

Abstract
Background and Objective:
Smoking is an important environmental risk factor involved in the causation and progression of periodontal disease. Smoking can impair various components of the host immune response and immune system. The virulence factors of periodontal pathogens stimulate inflammatory cytokine expression by mononuclear cells. IL-1β is the key mediator involved in the pathogenesis and disease progression. Therefore, whole gingival biopsy samples are assessed for this increased expression of IL-1.
Materials and Methods:
29 male subjects’ age and gender matched were divided into three groups based on the periodontal and smoking status (Group A:healthy, Group B: non-smokers suffering from chronic periodontitis, Group C: smokers suffering from chronic periodontitis). Periodontal parameters like plaque index, gingival index, probing pocket depth and clinical attachment level were recorded at baseline and post scaling. The mRNA expression of IL-1β was determined by real time polymerase chain reaction and correlated with the periodontal and smoking status.
Results:
The improvement in the periodontal parameters was statistically significant in the non- smokers (Group B) and there was a 2 fold increase in the mRNA expression in this group. The smokers (Group C) showed lesser improvement in the periodontal parameters and there was an 8 fold increase in the mRNA expression of IL-1β.
Conclusion:
Association of smoking status with periodontal destruction can thus be correlated with the increased mRNA expression of IL-1β in chronic periodontitis patients.
Keywords: Interleukin-1β, mRNA expression, periodontitis, smoking
INTRODUCTION
Periodontitis is a chronic infectious disease of multifactorial nature characterized by the irreversible destruction of collagen fibers and other matrix constituents of the gingiva, periodontal ligament and alveolar bone around teeth in conjunction with the formation of periodontal pockets due to apical migration of the junctional epithelium.[1] Multiple factors viz. microbial, host-related, environmental, host response, and genetic determine the risk of developing periodontal disease. Host-related factors are age and health status, and environmental factors are oral hygiene, exposure to oral pathogens and tobacco use. The host response and genetic factors are tissue repair mechanisms and the leucocytes of the immune system. These factors are inter-related, and the risk for developing periodontal disease is governed by their convergence.
Chemoattractant cytokines regulate the immune response by recruiting neutrophils and macrophages, and their expression is controlled by microbial and host factors. Interleukin-1 (IL-1) is a key regulator of tissue destruction and mediator of the local immune response.[2] Its varied biologic functions include prostaglandin E2 (PGE2) and collagenase synthesis by fibroblasts, increased production of IL-2 from T-lymphocytes and interferon, stimulation of bone resorption, and the induction and secretion of proteinases. All IL-1β dependent mechanisms contribute to inflammation and destruction of bone and attachment loss, which are characteristic features of periodontal disease.[3]
The presence of cytokines may be detected earlier than the appearance of acute symptoms of periodontal disease,[4] therefore, the measurement of IL-1β in adjacent tissues of periodontitis affected sites has been suggested as a sensitive and important aid in monitoring the clinical severity of the disease. Detection of the mRNA levels of the cytokine in diseased sites is an effective technique to study variations in cytokine levels.[5]
Smoking is a major risk factor in the development and progression of periodontal diseases,[6] and its effect on the extent and prevalence of periodontal destruction is dose-dependent. Measures of association of smoking status and periodontal destruction have been rather strong and consistent across studies and population.[7] Tobacco use is among the most important preventable risk factor in the incidence and progression of periodontal disease.[8] It impairs various components of the host response and immune system like inhibition of neutrophil chemotaxis and phagocytosis, inhibition of cellular immunity, and suppression of local antibody production.
Nicotine, the major constituent of tobacco smoke and its metabolites mask the early signs of periodontal disease by suppressing the inflammatory response. Soon after its intake in the body, much of the nicotine gets converted into cotinine, which is the major proximate metabolite of nicotine. The effects of nicotine thus, observed in the body are due to cotinine which affects several biological activities in the body.
In the periodontium, there is induced vasoconstriction. This contributes to impaired gingival blood flow and decreased the amount of oxygen and blood constituents that reach the gingiva. The reduced capacity to remove tissue waste products leads to tissue damage and a compromised immune response. Nicotine and cotinine get incorporated into the root surfaces and into fibroblasts which may inhibit reattachment of fibroblasts to the root surface.[9]
The present investigation was performed to study the effect of nicotine and its proximate metabolite cotinine. Thus, it was studied whether these have an influence on the periodontal status (plaque index [PI], gingival index [GI], probing pocket depth [PPD], and clinical attachment level [CAL]) and the mRNA expression of IL-1β in smokers suffering from chronic periodontitis.
MATERIALS AND METHODS
The study conformed to the ethical guidelines of the Helsinki Declaration and was evaluated and approved by the Institutional Ethical Committee. A written informed consent was obtained from all subjects participating in the study.
Patient selection
A total of 29 male individuals (12 smokers and 12 nonsmokers suffering from chronic periodontitis and 5 periodontally healthy nonsmokers) aged between 37 and 50 years were enrolled and a brief medical and dental history and information about the smoking status was obtained. The selection criteria were based on the following criteria.
Inclusion criteria
Male patients within the age group of 35–50 years
Patients who had smoked an average of >10 cigarettes/day for >2 years[10,11]
Patients having chronic periodontitis defined as having minimum of 20 remaining teeth, with periodontal disease as evidenced by at least 4 tooth sites with PPD >4 mm, CAL >2 mm;[12] and radiographic evidence of bone loss >2 mm from the cementoenamel junction[13]
Patients who were cooperative and committed to maintain oral hygiene
Patients with no contraindication to periodontal therapy.
Exclusion criteria
Beedi, cigar, pipe smokers, and paan masala chewers and passive smokers
Patients suffering from chronic systemic illness like diabetes, hypertension, active infection etc.; and taking medication for the same[14]
Patients taking agents or medication that may alter or hamper the salivary flow
Patients suffering from aggressive periodontitis, periodontal abscess, necrotizing ulcerative gingivitis or periodontitis[10]
Patients who have undergone any periodontal treatment or antibiotics within the preceding 6 months[14]
Known allergy or hypersensitivity to local anesthetic agents.
The study protocol was explained, and patients were grouped the following:
Group A: Periodontally healthy individuals undergoing therapeutic extraction for orthodontic treatment or extraction of impacted molars[15]
Group B: Nonsmokers suffering from chronic generalized periodontitis
Group C: Smokers suffering from chronic generalized periodontitis.
Study design
Periodontal parameter recording - PPD, CAL, GI[16] and PI[17] and scaling and root planning was performed at baseline and 1-month posttreatment. There was no restriction on the time spent on each tooth and the total time devoted to achieve total plaque control. At the 1-month recall visit, periodontal parameters were re-recorded, followed by flap procedures to obtain tissue samples. These tissue specimens were then subjected to mRNA analysis.
Smoking status
Twelve current smokers[18] suffering from chronic periodontitis were questioned about number of cigarettes smoked per day and number of years smoked. The pack-years were calculated accordingly.[8]
Estimation of cotinine in the salivary sample
The major proximate metabolite of nicotine is cotinine, and its levels were evaluated in saliva using Accutest® NicAlert™ test strip to assess nicotine exposure. Unstimulated saliva was collected at the 1-month posttreatment visit. All samples were collected between 9 am and 11 am, as diurnal variation in saliva viscosity has been observed. Eight drops of saliva were squeezed on the padded end, allowing each drop to soak into it before applying the next drop. The readings in the strip were recorded after 20 min.
For the purposes of assigning a semi-quantitative measure of tobacco exposure, the Accutest® NicAlert™ levels are expressed in terms of cotinine.
Each test strip is divided into six reactive chromo-graphic (color-change) levels of detection. The results are semi-quantitative and are based on the detection matrix provided by the manufacturer.
Gingival biopsies
Twenty-nine tissue specimens (12 from smokers, 12 from nonsmokers) and 5 from healthy individuals were harvested. From the periodontitis patients, biopsies were taken from sites with radiologic evidence of alveolar bone loss during Modified Widman flap surgery. For the healthy individuals, specimens were collected from tooth extraction sites.
All biopsies weighed about 15–30 mg and were washed with cold saline and immediately kept in vials containing TRIZOL solution and stored at −4°C until analyzed.
Interleukin-1β expression analysis
The frozen tubes containing the tissue samples were thawed, and the total RNA was isolated by following TRIZOL reagent technique. RNA concentration was quantified using spectrophotometer (PicoDrop®) and 250 ng of RNA sample was first reverse transcribed to cDNA with reverse transcriptase (Fermentas RevertAid™ H minus First Strand cDNA Synthesis kit #K1632) in a thermal cycler (Peltier Thermal cycler PTC-200). The reverse transcription reaction product was stored at −80°C for further investigation.
Real-time polymerase chain reaction
SYBR green-based quantitative-polymerase chain reaction (q-PCR) was done to check the expression of IL-1β with reference to β-actin (Forward primer CGACAGCAGTTGGTTGGAGC; Reverse primer GGTCTCAAGTCAGTGTACAG) gene as an internal control. Real-time PCR was performed using Takara reaction mix (Stratagene M × 3000®) in optically active tubes as per manufacturer's instructions.
1 μl each of forward (5’-AAACAGATGAAGTGCTCCTTCCAGG-3’) and reverse (5’-TGGAGAACACCACTTGTTGCTCCA-3’) primers of IL-1β (ACTIVE™-Oligos ILS) with 10 μl of SYBR, exTaq and 1 μl of cDNA was used as the reaction mix. The amplified q-PCR product was also confirmed by 2% agarose gel.
Statistical analysis
Three independent groups were compared by one-way analysis of variance (ANOVA), and the significance of the mean difference between the groups was done by Newman–Keuls post hoc test. Similarly, pre- and post-treatment levels of two independent Groups B and C were compared by repeated measures ANOVA using general linear models and the significance of mean difference within and between the groups was done by Newman–Keuls test. A two-tailed (α =2) P < 0.05 was considered to be statistically significant. The power of all statistical tests was 80.0%. All analyses were performed on SPSS (version 150.0, IBM SPSS Statistics) while graphs were made on GraphPad Prism (version 3.0).
The present study compares the periodonat status (PI, GI, PPD, and CAL) and mRNA expression of IL-1β in periodontally healthy nonsmokers, nonsmokers with chronic periodontits and smokers with chronic periodontits. A prior sample size based on an effect size of 2 (mean difference) and 1.1 pooled standard deviation (SD) in any of the variable among the groups with 5.0% margin of error (α = 0.05), 80.0% power (1− β =0.80) and 1:1:1 ratio, minimum 5 samples is required for one group or total 15 samples for three groups was calculated using online standard statistical software (MedCalc, version 9.4.2.0 MedCalc, Mariakerke, Belgium).
RESULTS
Twenty-nine subjects were included in the study with the mean age of 43.25 ± 4.16 years. In smokers, the number of cigarettes per day, number of years smoked and pack-years ranged from 8 to 20, 10–20 years, 4–20 years, respectively, with mean ± SD 14.92 ± 4.36, 14.58 ± 4.19 years and 11.13 ± 5.02 years, respectively, while it were NIL in control and nonsmokers.
Clinical parameters
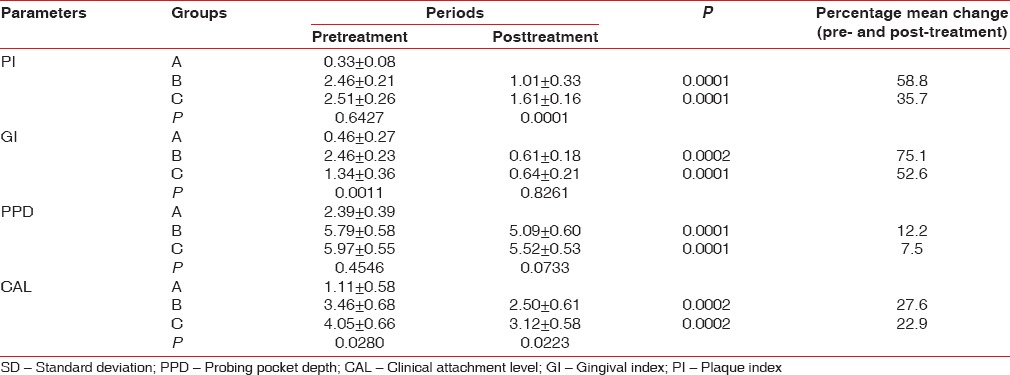
The mean PI in control, nonsmokers and smokers was 0.33 ± 0.08, 2.46 ± 0.21 and 2.51 ± 0.26, respectively, and it became 1.01 ± 0.33 and 1.61 ± 0.16 in group nonsmokers and smokers after 1-month (P = 0.0001).
Gingival index at baseline in control, nonsmokers and smokers was 0.46 ± 0.27, 2.46 ± 0.23 and 1.34 ± 0.36, respectively, P < 0.001 when control was compared to nonsmokers and smokers and P < 0.001 on intergroup comparison of nonsmokers and smokers. One month posttreatment the GI was 0.61 ± 0.18 and 0.64 ± 0.21 in nonsmoker group and smokers with chronic periodontitis, respectively. The percentage improvement in nonsmokers was 75.1% (P = 0.0002), and smokers were 52.6% (P = 0.0001) as smoking significantly decreases the vascularity of the gingiva. The baseline scores in smokers were significantly low as compared with the other groups and scores decreased after scaling but since smoking cessation did not occur the vascularity of the gingiva was still compromised and improvement was not as much as in the other groups.
Probing pocket depth in control group, nonsmokers and smokers was 2.39 ± 0.39, 5.79 ± 0.58 and 5.97 ± 0.55, respectively. Following periodontal therapy there was a 12.2% improvement in nonsmokers, as the mean PPD became 5.09 ± 0.60 (P = 0.0001) and for smokers it was 7.5% as the value came to 5.52 ± 0.53 (P = 0.0001). Improvements of 27.6% and 22.9% were seen in nonsmokers and smokers after treatment (P = 0.0002), as the CAL values became 2.50 ± 0.61 and 3.12 ± 0.58, respectively, though intergroup comparison posttreatment showed no statistical significance (P = 0.0223) [Table 1].
Table 1.
Pre- and post-treatment periodontal parameters and percentage improvement following periodontal treatment of each of the three groups (mean±SD)
Estimation of cotinine in saliva
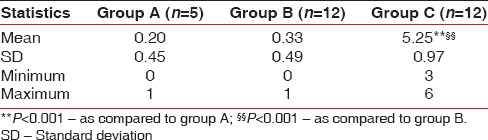
The scores of cotinine level in saliva of Group A, Group B, and Group C ranged from 0 to 1, 0–1 and 3–6, respectively, with mean ± SD 0.20 ± 0.45, 0.33 ± 0.49, and 5.25 ± 0.97, respectively. Comparing the scores of cotinine in saliva among the three groups, the mean scores of cotinine in healthy controls and nonsmokers with chronic periodontitis did not differ significantly, that is, found to be statistically the same (0.20 ± 0.45 vs. 0.33 ± 0.49; P = 0.7136), while the mean score in smokers was found to be significantly different and higher than both Group A (0.20 ± 0.45 vs. 5.25 ± 0.97, P = 0.0001) and Group B (0.33 ± 0.49 vs. 5.25 ± 0.97; P = 0.0001) [Table 2].
Table 2.
Scores of cotinine level in saliva summary of three groups
Expression analysis
The cycle threshold (ΔCt) levels were highest in control, followed by nonsmokers and smokers which indicates that that greatest number of PCR cycles were required to replicate in case of Group A and least in Group C. Therefore, relative quantity or the fold change in the expression of IL-1β showed that chronic periodontitis causes a two-fold increase and smoking associated with chronic periodontitis causes an eight-fold increase in the expression [Figure 1].
Figure 1.
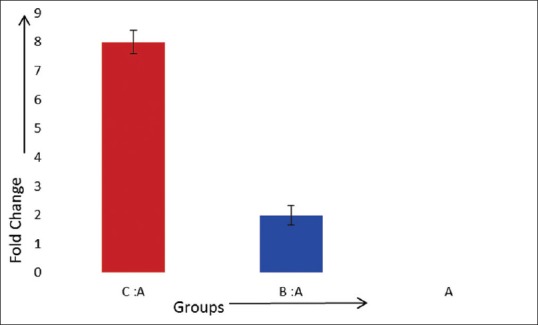
Fold change of interleukin-1β in comparison to healthy (controls) group
The correlation between smoking, mRNA expression, and periodontal parameters of all studied subjects [Table 3] showed that there was a positive and significant (P < 0.01) correlation among smoking related parameters (number of cigarettes/day vs. number of years smoked: r = 0.92, P < 0.01; number of cigarettes/day vs. pack years: r = 0.95, P < 0.01; number of years smoked vs. pack years: r = 0.95, P < 0.01). All smoking related parameters viz. number of cigarettes per day (r = 0.97, P < 0.01), number of years smoked (r = 0.95, P < 0.01), pack years (r = 0.93, P < 0.01) showed positive and significantly high correlation; while they showed negative and significant (P < 0.01) correlation with mRNA expression, signifying that the mRNA level is increased in smokers with periodontitis.
Table 3.
Correlation between smoking, cotinine, mRNA expression, and baseline periodontal parameters of all studied subjects (n=29)
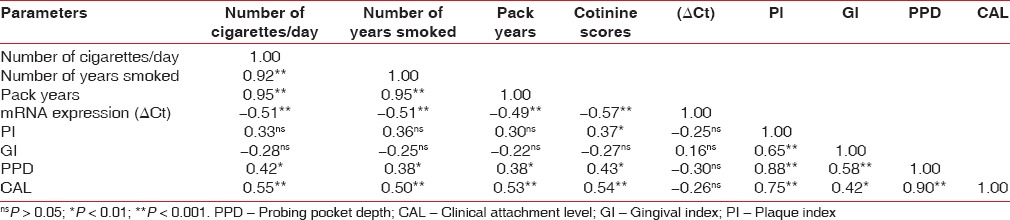
All smoking-related parameters did not show significant (P < 0.05) correlation with periodontal parameters such as PI and GI; while they showed positive and significant (P < 0.05 or P < 0.01) correlation with PPD (P < 0.05) and CAL (P < 0.01) [Figure 2].
Figure 2.
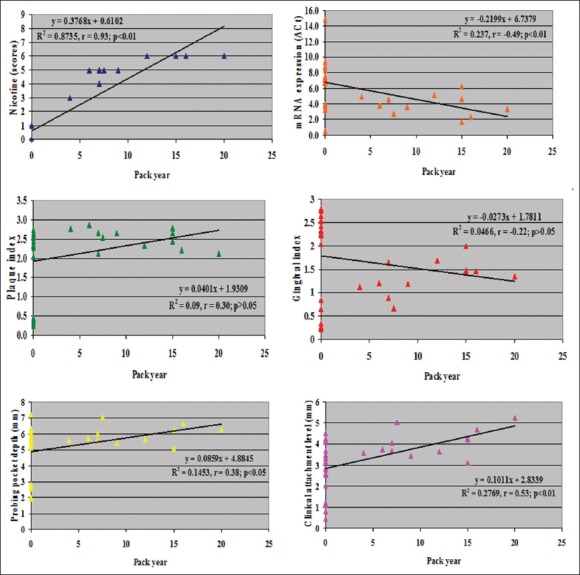
Correlation of gingival index, probing pocket depth, and clinical attachment level with pack years in all studied subjects (n = 29) and correlation of cotinine, mRNA expression and plaque index with pack years in all studied subjects (n = 29)
DISCUSSION
Interleukin-1 has proven to be the key regulator of tissue homeostasis, inflammation, immunity, and tissue breakdown. It stimulates gingival fibroblasts to produce collagenase and PGE2, which is a vasodilator and a mediator of bone destruction. Along-with tumor necrosis factor-α acts on endothelial cells to increase the attachment of neutrophils and monocytes and thereby aids in the recruitment of these cells into the sites of inflammation.[19]
Twenty-nine male patients were enrolled for the present study to assess the effect of nicotine and metabolites on periodontal disease status and the mRNA expression of IL-1β. All the individuals fell in the age range of 37–50 years and the groups were age-matched; as epidemiological studies have shown that smokers aged < 30 years are comparable to nonsmokers.[20] Individuals > 50 years have usually a considerable reduction in smoking due to development of medical problems aggravated by smoking and aging.[10] The mean age in the periodontally healthy group was 43.40 ± 4.67, nonsmokers with chronic periodontits group was 43.25 ± 4.16 and smokers suffering from chronic periodontitis C was 43.58 ± 4.36.
Only male patients were enrolled because the factor of sex affects the severity of periodontal disease, showing that male smoker patients have 17% higher recession, 6% greater probing depth, and 8% more clinical attachment loss than females.[21,22,23]
Smokers smoking >10 cigarettes/day for >2 years were included as nicotine and metabolites have their effect on periodontal tissues over a certain threshold. As the consumption increases so does the severity of periodontal disease, although the effect seems to clinically evident from >10 cigarettes/day.[10]
All periodontal parameters were found to be higher in Group B and C, compared to Group A. There was not much difference between nonsmokers and smokers suffering from chronic periodontitis at baseline, except the GI.
Higher plaque accumulation in Group B and C could be attributed to poor oral hygiene, pain and bleeding while brushing and improper brushing technique. Several studies have reported difference in the prevalence, bacterial counts of microflora in pockets of smokers and nonsmokers.[24,25,26,27]
0.7 times more improvement was observed in nonsmokers (75.1%) than smokers as smoking creates an environment that favors the colonization of anaerobic pathogens and smokers were also associated with poorer oral hygiene and suppressed hemorrhagic response.[28]
The gingival inflammatory response is dampened in smokers as compared to nonsmokers, evidenced by fibrotic appearance of the tissue fewer sites that bleed upon probing. The hemorrhagic responsiveness in patients with different levels of periodontal disease was found to be suppressed in smokers while in nonsmokers, an exponential hemorrhagic response to increasing plaque levels was suggested. When looking at the gingival vessel density smokers have a higher proportion of small blood vessels and a lower proportion of large blood vessels compared to nonsmokers. Thus, this difference in blood vessels may be associated with a suppression of the inflammatory response.[29]
One month posttreatment the percentage improvement in GI in nonsmokers was 75.1% (P = 0.0002) and smokers was 52.6% (P = 0.0001). Although improvement occurred in both the groups, patients in Group B improved 0.7 times more than C. Lesser improvement in Group C could be attributed also to the sustained vasoconstriction induced by nicotine, as none of the individuals quit smoking.[30] The gingival biopsies were obtained from the worst affected sites after 1-month posttreatment.
Nicotine is the principal alkaloid in tobacco and is responsible for causing dependence due to its psychoactive properties and capacity to induce self-administration behavior.[28] It is estimated that 5 mg of nicotine per day is a threshold level that can readily establish and sustain addiction.[31] Nicotine is a weak base (pKa 8.0), present mainly in the nonionized form in alkaline pH, and hence more quickly absorbed from saliva into the mucosa at increased pH levels.[32] Approximately 1–2 mg of the nicotine that is present in a cigarette is absorbed by the buccal mucosa and respiratory mucosa of smokers. Once circulating in the bloodstream, nicotine acts as a central and peripheral nervous system stimulant. Long-term tobacco use has been associated with increased incidence of carcinoma, atherosclerotic arterial disease, chronic obstructive pulmonary disease, hypertension, low birth weight of infants born to mothers who smoke and periodontal disease.[33]
When nicotine enters the blood stream, it is circulated to various body organs, including the liver and kidneys. The liver converts nicotine into several metabolites and a small percentage is excreted unchanged in the urine.
Cotinine is the major proximate metabolite and is used as a biomarker for estimating the exposure to nicotine. Determining the concentration of nicotine and cotinine in biological fluids is widely practiced in epidemiological and clinical smoking studies.[28]
Cotinine concentrations have been used more accurately in assessing population exposures to environmental tobacco smoke (ETS) and developing risk estimates.[31,34,35]
The Accutest® NicAlert™ test strip was used and the results were based on the detection matrix provided by the manufacturer. Unstimulated salivary samples were collected for all groups. The scores of cotinine level in saliva of Group A, Group B and Group C ranged from 0 to 1, 0–1 and 3–6, respectively, with mean ± SD 0.20 ± 0.45, 0.33 ± 0.49, and 5.25 ± 0.97, respectively. Scores for Group A and B were statistically the same (P = 0.7136) as the patients were nonsmokers. Minor detection levels could be attributed to the ETS exposure. Scores for Group C were significantly higher (P = 0.0001) as all the patients were chronic smokers, smoking 8–20 cigarettes/day for the past 10–20 years.
2000+ nicotine levels were found in smoker's saliva who smoked about 15–20 cigarettes/day. As the oral cavity is exposed to more and more nicotine, more cotinine would be produced owing to the detection levels so significant. None of the individuals quit smoking during the course of the study; in spite of the health implications explained them in detail at the commencement of the study.
The mean ΔCt value was found to be the highest in periodontally healthy, followed by nonsmokers and smokers, indicating that greater number of cycles were required to produce detectable levels of IL-1β for Group A and least for Group C. In other words, the mRNA expression was up-regulated in both Group B and Group C as compared to Group A and more evident in Group C than in Group B.
Relative quantity or the fold change in the expression of IL-1β showed that chronic periodontitis causes a two-fold increase and a risk factor; smoking associated with chronic periodontitis causes an eight-fold increase in the expression. Increased mRNA expression in periodontitis affected sites is due to the hyperresponsiveness of the gingival cells to lipopolysaccharides present in the subgingival plaque. Studies have shown that mononuclear cells, fibroblasts, and epithelial cells isolated from periodontitis patients tend to be primed to respond to bacterial lipopolysaccharide and show in vitro production of higher levels of IL-1β.[32]
When smoking, a confounding factor is present along with chronic periodontitis the expression increases eight-fold. It can be postulated that vasoconstrictive properties of nicotine impair the gingival blood flow, creating an anaerobic environment which is highly conducive for the growth of periodontopathogens. With the rapid increase in the number and quality of anaerobes, greater stimulation of the gingival monocytes and macrophages leads to an increased expression of IL-1β.[36] Furthermore, cigarette smoke stimulates the neutrophil oxidative burst with the release of potentially tissue destructive oxygen species such as superoxide, hydrogen peroxide and hydroxyl radical. These products of the oxidative burst not only have direct cytotoxic effects on cells of the periodontium, but also alter the release of cytokines from the inflammatory cells particularly monocytes and macrophages.[33]
The oral keratinocytes are vastly exposed to tobacco smoke and its components and respond to this exposure by producing pro-inflammatory cytokines such as PGE2 and IL-1. Elevated IL-1 production by oral keratinocytes plays a key role in the tissue injury.[29] Owing to these mechanisms, there is marked upregulation in the gene expression of Il-1β and greater tissue destruction in smokers suffering from chronic periodontitis.
The limitations of the study include small sample size, inability to perform subsequent sampling and their expression studies at different intervals. Furthermore, the topic of smoking cessation and former smokers was not addressed. Further studies with large sample sizes are needed for the generalization of the results.
CONCLUSION
The results of this study have demonstrated an upregulation of 2 times in the IL-1β gene expression in chronic periodontitis and 8 times more pronounced in smokers suffering from chronic periodontitis in a dose-dependent manner.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Page RC, Kornman KS. The pathogenesis of human periodontitis: An introduction. Periodontol 2000. 1997;14:9–11. doi: 10.1111/j.1600-0757.1997.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 2.Bickel M, Nöthen SM, Freiburghaus K, Shire D. Chemokine expression in human oral keratinocyte cell lines and keratinized mucosa. J Dent Res. 1996;75:1827–34. doi: 10.1177/00220345960750110301. [DOI] [PubMed] [Google Scholar]
- 3.Hou LT, Liu CM, Liu BY, Lin SJ, Liao CS, Rossomando EF. Interleukin-1beta, clinical parameters and matched cellular-histopathologic changes of biopsied gingival tissue from periodontitis patients. J Periodontal Res. 2003;38:247–54. doi: 10.1034/j.1600-0765.2003.02601.x. [DOI] [PubMed] [Google Scholar]
- 4.Kinane DF, Winstanley FP, Adonogianaki E, Moughal NA. Bioassay of interleukin 1 (IL-1) in human gingival crevicular fluid during experimental gingivitis. Arch Oral Biol. 1992;37:153–6. doi: 10.1016/0003-9969(92)90011-v. [DOI] [PubMed] [Google Scholar]
- 5.Bouaboula M, Legoux P, Pességué B, Delpech B, Dumont X, Piechaczyk M, et al. Standardization of mRNA titration using a polymerase chain reaction method involving co-amplification with a multispecific internal control. J Biol Chem. 1992;267:21830–8. [PubMed] [Google Scholar]
- 6.Tobacco use and the periodontal patient. J Periodontol. 1996;67:51–6. [PubMed] [Google Scholar]
- 7.Bregstrom J. Tobacco smoking and risk of periodontal disease. J Clin Periodontol. 2003;30:107–13. doi: 10.1034/j.1600-051x.2003.00272.x. [DOI] [PubMed] [Google Scholar]
- 8.Grossi SG, Zambon JJ, Ho AW, Koch G, Dunford RG, Machtei EE, et al. Assessment of risk for periodontal disease. I. Risk indicators for attachment loss. J Periodontol. 1994;65:260–7. doi: 10.1902/jop.1994.65.3.260. [DOI] [PubMed] [Google Scholar]
- 9.Sukumaran A. Gingival crevicular fluid immunoglobulins in smokers with chronic periodontitis. Pak Oral Dent J. 2008;28:51–6. [Google Scholar]
- 10.Grossi SG, Genco RJ, Machtei EE, Ho AW, Koch G, Dunford R, et al. Assessment of risk for periodontal disease. II. Risk indicators for alveolar bone loss. J Periodontol. 1995;66:23–9. doi: 10.1902/jop.1995.66.1.23. [DOI] [PubMed] [Google Scholar]
- 11.Boström L, Bergström J, Dahlén G, Linder LE. Smoking and subgingival microflora in periodontal disease. J Clin Periodontol. 2001;28:212–9. doi: 10.1034/j.1600-051x.2001.028003212.x. [DOI] [PubMed] [Google Scholar]
- 12.Lindhe J, Ranney R, Lamster I, Charles A, Chung CP, Flammig T, et al. Consensus report: Chronic periodontitis. Ann Periodontol. 1999;4:38. [Google Scholar]
- 13.Ng PY, Donley M, Hausmann E, Hutson AD, Rossomando EF, Scannapieco FA. Candidate salivary biomarkers associated with alveolar bone loss: Cross-sectional and in vitro studies. FEMS Immunol Med Microbiol. 2007;49:252–60. doi: 10.1111/j.1574-695X.2006.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tobón-Arroyave SI, Jaramillo-González PE, Isaza-Guzmán DM. Correlation between salivary IL-1beta levels and periodontal clinical status. Arch Oral Biol. 2008;53:346–52. doi: 10.1016/j.archoralbio.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Tokoro Y, Matsuki Y, Yamamoto T, Suzuki T, Hara K. Relevance of local Th2-type cytokine mRNA expression in immunocompetent infiltrates in inflamed gingival tissue to periodontal diseases. Clin Exp Immunol. 1997;107:166–74. doi: 10.1046/j.1365-2249.1997.d01-880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loe H, Silness J. Periodontal disease in pregnancy. I. Prevalence and severity. Acta Odontol Scand. 1963;21:533–51. doi: 10.3109/00016356309011240. [DOI] [PubMed] [Google Scholar]
- 17.Silness J, Loe H. Periodontal disease in pregnancy. II. Correlation between oral hygiene and periodontal condtion. Acta Odontol Scand. 1964;22:121–35. doi: 10.3109/00016356408993968. [DOI] [PubMed] [Google Scholar]
- 18.Hyattsville, MD: Centers for Disease Control and Prevention; 2004. U.S. Department of Health and Human Services NCIHS. NHANES III Reference Manuals and Reports. [Google Scholar]
- 19.Masada MP, Persson R, Kenney JS, Lee SW, Page RC, Allison AC. Measurement of interleukin-1 alpha and-1 beta in gingival crevicular fluid: Implications for the pathogenesis of periodontal disease. J Periodontal Res. 1990;25:156–63. doi: 10.1111/j.1600-0765.1990.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 20.Stoltenberg JL, Osborn JB, Pihlstrom BL, Herzberg MC, Aeppli DM, Wolff LF, et al. Association between cigarette smoking, bacterial pathogens, and periodontal status. J Periodontol. 1993;64:1225–30. doi: 10.1902/jop.1993.64.12.1225. [DOI] [PubMed] [Google Scholar]
- 21.Brown LF, Beck JD, Rozier RG. Incidence of attachment loss in community-dwelling older adults. J Periodontol. 1994;65:316–23. doi: 10.1902/jop.1994.65.4.316. [DOI] [PubMed] [Google Scholar]
- 22.Haber J, Wattles J, Crowley M, Mandell R, Joshipura K, Kent RL. Evidence for cigarette smoking as a major risk factor for periodontitis. J Periodontol. 1993;64:16–23. doi: 10.1902/jop.1993.64.1.16. [DOI] [PubMed] [Google Scholar]
- 23.Horning GM, Hatch CL, Cohen ME. Risk indicators for periodontitis in a military treatment population. J Periodontol. 1992;63:297–302. doi: 10.1902/jop.1992.63.4.297. [DOI] [PubMed] [Google Scholar]
- 24.Do LG, Slade GD, Roberts-Thomson KF, Sanders AE. Smoking-attributable periodontal disease in the Australian adult population. J Clin Periodontol. 2008;35:398–404. doi: 10.1111/j.1600-051X.2008.01223.x. [DOI] [PubMed] [Google Scholar]
- 25.Zambon JJ, Grossi SG, Machtei EE, Ho AW, Dunford R, Genco RJ. Cigarette smoking increases the risk for subgingival infection with periodontal pathogens. J Periodontol. 1996;67:1050–4. doi: 10.1902/jop.1996.67.10s.1050. [DOI] [PubMed] [Google Scholar]
- 26.Haffajee AD, Cugini MA, Dibart S, Smith C, Kent RL, Jr, Socransky SS. The effect of SRP on the clinical and microbiological parameters of periodontal diseases. J Clin Periodontol. 1997;24:324–34. doi: 10.1111/j.1600-051x.1997.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 27.Kamma JJ, Nakou M, Baehni PC. Clinical and microbiological characteristics of smokers with early onset periodontitis. J Periodontal Res. 1999;34:25–33. doi: 10.1111/j.1600-0765.1999.tb02218.x. [DOI] [PubMed] [Google Scholar]
- 28.Bergström J, Boström L. Tobacco smoking and periodontal hemorrhagic responsiveness. J Clin Periodontol. 2001;28:680–5. doi: 10.1034/j.1600-051x.2001.028007680.x. [DOI] [PubMed] [Google Scholar]
- 29.Tambwekar KR, Kakariya RB, Garg S. A validated high performance liquid chromatographic method for analysis of nicotine in pure form and from formulations. J Pharm Biomed Anal. 2003;32:441–50. doi: 10.1016/s0731-7085(03)00236-x. [DOI] [PubMed] [Google Scholar]
- 30.Ryder MI, Fujitaki R, Johnson G, Hyun W. Alterations of neutrophil oxidative burst by in vitro smoke exposure: Implications for oral and systemic diseases. Ann Periodontol. 1998;3:76–87. doi: 10.1902/annals.1998.3.1.76. [DOI] [PubMed] [Google Scholar]
- 31.Ciolino LA, McCauley HA, Fraser DB, Wolnik KA. The relative buffering capacities of saliva and moist snuff: Implications for nicotine absorption. J Anal Toxicol. 2001;25:15–25. doi: 10.1093/jat/25.1.15. [DOI] [PubMed] [Google Scholar]
- 32.Johnson GK, Organ CC. Prostaglandin E2 and interleukin-1 concentrations in nicotine-exposed oral keratinocyte cultures. J Periodontal Res. 1997;32:447–54. doi: 10.1111/j.1600-0765.1997.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 33.Mirbod SM, Ahing SI, Pruthi VK. Immunohistochemical study of vestibular gingival blood vessel density and internal circumference in smokers and non-smokers. J Periodontol. 2001;72:1318–23. doi: 10.1902/jop.2001.72.10.1318. [DOI] [PubMed] [Google Scholar]
- 34.Hatsukami DK, Hecht SS, Hennrikus DJ, Joseph AM, Pentel PR. Biomarkers of tobacco exposure or harm: Application to clinical and epidemiological studies 25-26 October 2001, Minneapolis, Minnesota. Nicotine Tob Res. 2003;5:387–96. doi: 10.1080/1462220031000094222. [DOI] [PubMed] [Google Scholar]
- 35.Hoofnagle AN, Laha TJ, Rainey PM, Sadrzadeh SM. Specific detection of anabasine, nicotine, and nicotine metabolites in urine by liquid chromatography-tandem mass spectrometry. Am J Clin Pathol. 2006;126:880–7. doi: 10.1309/LQ8U3UL956ET324X. [DOI] [PubMed] [Google Scholar]
- 36.Yoshinari N, Kawase H, Mitani A, Ito M, Sugiishi S, Matsuoka M, et al. Effects of scaling and root planing on the amounts of interleukin-1 and interleukin-1 receptor antagonist and the mRNA expression of interleukin-1beta in gingival crevicular fluid and gingival tissues. J Periodontal Res. 2004;39:158–67. doi: 10.1111/j.1600-0765.2004.00722.x. [DOI] [PubMed] [Google Scholar]