

Abstract
Alexander Disease (AxD) is a ‘gliopathy’ caused by toxic, dominant gain-of-function mutations in the glial fibrillary acidic protein (GFAP) gene. Two distinct types of AxD exist. Type I AxD affected individuals develop cerebral symptoms by four years of age and suffer from macrocephaly, seizures, and physical and mental delays. As detection and diagnosis have improved, approximately half of all AxD patients diagnosed have onset >4 years and brainstem/spinal cord involvement. Type II AxD patients experience ataxia, palatal myoclonus, dysphagia and dysphonia. No study has examined a mechanistic link between the GFAP mutations and caudal symptoms present in type II AxD patients. We demonstrate that two key astrocytic functions, the ability to regulate extracellular glutamate and to take up K+ via K+ channels, are compromised in hindbrain regions and spinal cord in AxD mice. Spinal cord astrocytes in AxD transgenic mice are depolarized relative to WT littermates, and have a threefold reduction in Ba2+-sensitive Kir4.1 mediated currents and six-fold reduction in glutamate uptake currents. The loss of these two functions is due to significant decreases in Kir4.1 (>70%) and GLT-1 (>60%) protein expression. mRNA expression for KCNJ10 and SLC1A2, the genes that code for Kir4.1 and GLT-1, are significantly reduced by postnatal day 7. Protein and mRNA reductions for Kir4.1 and GLT-1 are exacerbated in AxD models that demonstrate earlier accumulation of GFAP and increased Rosenthal fiber formation. These findings provide a mechanistic link between the GFAP mutations/overexpression and the symptoms in those affected with type II AxD.
Keywords: Kir4.1, GLT-1, potassium buffering, glutamate homeostasis, gliosis
Introduction
Alexander Disease (AxD) is a neurological disorder caused by dominant gain-of-function mutations in the gene encoding glial fibrillary acidic protein (GFAP) (reviewed in Brenner et al., 2009). Two distinct types, I & II exist. Individuals with type I, or early onset AxD, develop symptoms within the first few years of life and typically present with macrocephaly, seizures, and physical and mental delays, and display profound dysmyelination in the frontal lobes. As detection and diagnosis have improved, about half of AxD patients diagnosed have onset at any time during their lifespan and have signs of hindbrain involvement (Prust et al., 2011). Symptoms of these type II AxD patients include ataxia, palatal myoclonus, dysphagia and dysphonia; myelination defects may be restricted to more caudal brain regions or absent (Prust et al., 2011;Graff-Radford et al., 2014). A unifying pathological feature of AxD patients is the presence of GFAP protein aggregates, termed Rosenthal fibers (Wippold et al., 2006). Rosenthal fiber formation occurs in areas of highest GFAP expression: the perivascular and periventricular and subpial regions, and in the optic nerves, brainstem and spinal cord (Wippold et al., 2006;Rizzuto et al., 1980).
In healthy brain, GFAP expression is developmentally upregulated and demonstrates regional differences in promoter activity, mRNA, and protein expression (Palfreyman et al., 1979;Martin and O’Callaghan, 1995;Lein et al., 2007;Jany et al., 2013;Sosunov et al., 2013). Brain areas demonstrating most profound pathology and cellular changes in AxD correlate with those regions which express the highest levels of GFAP (Sosunov et al., 2013;Jany et al., 2013). In humans, caudal brain regions are the most profoundly affected in type II AXD, and in both humans and rodents these regions express the highest levels of GFAP protein and mRNA (Palfreyman et al., 1979;Martin and O’Callaghan, 1995). However, to date, astrocytes in caudal brain regions of animal models of AxD have not been examined.
In the current study, we examined two key astrocytic proteins, Kir4.1 and GLT-1, in brainstem and spinal cord in AxD mouse models. Kir4.1 is implicated in K+ channel-mediated K+ uptake while GLT-1 clears the extracellular space of neuronally released glutamate. Kir4.1 has particularly high expression in the spinal cord (Nwaobi et al., 2014), where it contributes to the high resting K+ conductance and the hyperpolarized resting membrane potential observed in mature astrocytes. GLT-1 is an electrogenic transporter and relies on the negative membrane potential provided by Kir4.1 for glutamate import (Kucheryavykh et al., 2007;Djukic et al., 2007). Dysfunction or inhibition of either protein results in increased neuronal excitability, which can lead to seizure activity. High concentrations of extracellular potassium [K+]o or glutamate [Glu−]o, for prolonged periods causes neuronal cell death (Kaiser et al., 2006;Balestrino et al., 1986;Bao et al., 2009;Choi, 1987;Reinert et al., 2000). Intriguingly, ‘reactive’ or gliotic astrocytes have a reduced capacity to perform these two homeostatic functions (Olsen et al., 2010;Dunlop et al., 2003;Tian et al., 2010). Part of the characterization of AxD astrocytes as ‘reactive’ (Sosunov et al., 2013) includes a loss of GLT-1 in hippocampus and cortex (Tang et al., 2010;Cho and Messing, 2009;Sosunov et al., 2013). In situ, the electrical properties of reactive astrocytes are similar to those seen in immature astrocytes; they have depolarized resting membrane potentials, reduced K+ conductance and reduced glutamate uptake (for review see, (Olsen and Sontheimer, 2008).
Using three transgenic AxD mouse models, we found that loss of Kir4.1 and GLT-1 proteins was most significant in animal models that presented with GFAP accumulation. Reduced Kir4.1 expression in AxD astrocytes was associated with decreased astrocytic whole-cell K+ currents and concomitant decrease in K+ uptake currents in response to puff application. Similarly, these cells demonstrated decreased glutamate transporter currents in response to a challenge of exogenously applied glutamate. These data suggest that glutamate homeostasis and Kir4.1-mediated channel uptake is altered in caudal brain regions, which provides a mechanistic link between the mutations in GFAP and the symptoms associated with type II AxD.
Methods
Animals
All animal procedures and protocols were performed in accordance with the National Institutes of Health guidelines and experiments described here were annually reviewed and approved by the University of Alabama Institutional Animal Care and Use Committee. Mouse lines have been described previously (Messing et al., 1998;Hagemann et al., 2006). Animals were housed with free access to food and water and were maintained on a 12hr light/dark cycle. Genotypes of offspring were confirmed by PCR of DNA isolated from tail (Terra Direct PCR, Clontech).
Slice preparation
Either gender postnatal day (PND) 13–18 pups were anesthetized with CO2 and decapitated, and the spinal cord was removed and placed for 30 s in ice-cold, low calcium, artificial cerebral spinal fluid (ACSF, containing in mM, 116 NaCl, 4.5 KCl, 1.0 MgCl2, 0.2 CaCl2, 26.2 NaHCO3, 11.1 glucose, and 5.0 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) sodium salt). The cord was then placed in liquid low-melt agarose at ~30°C, which was quickly solidified by placing on ice. Coronal sections were cut at 300 μm using a Vibratome 3000 (Ted Pella, Redding, CA) in low calcium ACSF. Before recording, slices were allowed to recover for ≥1 hr at room temperature in ACSF containing 2 mM CaCl2, which was continuously bubbled with 5% CO2-95% O2.
Slice electrophysiology
Whole cell voltage-clamp recordings were made as described previously (Olsen et al., 2006). Patch pipettes were made from thin-walled (outer diameter 1.5 mm, inner diameter 1.12 mm) borosilicate glass (TW150F-4 World Precision Instruments) and had resistances of 6 – 9 MΩ. Slices were transferred after the recovery period to a Zeiss Axio Examiner D1 microscope with a 40X water immersion lens to visualize astrocytes. Signals were acquired using an Axopatch 200B amplifier (Axon Instruments) controlled by Clampex 10.2 software via a Digidata 1440A interface (Molecular Devices). Signals were filtered at 2 kHz and digitized at 5 kHz. Data acquisition and storage were conducted with the use of pClamp 10.2 (Axon Instruments). Resting membrane potentials were measured directly from the amplifier in I = 0 mode ~1 min after whole cell access was obtained. Where described in the text, whole cell capacitance and series resistances were also measured directly from the amplifier, with the upper limit for series resistance being 10 MΩ and series resistance compensation adjusted to 80% to reduce voltage errors. The pipette solution contained (in mM) 125 K-gluconate, 10 KCl, 10 HEPES sodium salt, 10 creatine phosphate, 2 Mg-ATP, 0.2 Na-GTP, and 0.5 EGTA, pH adjusted to 7.3 with KOH and adjusted to 285–290 mOsm with sucrose. Cells were continuously superfused at 34°C with oxygenated ACSF containing 2 mM CaCl2. Drugs were added directly to these solutions.
Potassium/Glutamate Puffing
Potassium and glutamate solutions were applied for 400 msec using a Picospritzer (Warner Instruments) to astrocytes voltage clamped at −80 mV. The K+ puffing pipette solution contained 105 mM NaCl, 30 mM KCl, 2 mM MgCl2, 2 mM CaCl2, and 32.5 mM HEPES, pH adjusted to 7.4 with NaOH. The glutamate puffing solution contained 120 mM NaCl, 3.5 mM KCl, 1.3 mM MgCl2, 2.5 mM CaCl2, 25 mM HEPES, 10 mM glucose, 200 μM glutamate (pH adjusted to 7.4). Tetrodotoxin (TTX) 500 nM, 100 μM CdCl2, 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 20 μM bicuculline, and 20μM 2-amino-5-phosphonopentanoic acid (AP5) were added to the glutamate puffing and bath solutions to reduce neuronal excitability. Both glutamate and K+ puffing experiments were performed in normal ACSF. HEPES was used to maintain pH at 7.4 in the puffing pipette. For all puffing experiments, the puffer pipette was placed in the same focal plane as the voltage clamped cell and manipulated until a maximal response was elicited. All traces shown are the average of 3–4 consecutive applications of potassium or glutamate.
Immunoblotting
Following spinal cord extraction as described above, protein lysates were prepared by homogenization in lysis buffer (1% sodium dodecyl sulfate (SDS), 100 mM Tris(hydroxymethyl)aminomethane (Tris) buffer, pH 7.5, supplemented with protease and phosphatase inhibitors (Sigma) using glass dounce homogenizers, followed by 2 rounds of sonication for 10 seconds. Lysates were spun at 12,000xg for 5 minutes at 4°C. Protein concentration of supernatant was determined by bicinchoninic acid (BCA) assay (Thermo Scientific). Protein was heated to 60°C for 15 min in an equal volume of 2× loading buffer (100 mM Tris, pH 6.8, 4% SDS, in Laemmli-sodium dodecyl sulfate, 600 mM β-mercaptoethanol, 200 mM dithiothreitol (DTT), and 20% glycerol). Equal amounts (10 μg) of protein were loaded into each lane of a 4–20% gradient precast SDS polyacrylamide gel (Bio-Rad). Gels were transferred onto PVDF membrane (Millipore) at 100V for one hour. Membranes were blocked in blocking buffer (10% dried milk in Tris-buffered saline and Tween 20, (TBST) which contained 50 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH adjusted to 7.4 with HCl). Blots were incubated with Kir4.1 (Alomone #APC-035) primary antibody 1:750 in blocking buffer at room temperature for 90 minutes. The membrane was then rinsed 3× for 15 min each and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (Santa Cruz #SC2054, 1:2000) for 60 min at room temperature. After three 10 min washes with TBST, membranes were developed with Millipore Luminata Classic Western horseradish peroxidase substrate for visualization on CL-XPosure autoradiography film (Thermo Scientific). The blots were then stripped and re-probed with antibodies against GLT-1 (Millipore #AB1783, 1:10 000), GFAP (Millipore #MAB5628, 1:30,000), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Millipore #AB2302, 1:1000) for a loading control, and Millipore anti-guinea pig (#AP108P), Santa Cruz anti-mouse (#SC-2005), and anti-chicken (#SC-2901) secondary antibodies at 1:2000. ‘Densitometric analysis was performed utilizing Image J software. Relative density of GFAP, Kir4.1 and GLT-1 were normalized to GAPDH. Non-saturated exposures of Western blots were used for quantification. For Kir4.1 quantification, the immunoreactivity of the entire lane was analyzed.
Immunocytochemistry
Animals were anaesthetized with a peritoneal injection of ketamine (100mg/kg) and perfused with 4% paraformaldehyde solution in PBS for 25 minutes. The spinal cord was removed and stored in 4% paraformaldehyde overnight at 4°C. After washing in PBS, 100μm sections were cut using a Vibratome (Oxford instruments). Sections were incubated in blocking buffer (BB) for 1h in 10% goat serum and 0.2% Triton-X100 in phosphate buffered saline. Following blocking, antibody incubation and washes were carried out in BB diluted 1:3 in PBS (diluted BB). Slices were incubated in primary antibody (GFAP, Dako #Z0334, 1:1000), (Kir4.1, Proteintech #12503-1-AP, 1:500), (GLT-1, Millipore #AB1783 1:1000), & (NeuN, Millipore #MAB377 1:1000) in diluted BB overnight at 4°C with gentle agitation. The sections were then washed three times in diluted BB and incubated for 60 minutes at room temperature with tetramethyl rhodamine isothiocyanate-conjugated secondary antibodies obtained from Invitrogen (#A11008, #A11029, #A11010, #A11074) 1:500 in diluted blocking buffer. The slices were washed two times with diluted BB, then incubated with 4′6-diamidino-2-phenylindole (DAPI) 0.1μg/mL; (Sigma), and finally washed twice with phosphate buffered saline before being mounted onto glass coverslips. Fluorescent images were acquired with a Zeiss Axio Observer D1.
Quantitative Real-Time PCR
Following tissue removal as described above, for quantitative real-time polymerase chain reaction (qRT PCR), sequential isolation of total mRNA and genomic DNA were performed using Qiagen All Prep DNA/RNA Mini Kit. A total of 1.0μg of mRNA was converted to cDNA using Invitrogen Superscript VILO cDNA synthesis kit. Prior to PCR, cDNA was diluted 1:3 using DEPC treated water. Applied Biosystems Taqman probes were used with Taqman Universal Mastermix II, no UNG. An Applied Biosystems StepOne machine was used to perform qPCR and corresponding software was subsequently used for analysis of results. Cycling parameters were: 50°C for 2 min, 95°C for 10 min, 40 repeats of 95°C for 15 s and 60°C for 1 min. GAPDH was used as control housekeeping gene. ΔΔCt method was used to determine relative fold expression of mRNA.
Cultured spinal cord astrocytes
For culture experiments, newborn (PND0) pups were decapitated. Spinal cords were removed and dissected in ice-cold serum-free EMEM (Gibco, Grand Island, NY) containing 20 mM glucose. Meninges were stripped and cords were minced and placed into O2-saturated EMEM with papain (Worthington, Lakewood, NJ) for 20 min. Tissue was washed twice with spinal cord astrocyte media (EMEM supplemented with 10% fetal calf serum (FCS), 20 mM glucose, and penicillin/streptomycin) and triturated. Cells were plated at a density of 1.0 × 106 cells/ml on polyornithine- and laminin-coated 12-well tissue culture plates (Falcon). The media was changed the first and second days, and every fourth day thereafter. Mature or differentiated spinal cord astrocytes (>6 days in culture [DIC]) were used for all cultured experiments.
Statistical analysis
Current responses to varied voltage steps and ramps were analyzed and measured in Clampfit (Molecular Devices); the resulting raw data were graphed and plotted in Origin 8.51 (MicroCal). Two-tailed t-test, Tukey-Kramer Multiple Comparisons Test, and Mann-Whitney tests were performed using Graphpad software (San Diego, CA) and P values are reported in the text. Unless otherwise stated, all values are reported as means ± SE with n indicating the number of cells sampled.
Results
Elevated GFAP induces reduction of Kir4.1 and GLT-1 expression in AxD mouse models
To investigate the possibility that alterations in Kir4.1 and GLT-1 functions in caudal brain regions have a role in type II AxD, we first examined Kir4.1 and GLT-1 protein expression in caudal brain regions from three animal models of AxD: a line in which wild type (WT) human GFAP is overexpressed from a transgene (hGFAPTG, Messing et al., 1998); a line heterozygous for an R236H knock-in mutation, which is homologous to the common and particularly severe R239H mutation in human patients (R236H+/− Hagemann et al., 2006); and mice generated by crossing these two lines, (hGFAPTG/R236H+/− Hagemann et al., 2006). Both the hGFAPTG and R236H+/− mice have elevated GFAP expression in hippocampus and cortex, form Rosenthal fibers, display CNS oxidative stress and are hypersensitive to kainic acid induced seizures, but have a normal lifespan and no myelin deficits. The same observations hold for hGFAPTG/R236H+/− mice (Hagemann et al., 2006), except that the changes occur sooner, are more severe, and these mice die between P24–P35 (Hagemann et al., 2006). We have also observed a full body clasp when these mice were suspended by the tail in the days prior to death (unpublished observations), suggesting profound motor impairment (Yamamoto et al., 2000; Mangiarini et al., 1996; Filali et al., 2011). Additionally, hGFAPTG animals have significantly lower body weight and increased brain water volume (Meisingset et al., 2010). However, the absence of myelin deficits in these mice suggests they better model late onset, type II AxD, than the type I form.
The astrocytic proteins Kir4.1, GLT-1, and GFAP were quantified in the three models of AxD with GAPDH as a loading control (Fig. 1). At four weeks of age significant increases in GFAP were present in both hGFAP TG (72.3 ± 6.0%) and hGFAPTG/R236H+/− (36.7 ± 4.4%) in cervical spinal cord, whereas the amount of GFAP detected in R236H+/− animals actually decreased relative to WT. Corresponding with the upregulation of GFAP, hGFAPTG animals demonstrated significantly lower amounts of Kir4.1 (~75% reduction) and GLT-1 (~60% reduction) and the double transgenic hGFAPTG/R236H+/− animals exhibited a near complete loss of Kir4.1 and GLT-1 immunoreactivity (these animals were collected from P24-P28). This is despite the fact that the increase of GFAP expression in these animals was less than that in the hGFAPTG mice, indicating that the R236H mutation contributes to the molecular changes observed in hGFAPTG/R236H+/− mice. Of note, we and others have shown multiple bands on Kir4.1 Western blots that correspond with a monomer through tetramer (~50 kD – 200 kD) of the Kir4.1 protein (Kaiser et al., 2006; Olsen et al., 2010). These bands are completely absent in Kir4.1 KO animals and in human embryonic kidney cells that do not express Kir4.1 (Olsen et al., 2006). Therefore, the entire lane is used for quantification. The image shown for the Kir4.1 western blot has been overexposed to indicate the striking loss of Kir4.1 in hGFAPTG/R236H+/− animals compared to WT animals; non-saturated exposure images were used for quantification. Our Western blots demonstrate R236H+/− animals demonstrated no increase in GFAP protein (Fig. 1A,B) at the four-week time point compared to WT animals. A slight but significant reduction in Kir4.1 protein was observed in R236H+/− spinal cord (Fig. 1A,C), but no significant differences in GLT-1 immunoreactivity were observed (Fig.1A,D). Because pathological changes increase with time in these mice, we also examined R236H+/− animals at two months and one year of age, but found no change in GFAP (data not shown). It is important to note that these observations are specifically for brainstem and spinal cord, as elevated GFAP expression and Rosenthal fiber accumulation is observed in the hippocampus of R236H+/− animals (Hagemann et al., 2006). Given the high levels of GFAP expression observed in caudal brain structures in human patients with Type II Alexander (Wippold et al., 2006;Rizzuto et al., 1980) and the lack of observable changes in GFAP expression in this model we omitted the R236H+/− mice from the remainder of the study. Additionally, the early lethality of the double hGFAPTG/R236H+/− transgenic mice represents a severe form of AxD not representative of the later onset type II AxD. Therefore we focused on the hGFAPTGmodel in subsequent experiments as an appropriate model of type II AxD.
Figure 1. Kir4.1 and GLT-1 protein expression are reduced in spinal cord of AxD mice.
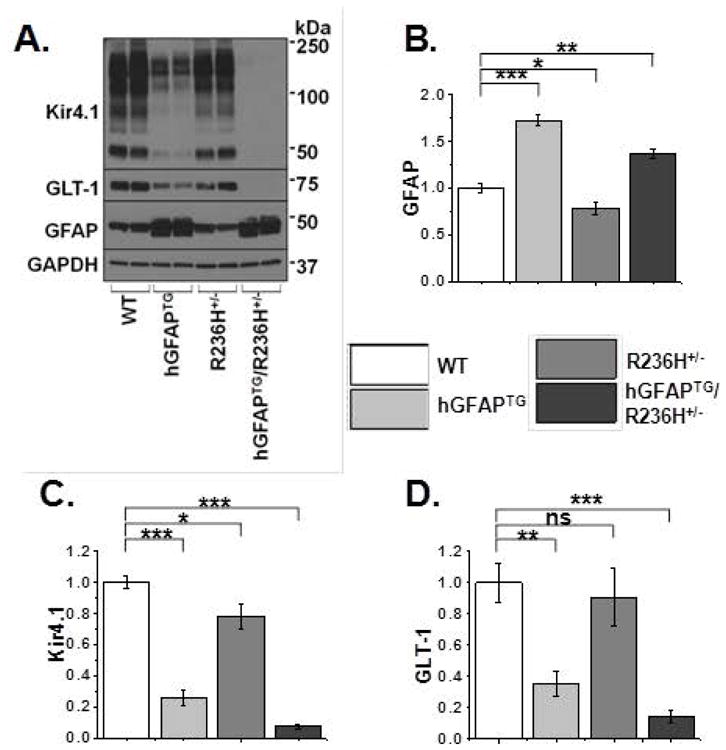
A. Western blot analysis of spinal cord from PND 24–28 WT, hGFAPTG, R236H+/−, and hGFAPTG/R236H+/− mice for Kir4.1, GLT-1 and GFAP and for GAPDH as a loading control. B–D. Quantification of western blot in (A) for GFAP (B), Kir4.1 (C), and GLT-1 (D), normalized to GAPDH levels and WT values. Data are ± SEM, n=4 in all groups, unpaired t-tests, *=p<0.05, **=p<0.01, ***=p<0.001.
To determine if the differences in Kir4.1 and GLT-1 in the hGFAP TG mice was transitory or of long duration, we examined protein expression in mice ranging in age from 2 weeks to one year (Fig. 2). Because both brainstem and spinal cord function are affected in Type II AxD, we examined tissue from both structures. Brainstem and spinal cord demonstrated a reduction in Kir4.1 and GLT-1 immunoreactivity by postnatal day (PND) 14 (Fig. 2A). This coincides with the postnatal developmental period during which these proteins typically demonstrate the most robust increases in expression (Jany et al., 2013; Nwaobi et al., 2014). Furthermore, Western blot analysis from brainstem and spinal cord tissue indicated that the loss of these proteins is maintained throughout adulthood (Fig. 2B). To assess the spatial distribution of astrocytic changes, we performed immunohistochemistry from 1-month old wild type and hGFAPTG mice (Fig. 3). Here we examined spinal cord expression of GFAP, Kir4.1, and GLT-1 proteins. We and others have shown that Kir4.1 and GLT-1 expression is most highly expressed in the gray matter neuropil and largely demarcates gray matter from white matter in transverse spinal cord sections (Olsen et al., 2007). Here we observed a similar pattern of expression of Kir4.1 and GLT-1 expression in four week old WT mice (Fig. 3). Kir4.1 immunoreactivity was markedly reduced throughout the gray matter in transverse spinal cord sections from hGFAP TG mice relative to WT littermates. These differences were observed in low magnification images which enabled visualization of the entire transverse section (left panels) and at higher magnification of the ventral horn (right panels). GFAP immunoreactivity was elevated throughout each transverse section, particularly in gray matter and subpial white matter. A similar pattern of loss for GLT-1 immunoreactivity was observed across the spinal cord and in the ventral horn (Fig. 3C,D). The loss of Kir4.1 and GLT-1 as well as the increase in GFAP expression was observed throughout cervical and lumbar regions of the spinal cord (data not shown). The decrease of Kir4.1 and GLT-1 protein surrounding motor neurons in cervical spinal cord may provide a mechanism by which astrocytes contribute to caudal brain symptoms in type II AxD.
Figure 2. Kir4.1 and GLT-1 protein expression patterns are similar in spinal cord and brainstem throughout development.
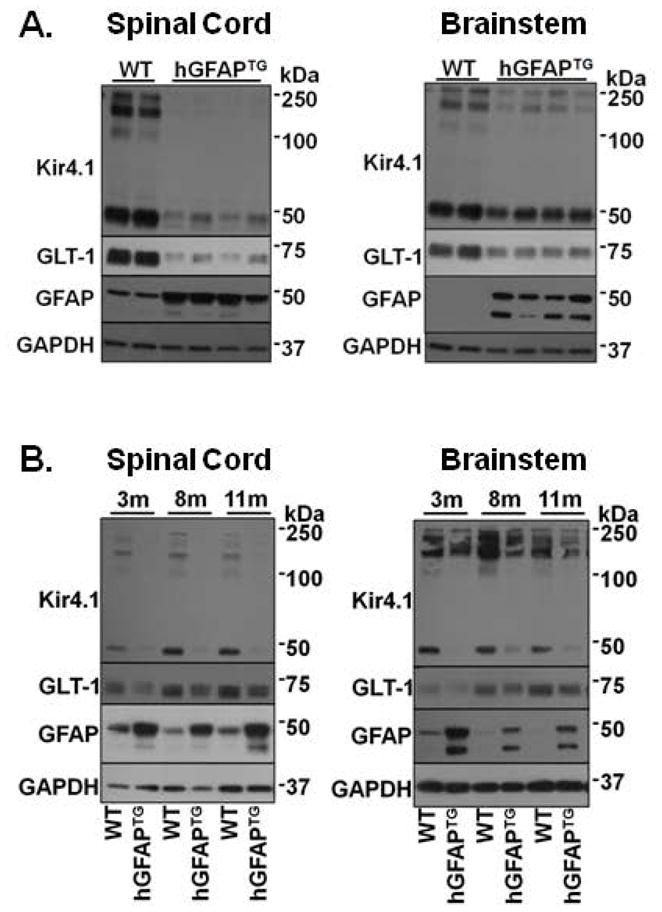
A. Representative Western blot for Kir4.1, GLT-1, and GFAP protein expression in PND 14 cervical spinal cord and brainstem. Data include 4 hGFAPTG mice and 2 littermate controls. B. Western blot analysis of spinal cord and brainstem from 3, 8, and 11 month old hGFAPTG mice and WT littermates are shown for Kir4.1, GLT-1 and GFAP and for GAPDH as a loading control. In brainstem, exposures were minimized to prevent saturation of GFAP in mutant animals.
Figure 3. Immunohistochemistry from transverse spinal cord sections demonstrates loss of Kir4.1 and GLT-1 of PND 28 hGFAPTG mice compared to WT littermates.
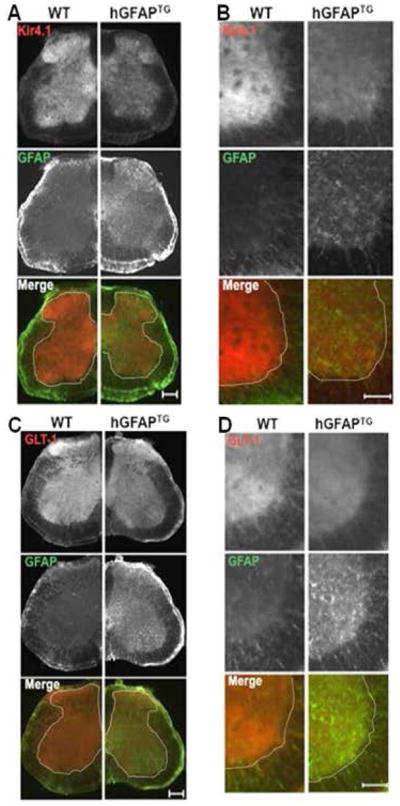
A. Low magnification images from transverse spinal cord sections indicate decreased Kir4.1 expression throughout the gray matter in hGFAPTG mice (top panel). GFAP is elevated in hGFAPTG sections (middle panel). Visualized together in the merged image, the loss of Kir4.1 (red) and increased GFAP (green) are apparent. B. As shown in A, higher magnification demonstrates protein changes in ventral horn. C. Low magnification images from transverse spinal cord sections indicate decreased GLT-1 expression throughout the gray matter in hGFAPTG mice (top panel). GFAP is elevated in hGFAPTG sections (middle panel). Visualized together in the merged image, the loss of GLT-1 (red) and increased GFAP (green) are apparent. D. As shown in C, higher magnification demonstrates protein changes in ventral horn. Dotted line demarcates gray from white matter. Scale bars (A,C = 200 μm, B,D = 100 μm)
hGFAPTG spinal cord astrocytes exhibit decreased Kir4.1 currents and reduced K+uptake
A primary cause of death in type II AxD patients is aspiration pneumonia due to dysphagia or an inability to swallow properly (Pareyson et al., 2008). This and other hindbrain symptoms in patients with type II AxD indicate profound motor dysfunction in cervical spinal cord regions. To better understand the implications of the loss of Kir4.1 for disease development, we assessed the capacity of cervical ventral horn astrocytes to take up K+ via Kir4.1. Whole-cell, electrophysiological recordings were performed from ventral horn astrocytes in acute cervical spinal cord sections from PND 14–18 WT and hGFAPTG animals. Astrocytes were identified based on morphology and electrophysiological properties. In some experiments, electrophysiological recordings were performed in hGFAPTG animals crossed with ALDH1L1-EGFP mice (green astrocytes) to assist with visualization (Yang et al., 2011). To activate Kir4.1 currents, we stepped astrocytes from −80 mV to 0 mV for 100 ms, and then from −180 mV to 100 mV in 20 mV increments as previously described (Olsen et al., 2007). Barium chloride (BaCl2, 100 μM) was used to isolate currents mediated by Kir4.1 channels as previously described (Ransom and Sontheimer, 1995). The resulting subtracted currents were identified as Kir4.1 currents. As depicted in Figure 4A, Ba2+-sensitive Kir4.1 currents were markedly smaller in hGFAPTG astrocytes. Indeed, they appeared to contribute little to whole cell currents as the currents in hGFAPTG animals were very similar to the post-Ba2+ trace in WT animals. A current-voltage (I–V) plot derived from voltage steps such as shown in Figure 4A indicates significant differences between WT and hGFAPTG Ba2+-sensitive, Kir4.1-mediated currents (Fig. 4B). This is represented graphically at one potential (−140 mV) in Figure 4C, which shows a change in current density from −81 ± 30 pA/pF in the WT to −17 ± 8 pA/pF in the hGFAPTG mice. This difference is current density is not a result of differences in whole cell capacitance as the mean whole cell capacitiance in WT and hGFAPTG cells was 11.35 +/− 1.1 (n = 14) and 11.4 +/− 1.0 (n = 10), respectively. The post Ba2+ currents were not significantly different in amplitude between the two cell populations and both demonstrate a similar, but small, rightward shift in reversal potential (data not shown). Astrocytes from hGFAPTGanimals were significantly depolarized (−62 ± 3 mV) relative to WT littermates (−70 ± 2 mV, Fig. 4D).
Figure 4. Kir4.1 mediated currents are smaller and astrocyte intrinsic membrane properties are altered in hGFAPTG mice compared to WT littermates.
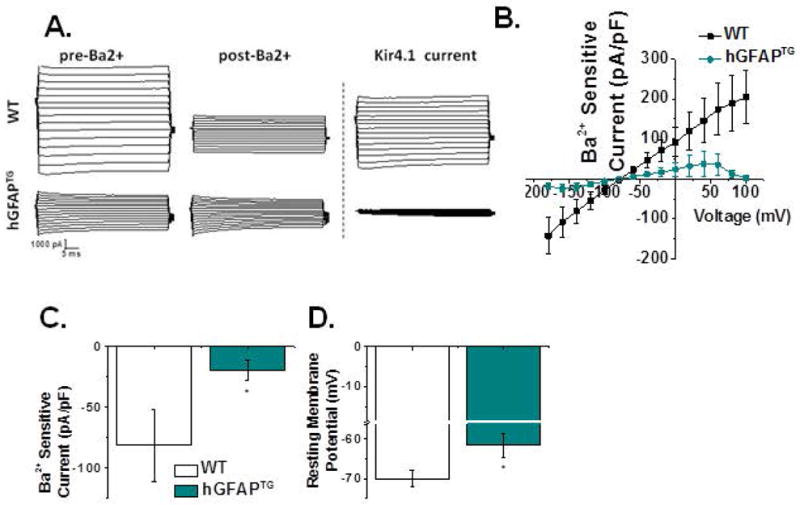
A. Representative whole-cell recordings in response to a voltage step protocol demonstrate a significant decrease in current amplitude in hGFAPTG ventral spinal cord astrocytes. B. I–V plot of cumulative data obtained from Ba2+-sensitive subtractions normalized to whole cell capacitance demonstrates diminished inward and outward currents in hGFAPTG astrocytes. C. Current density is significantly decreased at −140 mV in hGFAPTG astrocytes (WT, −81.07 ± 30.48 [n=14] hGFAPTG, −17.24 ± 7.59 [n=10] Mann-Whitney Test p= 0.0158). D. The mean resting membrane potential of astrocytes is significantly depolarized in hGFAPTG mice relative to WT littermates (WT, −70.02 ± 2.14 mV [n = 16], hGFAPTG, −61.68 ± 3.04 mV [n = 14] two-tailed t-test p= 0.0302).
To more directly measure the effects of reduced Kir4.1 on potassium uptake by hGFAPTG astrocytes, we applied 30 mM K+ for 400 ms using a picospritzer to mimic focal increases in [K+]o as would occur following neuronal activity. The puffer pipette was placed in the same focal plane as the voltage clamped cell at a distance that elicited a maximum response. Recordings were obtained from ventral horn astrocytes voltage-clamped at −80 mV. For clarity we show the response from each WT and hGFAPTG astrocyte (Fig. 5A). Mean data are summarized in Figure 5B. A scatter plot of peak current amplitudes in response to a potassium puff demonstrates a significant reduction in the overall amplitude of the K+ puff response in hGFAPTG astrocytes (−218 ± 44 pA) relative to astrocytes from WT littermates (−645 ± 15.4 pA). The variability in the scatter plot values for the WT animals may result from the recordings being performed between PND 14–18, the time when Kir4.1 expression increases most dramatically (Nwaobi et al., 2014), and thus likely would vary among cells. In contrast, there is little variability among the hGFAPTG cells, presumably because their Kir4.1 levels remain uniformly low. These data indicate reduced capacity for Kir4.1-mediated K+ uptake, consistent with the Kir4.1 protein loss indicated by Western blot.
Figure 5. [K+]o uptake by ventral spinal cord astrocytes is decreased in hGFAPTG mice relative to WT littermates.
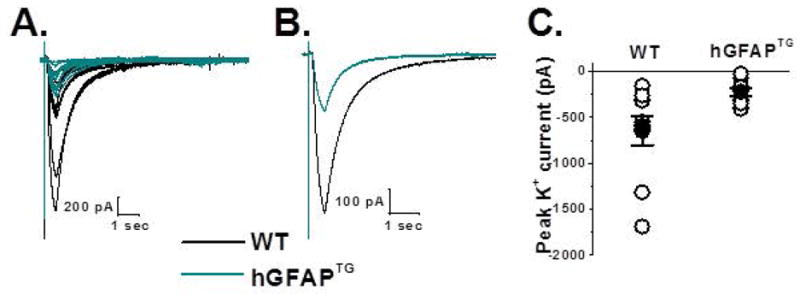
A. Superimposed traces of WT (black, n = 9) and hGFAPTG (cyan, n = 8) potassium currents in response to application of a brief (400 msec) application of 30mM potassium are indicated. B. Averaged traces from A are shown. C. Peak amplitudes of individual cells (open circles) from A and mean data (filled circles) indicate a significant decrease in potassium uptake in hGFAPTG mice (WT,−645 ± 15.4 pA, [n= 9], hGFAPTG, −218 ± 44 pA, [n = 8], two tailed t-test, p = 0.0264).
hGFAPTG spinal cord astrocytes exhibit decreased GLT-1 currents and reduced glutamate uptake
The possible contribution of GLT-1 reduction to disease development was evaluated by examining the response of astrocytes from the ventral horn to exogenously applied glutamate (Fig. 6). To isolate glutamate transporter currents and minimize neuronal activity and release of endogenous glutamate, we included 500 nM TTX, 100 μM CdCl2, 20 μM CNQX, 20 μM bicuculline and 20 μM AP5 in both the bathing solution and the glutamate puffing pipette (Bergles and Jahr, 1997; Grass et al., 2004). Using a picospritzer we pressure applied glutamate (200 μM, 400 ms) to ventral horn astrocytes voltage clamped at −80 mV. As with the K+ puffing experiments, the puffing pipette was manipulated until the maximum response was elicited from the voltage clamped cell. The resulting traces from 15 WT and 9 hGFAPTG astrocytes are shown in Figure 6A. Averaged data from all WT and hGFAPTG astrocytes are shown in Figure 6B. The summary scatter plot demonstrates a significant decrease in the glutamate uptake currents in response to a challenge of glutamate in ventral horn hGFAPTG astrocytes (−8.1 ± 4.1pA) relative to WT astrocytes (−53.2 ± 4.1 pA).
Figure 6. Glutamate uptake in ventral spinal cord astrocytes is reduced in hGFAPTG mice compared to WT littermates.
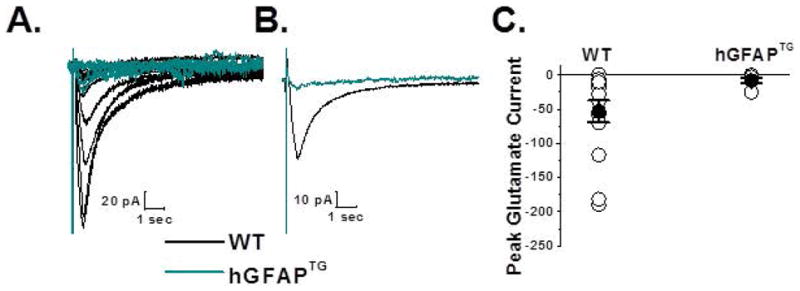
A. Superimposed traces of glutamate transporter currents recorded from ventral horn astrocytes from WT (black, n = 15) and hGFAPTG (cyan, n = 9) astrocytes are shown. Cells were voltage clamped at −80 mV and responses were evoked by a 200 μM, 400 msec puff of glutamate. Bathing and puffing solutions contained 20 μM AP5, 20 μM bicuculline, 20 μM CNQX, 500 nM TTX and 100 μM CdCl2. Each trace shown is an average of three individual traces for that astrocyte. B. Averaged data across all WT (black) and hGFAPTG (cyan) responses are shown. C. Summary scatter plot of the glutamate evoked responses indicates a decrease in the hGFAPTG mice (−8.1 ± 4.1 pA, [n = 9]) relative to WT littermate controls (−53.2 ± 4.1 pA, [n = 15], Two tailed t-test, p = 0.0416).
Reduced mRNA levels of KCNJ10 and SLC1A2 suggest a transcriptional mechanism
Mature astrocytic protein expression profiles are established by four weeks of age in wild type mice, with GFAP, Kir4.1 and GLT-1 protein expression upregulated during early postnatal development in all CNS regions. Figure 7A demonstrates the robust protein increase in each of these proteins during the first two postnatal weeks in WT mice. The dramatic upregulation of each of these proteins is mirrored by increased mRNA expression of KCNJ10 (Kir4.1), SLC1A2 (GLT-1) and GFAP in WT mice (white bars) during the same developmental time points, indicating a correlation between mRNA and protein expression for each gene (Fig. 7B,C,D). However, reductions in KCNJ10 mRNA and SLC1A2 were observed as early as PND7 in hGFAPTG mice relative to WT mice. Interestingly, reduced levels of KCNJ10 and SLC1A2 mRNA were observed prior to significant changes in mouse GFAP expression in hGFAPTG mice relative to WT mice (Fig. 7D), although there is a large increase in total GFAP (mouse plus human) relative to WT at PND7 (Fig. 7E,F). The decreased mRNA levels for KCNJ10 and SLC1A2 in the hGFAPTG mice relative to WT suggest a transcriptional component to their regulation.
Figure 7. Reduced KCNJ10 and SLC1A2 mRNA expression correlates with GFAP accumulation in hGFAPTG mice.

A. Western blot analysis of WT spinal cord demonstrates increased Kir4.1, GLT-1 and GFAP proteins during the first two postnatal weeks. B. Quantitative PCR demonstrates KCNJ10 mRNA increases during the first two postnatal weeks. KCNJ10 mRNA does not increase to the same level in hGFAPTG mice. C. Quantitative PCR demonstrates SLC1A2 mRNA increases during the first two postnatal weeks. SLC1A2 mRNA does not increase to the same level in hGFAPTG mice. D. Quantitative PCR demonstrates endogenous mouse GFAP mRNA increases during the first two postnatal weeks. At PND14 endogenous GFAP mRNA in hGFAPTG mice is elevated relative to WT mice. E. Human GFAP transcript also increases in hGFAPTG mice. F. Western blot demonstrates GFAP protein accumulation is evident at PND7 in hGFAPTG mice. *=p<0.05, **=p<0.01
We next queried whether the decreased Kir4.1 and GLT-1 protein in hGFAP TG mice was autonomous to the astrocyte, and thus possibly a primary cause of clinical signs, or was a secondary reaction to the disease process. To address this question, we cultured spinal cord astrocytes from WT and hGFAPTG mice, thereby removing them from the context of the diseased brain. Kir4.1 protein was found to be reduced in spinal cord astrocytes cultured from hGFAPTGmice relative to astrocyte cultures from WT littermates (Figure 8A). Similar results were observed for GLT-1 (Fig. 8B).
Figure 8. Cultured hGFAPTGastrocytes develop increased GFAP and decreased GLT-1 and Kir4.1 proteins in vitro.

Western blot analysis of two independent sets of primary astrocytic cultures obtained from WT and hGFAP TG PND0 spinal cord comparing GFAP, Kir4.1 (A), and GLT-1 (B) expression 10–12 days in vitro. GAPDH is shown as a loading control.
Kir4.1 protein expression profiles vary between areas affected in type I vs. type II AxD
While our study was in progress, Sosunov et al. (2013) published the observation that Kir4.1 levels were increased in the hippocampus of hGFAPTG mice, contrary to our results in caudal brain regions. To determine if the change in Kir4.1 levels was indeed CNS region specific, we performed Western blots for Kir4.1 and GLT-1 for hippocampal and cortical tissues isolated from 3, 8, and 11 month old hGFAPTG mice and WT littermates. As shown in Figure 9, we confirm the report of Sosunov et al. (2013) of an increase of Kir4.1 levels in the hippocampus, as well as their finding of a decrease in GLT-1.
Figure 9. Kir4.1 and GLT-1 protein expression in hippocampus and cortex of hGFAPTG differ from caudal brain regions.

Western blot of hippocampus (A) and cortex (B) from 3, 8, and 11-month-old hGFAPTG mice and WT littermates for Kir4.1, GLT-1 and GFAP and for GAPDH as a loading control.
Discussion
To our knowledge this is the first study to examine astrocytes in hindbrain regions in any model of AxD. In the current study we show when presented with equal challenges of glutamate and K+, GLT-1 mediated glutamate uptake and Ba2+-sensitive Kir4.1 mediated K+ uptake are dysregulated in the spinal cord and brainstem slices of AxD mice. This dysregulation is a result of significantly reduced GLT-1 and Kir4.1 protein levels, which manifest as early PND 14 in AxD mice and persist through adulthood (Fig. 1,2). The reductions in Kir4.1 and GLT-1 expression appear related to elevated GFAP expression, as the reductions are not observed in the R236H+/− animals, which do not exhibit elevated GFAP expression in the spinal cord. Studies performed in cultured spinal cord astrocytes indicate that these changes are autonomous to the astrocyte as they are observed when these cells are grown in the absence of other cell types. Finally, we demonstrate that reduced levels of Kir4.1 and GLT-1 are at least in part due to reduced transcription of each gene. We propose that the significantly reduced protein levels of Kir4.1 and GLT-1 and the changes in astrocyte physiology caused by loss of these proteins contribute to hindbrain-involved pathology and symptoms in type II AxD.
The role of Kir4.1 and GLT-1 in Type II AxD
The importance of Kir4.1 and GLT-1 to ‘normal’ CNS functioning is supported by Kir4.1 and GLT-1 mouse knockout studies (Djukic et al., 2007;Neusch et al., 2001;Kiryk et al., 2008;Tanaka et al., 1997). Both global and astrocyte-specific Kir4.1 knock out animals display ataxia, seizures, hind leg splaying/paralysis and early postnatal death (PND 12–25) (Djukic et al., 2007;Neusch et al., 2001). Interestingly, the most profound pathology in these animals is observed in spinal cord, which demonstrates vacuoles, demyelination, gliosis and motor neuron cell death (Neusch et al., 2001;Djukic et al., 2007). The importance of Kir4.1 in the spinal cord is not surprising given its high levels in this tissue relative to other brain regions at all developmental ages (Nwaobi et al., 2014). Importantly, humans with loss of function mutations in Kir4.1 exhibit ataxia, and extreme lower motor extremity weakness resulting in a loss of ambulation (Scholl et al., 2009;Bockenhauer et al., 2009). Other CNS abnormalities include early onset seizures (3 months of age), sensorineural deafness, ataxia, and mental retardation, all of which indicate a critical role for Kir4.1 in brain development. Humans displaying mutations in GLT-1 have not been identified; however, GLT-1 knockout mice die prematurely (average PND 23), have reduced body and brain weight, focal gliosis, and neuronal cell loss (Kiryk et al., 2008). Behaviorally, these animals also display hind limb clasping, a sign of motor impairment/motor degeneration, and seizures prior to death (Tanaka et al., 1997;Kiryk et al., 2008). Indeed, behavioral testing in hGFAPTG mice indicates significant reductions in forepaw grip strength (Meisingset et al., 2010), a motor skill dependent on cervical spinal cord function. Loss of Kir4.1 and GLT-1 protein expression and function in this region may contribute to motor dysfunction observed in this region.
Previous work demonstrates DHK-sensitive, GLT-1 mediated glutamate uptake accounts for nearly all glutamate uptake in ventral spinal cord astrocytes (Regan et al., 2007). We expect the significant reduction in protein and associated GLT-1 transporter function in AxD mice would contribute to less efficient glutamate clearance in spinal cord of AxD mice. The result of a significant reduction in Kir4.1 channel protein and function is less clear. In astrocytes specifically, Kir4.1 has several putative functions, including astrocyte volume regulation, maintenance or contribution to resting membrane potential and K+ homeostasis (Djukic et al., 2007, Olsen et al., 2006, Neusch et al., 2006, Pannicke et al., 2004, Dibaj et al., 2007). In terms of K+ regulation, experimental evidence demonstrates blockade of Na+K+ATPase plays a more significant role in K+ accumulation following electrical stimulation (D’Ambrosio et al., 2002, Djukic et al., 2007, Haj-Yasein et al., 2011; Neusch et al., 2006). While pharmacological blockade or KO of Kir4.1 results in a prolonged undershoot following stimulation and time to recover from this undershoot and return to baseline K+ levels (Haj-Yasein et al., 2011, Chever et al., 2011, Djukic et al., 2007, Neusch et al., 2006). Because of the weakly rectifying nature of this channel, it is capable of bidirectional potassium flux. It is thought that Kir4.1 provides a return pathway for K+ into the ECS following the undershoot in [K+]o driven by neuronal Na-KATPase (D’Ambrosio et al., 2002). Given the experimental findings upon blockade or KO of both GLT-1 and Kir4.1, we presume the loss of these two proteins would modulate the extracellular environment resulting in changes in neuronal membrane properties and network excitability in hGFAPTG and hGFAPTG/R236H+/− animals. Supporting this, it was recently shown that a loss of Kir4.1 protein expression in Huntington’s disease mice contributes to medium spiny neuron hyperexcitability (Tong et al., 2014). Further, restoration of Kir4.1 into astrocytes by AAV transduction ameliorated motor abnormalities in these mice. In the current study we demonstrate a striking loss of Kir4.1, which contributes to a significantly depolarized astrocyte resting membrane potential. This depolarization of the resting membrane potential would render glutamate uptake through the remaining GLT-1 glutamate transporters less effective, given the dependence of the transporter on a negative resting membrane potential. Future work directed at understanding the relationship between loss of function of Kir.4.1 and GLT-1 and neuronal function may shed light on hindbrain pathology in type II AxD as well as other CNS pathologies which present with gliosis in the brainstem and spinal cord.
Phenotypes of the R236H+/− and the HGFAPTG X R236H+/− mice
The small body weight, posture, hind limb clasping and premature death (P22–P30) observed in the HGFAPTG X R236H+/− (double cross) mice is not surprising given the striking loss of both Kir4.1 and GLT-1 observed in these animals at PND 24–28 (Figure 1). Indeed we began collecting the HGFAPTG X R236H+/− mice at PND24 because they were dying before the four week collection time point. Interestingly, Western blots indicated that total GFAP protein expression was not higher in these mice relative to HGFAPTG alone. However, immunohistochemistry performed on these animals at PND24 revealed significant accumulation of aggregated GFAP in the brainstem and spinal cord (data not shown). This supports previous work in whole brain which demonstrated increased Rosenthal fiber accumulation and high levels of insoluble GFAP in HGFAPTG X R236H+/− mice relative to the HGFAPTG or R236H−/+ animals (Hageman et al., 2006). These data indicate that both the total level of GFAP and its form influence the expression of Kir4.1 and GLT-1. This may have important consequences for the relative expression of these proteins in CNS pathologies which present with severe reactive gliosis and/or permanent glial scarring.
In contrast to the severe pathology of the HGFAPTG X R236H+/− mice, no increase in GFAP was found in the brainstem or spinal cord of R236H+/− mice between 4 weeks and 1 year of age. At 4 weeks there was actually a modest decrease in GFAP levels in spinal cord compared to wild type; a result recently reported for R236H+/− mice at 8 weeks of age (Jany et al., 2013). These mice do have a significant increase in GFAP levels in more rostral brain regions (Jany et al., 2013). The corresponding R239H mutation in humans produces severe, early onset AxD, characterized by massive pathology in the rostral brain regions (Brenner et al., 2009). Contrary to the observations in mice, human R239H patients also show striking pathology in their brainstem and spinal cord (Klein and Anzil, 1994; Gingold et al., 1999). Thus while rostral pathology predominates in the description of type I AxD, these patients may also have pathology in the brainstem and spinal cord. Accordingly, our findings may have relevance to the Type I disease form as well as to the Type II form
Kir4.1 and GLT-1 expression levels in gliosis and AxD
The association between reduced Kir4.1 and/or GLT-1 protein expression and gliosis has been explored by many groups, including our own. For instance, we have demonstrated that both proteins were significantly downregulated in association with gliosis in the spinal cord following a traumatic spinal cord injury (Olsen et al., 2010). While this study examined pathology in the context of acute injury, similar results have been observed in chronic neurodegenerative diseases such as ALS (Kaiser et al., 2006). Interestingly, ALS shares many features with type II AxD, including spasticity, ataxia, pyramidal involvement and difficulty with speech, swallowing and chewing. Indeed, type II AxD is sometimes misdiagnosed clinically as ALS (Pareyson et al., 2008). In the rat model of ALS, loss of GLT-1 protein is associated with gliosis and precedes motor neuron degeneration (Howland et al., 2002). Gliosis and reduced GLT-1 expression levels are seen in the human ALS population as well (Bendotti et al., 2001;Rothstein et al., 1992;Rothstein et al., 1995;Ye et al., 1999); and glutamate is elevated in the CSF of ALS patients (Tarasiuk et al., 2012), suggesting a dysregulation of glutamate homeostasis. Similarly, Kir4.1 protein expression was decreased by nearly 30% in pre-symptomatic ALS mice, and reached a final decrease of 80% in end stage disease (Kaiser et al., 2006). In these mice, an inverse relationship between Kir4.1/GLT-1 and GFAP was observed. These are but a few examples that demonstrate reduced Kir4.1 and GLT-1 protein levels with gliosis, typically defined by elevated GFAP levels. Indeed, we had assumed this to be a generalization and were surprised by the results presented here and those by Sosunov et al. (2013) demonstrating an increase in Kir4.1 protein expression in frontal brain regions of AxD mice. This increase in Kir4.1, associated with elevated GFAP expression are the only instances currently reported. It is unclear why there is such a sharp contrast between frontal and hindbrain regions in the AxD models. This is particularly intriguing given that increased Kir4.1 is not observed in other forms of gliosis, including animal models of chronic disease and acute injury in forebrain regions (for review see, Olsen et al 2008).
Gene regulation and gliosis
We and others have demonstrated that developmental expression of astrocytic proteins GFAP, Kir4.1, and GLT-1 share a similar temporal induction in early postnatal development which tightly parallels increasing transcript levels for each gene. Further, we show a direct correlation between decreased mRNA and protein expression for both Kir4.1 and GLT-1, associated with elevated GFAP expression. Our data indicate that prior to changes in GFAP mRNA in the AxD model mice, mRNA expression for the genes which encode Kir4.1 and GLT-1 are decreased, suggesting these two genes are more sensitive to the factors initiating a gliotic response than GFAP itself. These data suggest, at least in part, that transcriptional mechanisms are involved in controlling the reduction of total levels of GLT-1 and Kir4.1 proteins. The mechanisms by which GFAP forms specific interactions leading to transcriptional changes are unknown, but studies of other neurodegenerative diseases featuring protein aggregates, including ALS and frontotemporal dementia (FTLD-TDP) (Neumann et al., 2006), suggest a possible role for TAR DNA-binding protein 43 (TDP-43). TDP-43 is an RNA and DNA binding protein with many functions affecting both transcription and translation (for review see Lee et al. (2012). Originally identified for its binding to chromosomally integrated trans-activation response element (TAR) DNA and transcriptional repression of HIV-1, TDP-43 has also been shown to be involved with pre-mRNA splicing and translational regulation. In ALS (Neumann et al., 2006) and other neurodegenerative conditions including Alzheimer’s, Parkinson’s, and Huntington’s diseases (Nakashima-Yasuda et al., 2007; Schwab et al., 2008; Uryu et al., 2008), TDP-43 has been shown to bind to cytoplasmic protein aggregates, resulting in its depletion from the nucleus. Interestingly, cytoplasmic aggregate localization of TDP-43 has also been identified in Rosenthal fibers in pilocytic astrocytoma (Lee et al., 2008) and, very recently, AxD (Walker et al., 2014). Since GFAP is the primary constituent of Rosenthal fibers, this raises the possibility that TDP-43 interacts with GFAP, providing a possible link between the increased GFAP levels in gliosis and persistent downregulation of Kir4.1 and GLT-1 gene transcripts Knockdown of TDP-43 in fact does decrease GLT-1 mRNA levels, but only modestly (Polymenidou et al., 2011); perhaps other changes associated with reactive gliosis synergize with the decrease in nuclear TDP-43 to produce a more profound and persistent alteration in gene expression.
Conclusions
We have observed profound decreases in Kir4.1 and GLT-1 mRNA, protein and function in the brainstem and spinal cord of AxD model mice. These changes may compromise neuronal activity, and could explain many of the neurological signs of type II AxD. More generally, these findings may provide insight to more global mechanisms of astrocyte gliosis, which was the original intention of the creation of the hGFAPTG mice (Messing et al., 1998). Our data indicate that elevated GFAP is sufficient to prevent ‘normal’ levels of Kir4.1 and GLT-1 protein expression in at least some populations of astrocytes, in the absence of any other underlying disease mechanism or precipitating insult. Thus GFAP may act as a ‘master regulator’ in gliosis, at least in terms of K+ and glutamate homeostasis, and provide a therapeutic gene target that would benefit a wide variety of CNS insults.
Main Points.
AxD mice have profoundly reduced Kir4.1 and GLT-1 protein and function in caudal brain regions.
Loss of glutamate and channel-mediated K+ uptake may contribute to type II AxD pathology.
These changes may be a general consequence of the elevated GFAP levels associated with reactive gliosis
Acknowledgments
This work was supported in part by NINDS R01NS075062 to MLO and NINDS P01NS42803 to MB.
Reference List
- Balestrino M, Aitken PG, Somjen GG. The effects of moderate changes of extracellular K+ and Ca2+ on synaptic and neural function in the CA1 region of the hippocampal slice. Brain Res. 1986;377:229–239. doi: 10.1016/0006-8993(86)90863-2. [DOI] [PubMed] [Google Scholar]
- Bao X, Pal R, Hascup KN, Wang Y, Wang WT, Xu W, Hui D, Agbas A, Wang X, Michaelis ML, Choi IY, Belousov AB, Gerhardt GA, Michaelis EK. Transgenic Expression of Glud1 (Glutamate Dehydrogenase 1) in Neurons: In Vivo Model of Enhanced Glutamate Release, Altered Synaptic Plasticity, and Selective Neuronal Vulnerability. Journal of Neuroscience. 2009;29:13929–13944. doi: 10.1523/JNEUROSCI.4413-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A, Rizzi M, Rattray M, Mennini T. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem. 2001;79:737–746. doi: 10.1046/j.1471-4159.2001.00572.x. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Goldman JE, Quinlan RA, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon P, editors. Astrocytes in (patho)physiology of the nervous system. New York, NY: Springer; 2009. pp. 591–648. [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 Channel in Excess Potassium Clearance: An In Vivo Study on Anesthetized Glial-Conditional Kir4.1 Knock-Out Mice. Journal of Neuroscience. 2010;30:15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W, Messing A. Properties of astrocytes cultured from GFAP over-expressing and GFAP mutant mice. Exp Cell Res. 2009;315:1260–1272. doi: 10.1016/j.yexcr.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Dibaj P, Kaiser M, Hirrlinger J, Kirchhoff F, Neusch C. Kir4.1 channels regulate swelling of astroglial processes in experimental spinal cord edema. J Neurochem. 2007;103:2620–2628. doi: 10.1111/j.1471-4159.2007.04979.x. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J, Beal MH, She Y, Howland DS. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:1688–1696. doi: 10.1523/JNEUROSCI.23-05-01688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali M, Lalonde R, Rivest S. Sensorimotor and cognitive functions in a SOD1(G37R) transgenic mouse model of amyotrophic lateral sclerosis. Behav Brain Res. 2011;225:215–221. doi: 10.1016/j.bbr.2011.07.034. [DOI] [PubMed] [Google Scholar]
- Gingold MK1, Bodensteiner JB, Schochet SS, Jaynes M. Alexander’s disease: unique presentation. J Child Neurol. 1999;14(5):325–9. doi: 10.1177/088307389901400510. [DOI] [PubMed] [Google Scholar]
- Graff-Radford J, Schwartz K, Gavrilova RH, Lachance DH, Kumar N. Neuroimaging and clinical features in type II (late-onset) Alexander disease. Neurology. 2014;82:49–56. doi: 10.1212/01.wnl.0000438230.33223.bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass D, Pawlowski PG, Hirrlinger J, Papadopoulos N, Richter DW, Kirchhoff F, Hulsmann S. Diversity of functional astroglial properties in the respiratory network. J Neurosci. 2004;24:1358–1365. doi: 10.1523/JNEUROSCI.4022-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby +, Nagelhus EA. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human kir4.1 gene (KCNJ10) Glia. 2011;59:1635–1642. doi: 10.1002/glia.21205. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jany PL, Hagemann TL, Messing A. GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro. 2013;5:e00109. doi: 10.1042/AN20130003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M, Maletzki I, Hulsmann S, Holtmann B, Schulz-Schaeffer W, Kirchhoff F, Bahr M, Neusch C. Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;99:900–912. doi: 10.1111/j.1471-4159.2006.04131.x. [DOI] [PubMed] [Google Scholar]
- Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, Knapska E, Filipkowski RK, Kaczmarek L, Danysz W. Behavioral characterization of GLT1 (+/−) mice as a model of mild glutamatergic hyperfunction. Neurotox Res. 2008;13:19–30. doi: 10.1007/BF03033364. [DOI] [PubMed] [Google Scholar]
- Klein EA, Anzil AP. Prominent white matter cavitation in an infant with Alexander’s disease. Clin Neuropathol. 1994;13(1):31–8. [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–281. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojamowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol. 2008;23(4):1233–40. doi: 10.1007/s00401-007-0331-5. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojamowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012;13(1):38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Martin PM, O’Callaghan JP. A direct comparison of GFAP immunocytochemistry and GFAP concentration in various regions of ethanol-fixed rat and mouse brain. J Neurosci Methods. 1995;58:181–192. doi: 10.1016/0165-0270(94)00175-g. [DOI] [PubMed] [Google Scholar]
- Meisingset TW, Risa O, Brenner M, Messing A, Sonnewald U. Alteration of glial-neuronal metabolic interactions in a mouse model of Alexander disease. Glia. 2010;58:1228–1234. doi: 10.1002/glia.21003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114(3):221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, Hulsmann S. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 Potassium Channel Subunit Is Crucial for Oligodendrocyte Development and In Vivo Myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Lin E, Peramsetty SR, Olsen ML. DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia. 2014;62:411–427. doi: 10.1002/glia.22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Campbell SL, Sontheimer H. Differential Distribution of Kir4.1 in Spinal Cord Astrocytes Suggests Regional Differences in K+ Homeostasis. Journal of Neurophysiology. 2007;98:786–793. doi: 10.1152/jn.00340.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Campbell SC, McFerrin MB, Floyd CL, Sontheimer H. Spinal cord injury causes a wide-spread, persistent loss of Kir4.1 and glutamate transporter 1: benefit of 17 beta-oestradiol treatment. Brain. 2010;133:1013–1025. doi: 10.1093/brain/awq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia. 2006;53:516–528. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem. 2008;107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfreyman JW, Thomas DG, Ratcliffe JG, Graham DI. Glial fibrillary acidic protein (GFAP): purification from human fibrillary astrocytoma, development and validation of a radioimmunoassay for GFAP-like immunoactivity. J Neurol Sci. 1979;41:101–113. doi: 10.1016/0022-510x(79)90144-8. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A, Bringmann A. A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci. 2004;26:493–502. doi: 10.1016/j.mcn.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F, Girotti F, Gattellaro G, Carriero MR, Farina L, Ceccherini I, Savoiardo M. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131:2321–2331. doi: 10.1093/brain/awn178. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43 Nat. Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prust M, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology. 2011;77:1287–1294. doi: 10.1212/WNL.0b013e3182309f72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. Journal of Neurophysiology. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert M, Khaldi A, Zauner A, Doppenberg E, Choi S, Bullock R. High level of extracellular potassium and its correlates after severe head injury: relationship to high intracranial pressure. J Neurosurg. 2000;93:800–807. doi: 10.3171/jns.2000.93.5.0800. [DOI] [PubMed] [Google Scholar]
- Rizzuto N, Ferrari G, Piscioli A. Diffuse Rosenthal fiber formation in adults. A case report. Acta Neuropathol. 1980;50:237–240. doi: 10.1007/BF00688761. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van KM, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008;67(12):1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- Sosunov AA, Guilfoyle E, Wu X, McKhann GM, Goldman JE. Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J Neurosci. 2013;33:7439–7450. doi: 10.1523/JNEUROSCI.4506-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tang X, Hang D, Sand A, Kofuji P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem Biophys Res Commun. 2010;399:537–541. doi: 10.1016/j.bbrc.2010.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasiuk J, Kulakowska A, Drozdowski W, Kornhuber J, Lewczuk P. CSF markers in amyotrophic lateral sclerosis. J Neural Transm. 2012;119:747–757. doi: 10.1007/s00702-012-0806-y. [DOI] [PubMed] [Google Scholar]
- Tian R, Wu X, Hagemann TL, Sosunov AA, Messing A, McKhann GM, Goldman JE. Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol. 2010;69:335–345. doi: 10.1097/NEN.0b013e3181d3cb52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XA, Ao Y, Nwaobi SE, Xu J, Haustein MA, Anderson MA, Mody I, Olsen ML, Sofronview MV, Khakh BS. Huntington’s disease model mice exhibit astrocyte Kir4.1 ion channel dysfunction that contributes to medium spiny neuron dysfunction. 2014 doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Trojanowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008;67(6):555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A. Astrocytic TDP-43 pathology in Alexander disease. J. Neurosci. 2014;34(19):6448–6458. doi: 10.1523/JNEUROSCI.0248-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wippold FJ, Perry A, Lennerz J. Neuropathology for the neuroradiologist: Rosenthal fibers. AJNR Am J Neuroradiol. 2006;27:958–961. [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]