across all frequencies showed a noticeable decrease compared with 4 day group (Figure 2B, P < 0.05). Correspondingly, significant restoration of ABR thresholds was also found on post-exposure Day 14, with thresholds at 4, 8, 16 and 32 kHz showing no significant difference in comparison to before exposure (Figure 2A, P > 0.05), indicating temporary threshold shifts (TTSs) following 100 dB SPL white noise exposure.
A dominant component of hearing loss was a severe reduction of DPOAEs instantly after noise exposure, as shown in Figure 2B. However, DPOAEs began to return to normal levels 4 days after exposure in 16 kHz, 32 kHz (p < 0.05, one-way ANOVA). For the 7th noise exposure, no loss of DPOAEs was detected (p > 0.05).
Reduction of ABR wave 1 peak amplitude after TTS noise exposures
After 2 h 100 dB sound exposure, quantification of wave I peak reduced quickly in click (Figure 3B, p < 0.05), accompanied with hearing threshold elevating, wave I peak amplitude appeared maximal reduction 4 days after noise (p < 0.01). Even though ABR thresholds completely restored in the 14 days group, the ABR wave I peak didn’t return to normal (Table 1), suggesting auditory function didn’t fully recovered.
Figure 3.

Morphology of cochlear hair cells and noise exposure. Both OHCs and IHCs display a normal array of three rows of OHCs and one row of IHCs (A-E, Scale bar = 10 um). SEM showes no significant changes of the stereocilia of IHCs (F-J, Scale bar = 10 um) before and after the noise exposure.
Table 1.
Amplitudes of wave I in click of the 5 groups from hearing threshold to 90 dB SPL
| SPL (dB) | Control group | 0 day group | 4 days group | 7 days group | 14 days group | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| A of wave IP | Mean of IP | A of wave IP | Mean of IP | A of wave IP | Mean of IP | A of wave IP | Mean of IP | A of wave IP | Mean of IP | |
| 20 | 0.92 ± 0.3 | 1.78 | 0.67 ± 0.18 | 1.79 | ||||||
| 25 | 1.06 ± 0.28 | 1.74 | 0.93 ± 0.12 | 1.75 | ||||||
| 30 | 1.42 ± 0.21 | 1.71 | 1.26 ± 0.41 | 1.72 | ||||||
| 35 | 2.02 ± 0.17 | 1.68 | 0.63 ± 0.23 | 1.76 | 0.98 ± 0.11 | 1.72 | 1.75 ± 0.51 | 1.68 | ||
| 40 | 2.97 ± 0.66 | 1.65 | 0.88 ± 0.58 | 1.72 | 1.02 ± 0.47 | 1.70 | 2.36 ± 0.61 | 1.65 | ||
| 45 | 3.76 ± 0.48 | 1.62 | 1.10 ± 0.33 | 1.69 | 0.46 ± 0.08 | 1.67 | 1.86 ± 0.64 | 1.67 | 3.41 ± 0.72 | 1.62 |
| 50 | 4.51 ± 0.62 | 1.60 | 1.65 ± 0.74 | 1.66 | 0.64 ± 0.52 | 1.63 | 2.71 ± 1.71 | 1.62 | 3.82 ± 0.63 | 1.59 |
| 55 | 4.86 ± 1.02 | 1.55 | 1.92 ± 0.39 | 1.63 | 1.01 ± 0.35 | 1.59 | 3.22 ± 1.63 | 1.58 | 4.51 ± 0.68 | 1.56 |
| 60 | 5.24 ± 1.73 | 1.52 | 2.24 ± 0.74 | 1.60 | 1.32 ± 0.13 | 1.56 | 3.46 ± 1.21 | 1.55 | 4.69 ± 0.75 | 1.52 |
| 65 | 5.57 ± 1.08 | 1.47 | 2.86 ± 0.89 | 1.57 | 1.5 ± 0.69 | 1.52 | 4.01 ± 0.94 | 1.50 | 5.16 ± 1.82 | 1.49 |
| 70 | 5.90 ± 1.26 | 1.43 | 3.01 ± 1.18 | 1.54 | 1.68 ± 0.51 | 1.50 | 4.17 ± 1.17 | 1.46 | 5.45 ± 0.66 | 1.47 |
| 75 | 6.22 ± 0.97 | 1.40 | 3.17 ± 0.86 | 1.49 | 1.96 ± 0.43 | 1.48 | 4.51 ± 0.35 | 1.44 | 5.95 ± 1.37 | 1.45 |
| 80 | 6.48 ± 1.76 | 1.38 | 3.46 ± 1.24 | 1.44 | 2.03 ± 0.45 | 1.45 | 4.67 ± 0.47 | 1.41 | 6.03 ± 0.71 | 1.42 |
| 85 | 6.53 ± 1.16 | 1.34 | 3.72 ± 0.65 | 1.40 | 2.26 ± 0.73 | 1.42 | 4.91 ± 1.42 | 1.38 | 6.29 ± 1.51 | 1.38 |
| 90 | 6.87 ± 1.36 | 1.32 | 4.16 ± 1.38 | 1.36 | 2.68 ± 0.82 | 1.39 | 5.06 ± 1.20 | 1.35 | 6.42 ± 2.17 | 1.35 |
| Sig | ** | ** | * | * | ||||||
A stands for amplitude (μV), Ip stands for the latency of wave I (ms). Sig stands for significant difference in amplitudes of wave I was observed among the 5 groups;
p < 0.05;
p < 0.01.
Morphology of cochlear hair cells and spiral ganglion cells counting
According to Dapi stain, our study showed that cochlear hair cells (IHCs and OHCs) displayed a normal array of three rows of OHCs and one row of IHCs (Figure 3A-E). SEM observations found that there were no significant changes of IHC stereocilia in response to the sound exposure (Figure 3F-J), suggesting that the 100 dB SPL white noise stimulation of 2 hours was not sufficient to cause morphological changes of cochlear hair cells in these mice. By observing and counting numbers of SGN, the differences among the 5 groups didn’t reach statistical significance (p = 0.07, Figure 4A, 4B).
Figure 4.
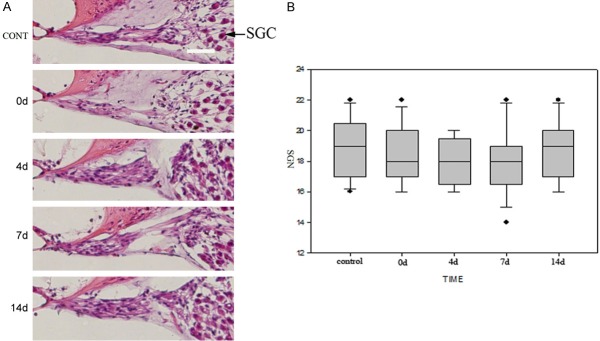
Morphology of spiral ganglion cells (SGCs) and noise exposure. A. HE staining and light microscopy show no significant differences in the density of SGCs and innervations after noise exposure compared to before exposure. Arrow heads indicate SGCs and SGNs. Bar = 10 um. B. SGN counts in the 5 groups were 18.86 ± 1.88, 18.38 ± 1.86, 18.14 ± 1.53, 18.04 ± 2.22, 18.71 ± 1.90 respectively. Statistical analysis by the Mann-Whitney U test indicated that no significant difference exists among 5 groups (p > 0.05). Dashed lines showed means.
Change in the numbers of pre-synapse ribbons and PSDs
Acceptable recordings of sufficient duration for data analysis were obtained from 15 cells in each animal at each time point. We identified RIBEYE/CtBP2, a specific pre-synaptic protein of ribbon synapses [6]. In this study, the readings of number of pre-synaptic RIBEYE/CtBP2 puncta were 16.82 ± 1.80/IHC before exposure, and 12.60 ± 2.59, 2.77 ± 0.62, 10.56 ± 4.06 and 17.17 ± 1.34 per IHC on post-exposure Days 0, 4, 7 and 14, respectively (Figure 5). The number decreased after noise exposure (Figures 5, 6, p < 0.05), with the maximal reduction occurring on Post-exposure Day 4 (Figures 5, 6, p < 0.01). Partial restoration of RIBEYE/CtBP2 puncta was seen on post-exposure Day 7, although still less than before exposure (Figure 6, p < 0.05). Complete RIBEYE/CtBP2 number recovery was seen on post-exposure Day 14, showing no significant difference from the control (Figure 6, p > 0.05).
Figure 5.
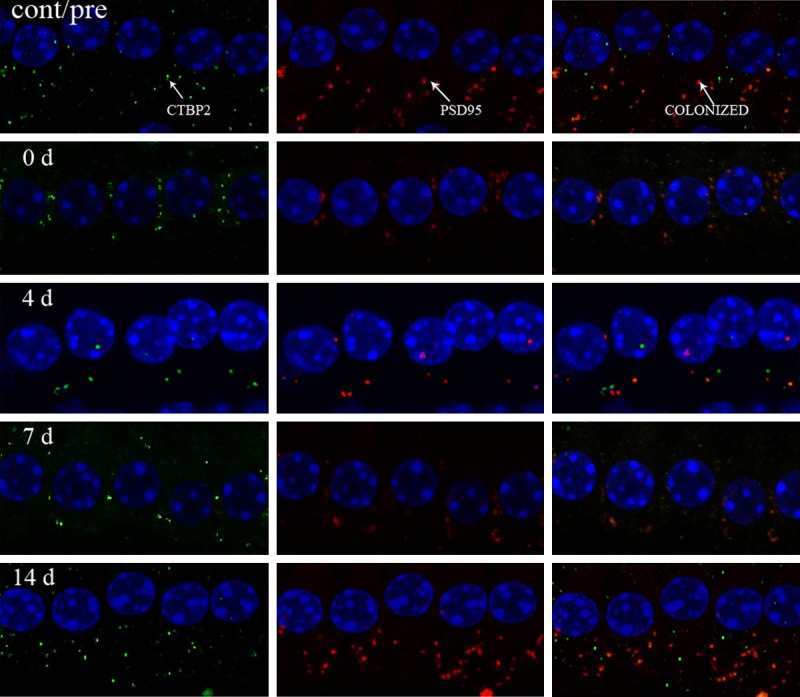
Change of RIBEYE/CtBP2 in confocal microscopy in control group (cont/pre), 0 day group (0 d), 4 day group (4 d), 7 day group (7 d), 14 day (14 d). The number of RIBEYE/CtBP2 shows a decrease instantly after noise exposure (p < 0.05). Maximal reduction of RIBEYE/CtBP2 staining appears on the 4th day after exposure as compared with before exposure (p < 0.01). An increase of RIBEYE/CtBP2 spots is seen on the 7th post-exposure day compared with the 4th day (p < 0.05), although still less than that before exposure (p < 0.05). No significant difference is seen between the 14th post-exposure day and control (before exposure) (p > 0.05).
Figure 6.
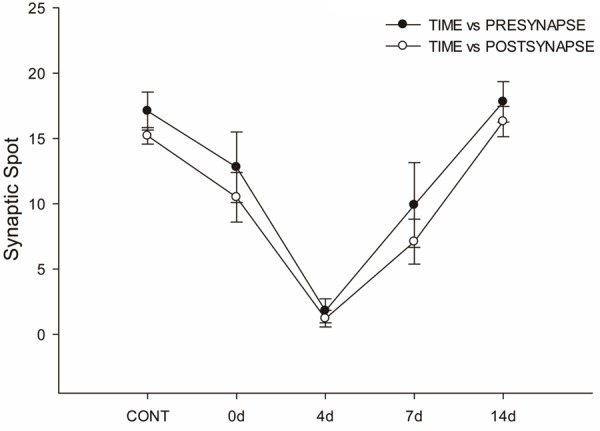
Parallel changes of synaptic ribbons and PSD95 in response to the noise exposure. The curve representing synaptic ribbons (dark circle) shows a decrease instantly after noise exposure (on the day of exposure). Maximal reduction of ribbon numbers is seen on the 4th post-exposure day, and recovery of ribbon number is seen on the 7th day. Complete restoration of ribbon numbers is seen on the 14th post-exposure day. The curve representing PSD95 (white circle) shows changes parallel to those of synaptic ribbons.
Also, we labeled the postsynaptic structures, using the antibody against Post-Synaptic Density protein (PSD95) [7,8]. The number of PSD95 was 16.52 ± 2.02/IHC before exposure, and 13.67 ± 2.63, 2.78 ± 0.57, 10.26 ± 2.24, and 16.67 ± 1.58 per IHC on post-exposure Days 0, 4, 7 and 14, respectively (Figure 5). The maximal PSD95 number loss occurred on post-exposure Day 4 (Figures 5, 6, p < 0.05), suggesting significant effects on the number of postsynaptic receptors by the noise exposure. It also paralleled the elevation of ABR thresholds on post-exposure Day 4 (Figure 2A). The number of colocalized pre and postsynaptic puncta was 15.73 ± 0.95/IHC before exposure, and 10.27 ± 4.84, 2.2 ± 0.43, 9.98 ± 3.39, and 16.85 ± 0.56 per IHC on post-exposure Days 0, 4, 7 and 14, respectively (Figures 5, 6).
Discussion
The effect of overexposure to noise on hearing thresholds
The degree of hearing loss produced by acoustic trauma is dependent on the loudness of the sound and the duration of the exposure. Temporary Threshold Shifts may be observed immediately following exposure, and if the sound is loud enough it may produce permanent elevations in thresholds resulting in permanent threshold shifts (PTS) [9].
The hearing loss produced by our stimulus can be characterized by significant threshold shifts across all frequencies and all time points. Hearing loss was observed instantly after 2-hour-exposure, while the maximal threshold shift appeared in the 4th day , which indicated an acute hearing function damage. It seems contrary to most previous study [5,10] which indicated the maximal hearing loss appeared in 24 hours following noise exposure. However, in the studies mentioned above, they didn’t observe the 4th day hearing threshold. Our study consistent with Brown’s study [9] which showed persist hearing loss in 4 days after noise exposure. Day 7 suggests that some hearing function has recovered after the acoustic trauma. We postulates ribbon synapses have time-delay reaction to acoustic trauma, it might be for self protection. The ribbon structure is responsible for both quick responses to rapidly changing signals and long-lasting responses to continuous stimuli. Glutamate over releasing due to sound overexposure may produce a long lasting damage to ribbon synapses which causes proceeding hearing loss until the 4th day.
The initial threshold shifts may be a result of peripheral damage to the auditory system. It is known that IHC and OHC cannot regenerate in humans and most mammals [11]. Our study shows normal array and morphology of OHC and IHC, that’s why the acoustic trauma produce TTS not PPS. By analyzing amplitude I wave of click, a dramatic I wave reduction seen clearly, which indicates the synapses between cochlear inner hair cells (IHCs) and type I spiral ganglion neurons (SGNs) injuries.
Changes of cochlear ribbon synapses might be molecular basis of temporary hearing threshold shifts in noise induced hearing loss
Synaptic ribbons are sites of intense synaptic vesicle turnover, a process that requires bending of membranes during fusion and fission with the plasma membrane. Normal hearing relies on faithful synaptic transmission at the ribbon synapse of IHCs [12,13]. Pathological changes to the afferent innervation in IHCs or adjacent areas will certainly disrupt synaptic transmission [14]. Prior reports showed that primary noise-induced neural degeneration did not affect ABR thresholds but reliably led to a reduction in the amplitude of ABR wave I in mice [14,15], which reflects the summed activity of the remaining auditory nerve fibers. Liu et al once reported that cochlear ribbon synapse was the primary target of ototoxicity exposure [4], supporting that the ribbon synapse is a sensitive nanostructure which can be easily affected by external factors. In this study, we further explored the effects of noise exposure on the temporal processing ability of the auditory system. Beside observing the ABR, we observed the damage and the repair of ribbon synapses at both pre- and post-synaptic sites in an attempt to provide insight into the plastic changes of ribbon synapses during noise exposure.
Previous studies show acute noise-induced excitability results in little or no terminal regeneration or synaptogenesis, and cochlear thresholds can still recover despite a loss of 50% of the auditory nerve terminals [16-18]. In contrast, our study found that ABR thresholds could undergo 100% recovery despite a loss of more than 80-90% synaptic stumps. The difference is demonstrated in several ways. Firstly, our study use C57BL/6J mice as animal models because they are more sensitive to noise and are more likely to bite than the more docile laboratory strains such as BALB/c [19]. However, as seen in this report, ribbon loss in C57BL/6J mice is much more reversible when compared to that previously reported in BALB/c mice. It may due to the species used, the age of the animals, method of acoustic trauma, method of quantification and assessing ribbon synapses at differing time. Secondly, this is the first study that has investgated changes of ribbon synapses with no significant OHC or IHC loss, although DPOAEs showed transient change, but morphology of OHC and IHC were normal, suggesting ribbon synapse might be the primary target in noise induced hearing loss.
Except the loss and recovery of hair cell ribbons and post-synaptic densities (PSDs) occurring in parallel, previous study reported that the recovery was not complete, resulting in a permanent loss of less than 10% synapses [2]. A plastic interaction between ribbons and postsynaptic terminals may be involved in the reconstruction of synaptic contact between ribbons and PSDs, as shown by location changes in both structures. Synapse restoration is associated with a breakdown in temporal processing, hence the deterioration in temporal processing originated from the cochlea. This deterioration develops with the recovery in hearing threshold and ribbon synapse counts, suggesting that repaired synapses have deficits in temporal processing [2,20].
However, our study seems to show that synaptic repair is complete as indicated by lack of significant differences in both ABR thresholds and synapse numbers between the 14th day after noise exposure and before exposure. Very differently, previous studies proposed that this damage was largely irreparable in mice [5,17,20]. Possible reason may come from the large-scale degenerative SGN death that developed slowly after the initial damage in those studies [17]. Although the present study does not observe the SGN loss in longer time line, it at least confirm that synapses change prior to SGN. In guinea pigs, however, initial damage of a similar degree was found to be largely repairable [5], suggesting a cross-species difference in the repair process in the ribbon synapses. In consist with our study, we analyzed click-evoked ABR wave I amplitude and found reduction at 20 dB above hearing threshold in 14 day group compared with control, which indicated incomplete recovery of ribbon synapses function.
Dramatic changes of PSDs were observed in this study. Supporting data have also been found in guinea pigs, in which the degree of swelling can be reduced by cochlear perfusion with glutamate antagonists during the noise exposure [21], and can be mimicked, in the absence of noise exposure, by cochlear perfusion with the glutamate analog, kainite [22]. The observation, that targeted deletion of a key gene (GLAST) in the glutamate re-uptake system in supporting cells increases noise-induced threshold shift, is consistent with the view that glutamate excitability is an important component of noise-induced cochlear pathology [23].
Similar with our findings, noise exposure at relatively low levels or doses has been found to cause “silent” damage to the afferent cochlear innervation [5,17,20]. It has been called “silent” damage because the noise exposure does not cause a permanent threshold shift in hearing. The loss and recovery of hair cell ribbons and post-synaptic densities (PSDs) occurred in parallel in our study. Our study once again confirmed that synaptic ribbons and PSDs display identical changes during the acoustic exposure. It seems likely that the noise-induced damage to the IHC-SGN synapse can cause deterioration of temporal processing. Housley et al [24] found purinergic signaling contributes to cochlear adaption to elevated sound levels and protection from overstimulation. Their study shows acoustic overstimulation leads over release of ATP into the cochlear partition, which activate P2X2 receptors and reduces sound transduction and synaptic transmition. According to their study, loss of synapses is to some extent otoprotective against noise damage. That’s why recovery of hearing threshold was detected after synapses loss. We hypothesis the purinergic signal might be the mechanism of noise induced synapses loss.
In our study, we divide each basilar membrane into parietal gyrus, middle gyrus and basal gyrus. We find there is no difference of ribbon synapse loss among the gyri. We speculate it might because white noise which is a random signal with a constant power spectral density (Carter, 2009) makes influence to all frequencies. However, with the same synapses loss, it appears more dramatic hearing loss in 4, 8 KHZ. Whether it indicates different compensation function in different frequencies is not clear.
Because the chance of being exposed to low-level noise is widespread encounters in modern society, the accumulations of such damage in cochlea can become one of the mechanisms for the reduced temporal resolution in aging subjects [25]. The deficits are considered a major obstacle in signal coding and the major reason for emerging problems in speech perception with aging [21,23,26-29].
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (81503372) and the National Basic Research Program of China (973Program) (2012CB967900; 2012CB967901).
Disclosure of conflict of interest
None.
References
- 1.Cui Y, Sun GW, Yamashita D, Kanzaki S, Matsunaga T, Fujii M, Kaga K, Ogawa K. Acoustic overstimulation-induced apoptosis in fibrocytes of the cochlear spiral limbus of mice. Eur Arch Otorhinolaryngol. 2011;268:973–978. doi: 10.1007/s00405-011-1484-3. [DOI] [PubMed] [Google Scholar]
- 2.Shi L, Liu L, He T, Guo X, Yu Z, Yin S, Wang J. Ribbon synapse plasticity in the cochleae of Guinea pigs after noise-induced silent damage. PLoS One. 2013;8:e81566. doi: 10.1371/journal.pone.0081566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 4.Liu K, Jiang X, Shi C, Shi L, Yang B, Shi L, Xu Y, Yang W, Yang S. Cochlear inner hair cell ribbon synapse is the primary target of ototoxic aminoglycoside stimuli. Mol Neurobiol. 2013;48:647–654. doi: 10.1007/s12035-013-8454-2. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Wang H, Shi L, Almuklass A, He T, Aiken S, Bance M, Yin S, Wang J. Silent damage of noise on cochlear afferent innervation in guinea pigs and the impact on temporal processing. PLoS One. 2012;7:e49550. doi: 10.1371/journal.pone.0049550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu K, Ji F, Xu Y, Wang X, Hou Z, Yang S. Myosin VIIa and otoferlin in cochlear inner hair cells have distinct response to ototoxic exposure. Acta Otolaryngol. 2014;134:564–70. doi: 10.3109/00016489.2014.892631. [DOI] [PubMed] [Google Scholar]
- 7.Webster M, Webster DB. Spiral ganglion neuron loss following organ of Corti loss: a quantitative study. Brain Res. 1981;212:17–30. doi: 10.1016/0006-8993(81)90028-7. [DOI] [PubMed] [Google Scholar]
- 8.Grant L, Yi E, Goutman JD, Glowatzki E. Postsynaptic recordings at afferent dendrites contacting cochlear inner hair cells: monitoring multivesicular release at a ribbon synapse. J Vis Exp. 2011 doi: 10.3791/2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browne CJ, Morley JW, Parsons CH. Tracking the expression of excitatory and inhibitory neurotransmission-related proteins and neuroplasticity markers after noise induced hearing loss. PLoS One. 2012;7:e33272. doi: 10.1371/journal.pone.0033272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda Y, Fukushima K, Omichi R, Kariya S, Nishizaki K. Time courses of changes in phospho- and total- MAP kinases in the cochlea after intense noise exposure. PLoS One. 2013;8:e58775. doi: 10.1371/journal.pone.0058775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edge AS, Chen ZY. Hair cell regeneration. Curr Opin Neurobiol. 2008;18:377–382. doi: 10.1016/j.conb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neef A, Khimich D, Pirih P, Riedel D, Wolf F, Moser T. Probing the mechanism of exocytosis at the hair cell ribbon synapse. J Neurosci. 2007;27:12933–12944. doi: 10.1523/JNEUROSCI.1996-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobkowicz HM, August BK, Slapnick SM. Synaptic arrangements between inner hair cells and tunnel fibers in the mouse cochlea. Synapse. 2004;52:299–315. doi: 10.1002/syn.20026. [DOI] [PubMed] [Google Scholar]
- 14.Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melcher JR, Kiang NY. Generators of the brainstem auditory evoked potential in cat. III: Identified cell populations. Hear Res. 1996;93:52–71. doi: 10.1016/0378-5955(95)00200-6. [DOI] [PubMed] [Google Scholar]
- 16.Weisz C, Glowatzki E, Fuchs P. The postsynaptic function of type II cochlear afferents. Nature. 2009;461:1126–1129. doi: 10.1038/nature08487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberman LD, Wang H, Liberman MC. Opposing gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci. 2011;31:801–808. doi: 10.1523/JNEUROSCI.3389-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maison SF, Casanova E, Holstein GR, Bettler B, Liberman MC. Loss of GABAB receptors in cochlear neurons: threshold elevation suggests modulation of outer hair cell function by type II afferent fibers. J Assoc Res Otolaryngol. 2009;10:50–63. doi: 10.1007/s10162-008-0138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conner DA. Transgenic mouse colony management. In: Ausubel FM, et al., editors. Current protocols in molecular biology. 2005. Chapter 23: Unit 23 10. [DOI] [PubMed] [Google Scholar]
- 20.Lin HW, Furman AC, Kujawa SG, Liberman MC. Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J Assoc Res Otolaryngol. 2011;12:605–616. doi: 10.1007/s10162-011-0277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puel JL, Pujol R, Tribillac F, Ladrech S, Eybalin M. Excitatory amino acid antagonists protect cochlear auditory neurons from excitotoxicity. J Comp Neurol. 1985;341:241–256. doi: 10.1002/cne.903410209. [DOI] [PubMed] [Google Scholar]
- 22.Pujol R, Lenoir M, Robertson D, Eybalin M, Johnstone BM. Kainic acid selectively alters auditory dendrites connected with cochlear inner hair cells. Hear Res. 1985;18:145–151. doi: 10.1016/0378-5955(85)90006-1. [DOI] [PubMed] [Google Scholar]
- 23.Hakuba N, Koga K, Gyo K, Usami SI, Tanaka K. Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. J Neurosci. 2013;20:8750–8753. doi: 10.1523/JNEUROSCI.20-23-08750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Housley GD, Morton-Jones R, Vlajkovic SM, Telang RS, Paramananthasivam V, Tadros SF, Wong AC, Froud KE, Cederholm JM, Sivakumaran Y, Snguanwongchai P, Khakh BS, Cockayne DA, Thorne PR, Ryan AF. ATP-gated ion channels mediate adaptation to elevated sound levels. Proc Natl Acad Sci U S A. 2013;110:7494–7499. doi: 10.1073/pnas.1222295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parthasarathy A, Cunningham PA, Bartlett EL. Age-related differences in auditory processing as assessed by amplitude-modulation following responses in quiet and in noise. Front Aging Neurosci. 2010;2:152. doi: 10.3389/fnagi.2010.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walton JP. Timing is everything: temporal processing deficits in the aged auditory brainstem. Hear Res. 2010;264:63–69. doi: 10.1016/j.heares.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grose JH, Mamo SK, Hall JW 3rd. Age effects in temporal envelope processing: speech unmasking and auditory steady state responses. Ear and Hear. 2009;30:568–575. doi: 10.1097/AUD.0b013e3181ac128f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon-Salant S. Hearing loss and aging: new research findings and clinical implications. J Rehabil Res Dev. 2005;42:9–24. doi: 10.1682/jrrd.2005.01.0006. [DOI] [PubMed] [Google Scholar]
- 29.Simon H, Frisina RD, Walton JP. Age reduces response latency of mouse inferior colliculus neurons to AM sounds. J Acoust Soc Am. 2004;116:469–477. doi: 10.1121/1.1760796. [DOI] [PubMed] [Google Scholar]