

The six subunits of human prefoldin were overexpressed in E. coli. Prefoldin was reconstituted, purified and crystallized. X-ray diffraction data were collected to 4.7 Å resolution.
Keywords: protein folding, molecular chaperone, prefoldin
Abstract
Proper protein folding is an essential process for all organisms. Prefoldin (PFD) is a molecular chaperone that assists protein folding by delivering non-native proteins to group II chaperonin. A heterohexamer of eukaryotic PFD has been shown to specifically recognize and deliver non-native actin and tubulin to chaperonin-containing TCP-1 (CCT), but the mechanism of specific recognition is still unclear. To determine its crystal structure, recombinant human PFD was reconstituted, purified and crystallized. X-ray diffraction data were collected to 4.7 Å resolution. The crystals belonged to space group P21212, with unit-cell parameters a = 123.2, b = 152.4, c = 105.9 Å.
1. Introduction
Proper protein folding is an essential process for all organisms. Several proteins require assistance by molecular chaperones to attain their native conformations (Hartl & Hayer-Hartl, 2002 ▸). Prefoldin (PFD), a molecular chaperone that is present in archaea and eukaryotes, facilitates the folding of non-native proteins by transferring them to group II chaperonin (Vainberg et al., 1998 ▸). Crystal structures of archaeal PFD revealed a jellyfish-like structure with six coiled coils consisting of two α subunits and four β subunits (Siegert et al., 2000 ▸; Ohtaki et al., 2008 ▸; Kida et al., 2008 ▸). It has been suggested that hydrophobic patches on the coiled-coil tentacles of archaeal PFD interact with various target proteins to prevent aggregation (Leroux et al., 1999 ▸; Siegert et al., 2000 ▸; Martín-Benito et al., 2007 ▸; Ohtaki et al., 2008 ▸). In contrast, eukaryotic PFD consists of six different subunits (PFD1–PFD6; Vainberg et al., 1998 ▸). PFD3 and PFD5 are homologous to the archaeal α subunit, and the others are homologous to the β subunit. Eukaryotic PFD specifically recognizes nascent polypeptide chains of actin and tubulin, and delivers them to the chaperonin-containing TCP-1 (CCT), a eukaryotic group II chaperonin (Vainberg et al., 1998 ▸; Hansen et al., 1999 ▸). Although cryoelectron microscopy structures of PFD–actin and PFD–CCT complexes suggested a handoff mechanism for the delivery of non-native actin (Martín-Benito et al., 2002 ▸), a higher resolution structure of eukaryotic PFD is necessary to elucidate how PFD recognizes non-native actin and tubulin. Here, we report the expression, purification, crystallization and preliminary X-ray diffraction studies of human PFD.
2. Materials and methods
2.1. Cloning and expression
The DNA fragment encoding each PFD subunit was amplified by PCR from a human liver cDNA library (Takara Bio, Shiga, Japan) using the primers listed in Table 1 ▸. The N-terminal 37 residues of PFD3 and four residues of PFD4 were deleted. The fragments were individually cloned into the NdeI/BamHI sites of pET-21a (Novagen, Darmstadt, Germany) for PFD1, PFD3, PFD5 and PFD6 and of pET-28a for PFD2 and PFD4. The resulting sequences contained N-terminal hexahistidine tags followed by thrombin cleavage sites on PFD2 and PFD4. The plasmids for PFD1, PFD4, PFD5 and PFD6 were individually transformed into Escherichia coli Rosetta2 (DE3) pLysS cells (Novagen). The cells were grown to an OD600 of 0.6 in Luria–Bertani broth with appropriate antibiotics. Expression of these subunits was induced for 8 h at 293 K, for 4 h at 310 K, for 8 h at 293 K and for 2 h at 310 K, respectively, by adding 1 mM isopropyl β-d-1 thiogalactopyranoside. The plasmids for PFD2 and PFD3 were co-transformed into E. coli Rosetta2 (DE3) pLysS cells. Cell culture and induction of expression were carried out similarly with induction for 4 h at 310 K.
Table 1. Macromolecule-production information.
The restriction sites used in the primer sequences and the hexahistidine tags removed by thrombin are underlined.
| PFD1 | |
| Source organism | Homo sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | ATATCATATGGCCGCCCCCGTGGATCTAG |
| Reverse primer | CGGGATCCCTACTGGGCCCTTCGTGCCATCAG |
| Cloning vector | pET-21a |
| Expression vector | pET-21a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MAAPVDLELKKAFTELQAKVIDTQQKVKLADIQIEQLNRTKKHAHLTDTEIMTLVDETNMYEGVGRMFILQSKEAIHSQLLEKQKIAEEKIKELEQKKSYLERSVKEAEDNIREMLMARRAQ |
| PFD2 | |
| Source organism | H. sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | ATATCATATGGCGGAGAACAGCGGTCGC |
| Reverse primer | CGGGATCCCTAGGAGACCAACACTCCAGCTGAG |
| Cloning vector | pET-28a |
| Expression vector | pET-28a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MGSSHHHHHHSSGLVPRGSHMAENSGRAGKSSGSGAGKGAVSAEQVIAGFNRLRQEQRGLASKAAELEMELNEHSLVIDTLKEVDETRKCYRMVGGVLVERTVKEVLPALENNKEQIQKIIETLTQQLQAKGKELNEFREKHNIRLMGEDEKPAAKENSEGAGAKASSAGVLVS |
| PFD3 | |
| Source organism | H. sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | GGAATTCCATATGAAACAGCCTGGGAATGAGACTGCAG |
| Reverse primer | CGGGATCCTTATGCTTTGTTCTTGGTAGAGTCATCCTTGTTTCTTC |
| Cloning vector | pET-21a |
| Expression vector | pET-21a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MKQPGNETADTVLKKLDEQYQKYKFMELNLAQKKRRLKGQIPEIKQTLEILKYMQKKKESTNSMETRFLLADNLYCKASVPPTDKVCLWLGANVMLEYDIDEAQALLEKNLSTATKNLDSLEEDLDFLRDQFTTTEVNMARVYNWDVKRRNKDDSTKNKA |
| PFD4 | |
| Source organism | H. sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | GGAATTCCATATGAAGAAGGCGGCTGCAGAAGATGTCAATG |
| Reverse primer | CGGGATCCTTAACTTTCATCAGCTTCAAGGTTTATGTTGCTCCCG |
| Cloning vector | pET-28a |
| Expression vector | pET-28a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MGSSHHHHHHSSGLVPRGSHMKKAAAEDVNVTFEDQQKINKFARNTSRITELKEEIEVKKKQLQNLEDACDDIMLADDDCLMIPYQIGDVFISHSQEETQEMLEEAKKNLQEEIDALESRVESIQRVLADLKVQLYAKFGSNINLEADES |
| PFD5 | |
| Source organism | H. sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | GGAATTCCATATGGCGCAGTCTATTAACATCACGGAGC |
| Reverse primer | CGGATCCTCAGGCCTTAGCAGTAGCCTGAG |
| Cloning vector | pET-21a |
| Expression vector | pET-21a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MAQSINITELNLPQLEMLKNQLDQEVEFLSTSIAQLKVVQTKYVEAKDCLNVLNKSNEGKELLVPLTSSMYVPGKLHDVEHVLIDVGTGYYVEKTAEDAKDFFKRKIDFLTKQMEKIQPALQEKHAMKQAVMEMMSQKIQQLTALGAAQATAKA |
| PFD6 | |
| Source organism | H. sapiens |
| DNA source | Human liver cDNA library |
| Forward primer | GGAATTCCATATGGCGGAGCTGATCCAGAAGAAGCTACAG |
| Reverse primer | GGGATCCCTCAGGCCTTGCCAGGAGCCCC |
| Cloning vector | pET-21a |
| Expression vector | pET-21a |
| Expression host | E. coli Rosetta2 (DE3) pLysS |
| Complete amino-acid sequence of the construct produced | MAELIQKKLQGEVEKYQQLQKDLSKSMSGRQKLEAQLTENNIVKEELALLDGSNVVFKLLGPVLVKQELGEARATVGKRLDYITAEIKRYESQLRDLERQSEQQRETLAQLQQEFQRAQAAKAGAPGKA |
2.2. Purification
All purification steps were carried out at 277 K. Cells expressing PFD subunits were mixed and lysed by sonication in buffer A (50 mM sodium phosphate pH 7.0, 300 mM NaCl, 5 mM β-mercaptoethanol). For affinity purification using hexahistidine tags on PFD2 and PFD4, the supernatant was applied onto Talon Superflow Metal Affinity Resin (Clontech, California, USA) equilibrated with buffer A. The protein was eluted with buffer A containing 150 mM imidazole. This step excludes the mis-assembled specimens that carry no hexahistidine tags. The eluted sample was treated with thrombin for 16 h for tag removal, and was further purified using a Mono S cation-exchange column (GE Healthcare, Little Chalfont, England) by eluting with a linear gradient of 0.1–1 M NaCl in buffer B [20 mM 2-(N-morpholino)ethanesulfonic acid pH 6.0, 1 mM dithiothreitol (DTT)]. Finally, the sample was purified using a Superdex 200 10/300 gel-filtration column (GE Healthcare) equilibrated with buffer C (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM DTT). The purity of the sample was confirmed by SDS–PAGE followed by Coomassie staining (Fig. 1 ▸ a). The band pattern of the sample was similar to that previously reported for bovine PFD (Vainberg et al., 1998 ▸), suggesting the successful reconstitution and purification of recombinant human PFD.
Figure 1.

Biochemical characterization of human PFD. (a) SDS–PAGE analysis of purified human PFD. Lane M, molecular-weight marker (labelled in kDa); lane 1, peak fraction from Superdex 200 column. PFD subunits are assigned to the bands according to the molecular weights calculated from the sequences. (b) SEC-MALS of purified PFD. Red, molecular mass; blue, absorbance at 280 nm. (c) CD spectra. Red, spectrum of human PFD (HsPFD); blue, spectrum of P. horikoshii PFD (PhPFD). (d) MALDI-TOF MS. Peaks are annotated with the observed molecular mass (black) and the calculated mass from the sequence (blue, in parentheses).
2.3. SEC-MALS
The molecular mass of purified PFD was determined by size-exclusion chromatography with multi-angle light scattering (SEC-MALS) using a Superdex 200 10/300 column connected to a miniDAWN detector (Wyatt Technology, California, USA; Fig. 1 ▸ b). The purified sample was diluted to 0.5 mg ml−1 in buffer D (100 mM Tris–HCl pH 7.5, 300 mM NaCl). A 100 µl aliquot was loaded onto the column and eluted with buffer D at a flow rate of 0.5 ml min−1. The signal at a 90° angle was analyzed using the Astra software (Wyatt Technology). The average molecular masses of the first and second peaks were 204.8 and 100.6 kDa, corresponding to a dimer and a monomer of hexameric PFD (97.0 kDa), respectively. Although dimerization of PFD was also observed, this result supports the proper reconstitution of recombinant PFD.
2.4. CD spectroscopy
Circular-dichroism (CD) spectra of PFD were measured in a quartz cuvette with a path length of 1 mm using a J-805 spectropolarimeter (Jasco, Tokyo, Japan) at 298 K (Fig. 1 ▸ c). Protein samples were diluted to 0.1 mg ml−1 in buffer E (10 mM Tris–HCl pH 7.5, 1 mM DTT) before measurements. The buffer baseline was subtracted from the spectra and five scans were averaged. The mean residue molar ellipticities (ΘMRW) were calculated using the equation
where n is the number of amino-acid residues, c is the molar concentration and l is the path length. The secondary structure of human PFD consisted of 79% α-helix and 21% β-sheet. The spectrum of human PFD was similar to that of the archaeal jellyfish-like PFD from Pyrococcus horikoshii (PhPFD; Ohtaki et al., 2008 ▸). These results suggest that human PFD has a jellyfish-like structure similar to that of PhPFD.
2.5. MALDI-TOF MS
To confirm the correct assembly of PFD subunits, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed (Fig. 1 ▸ d). Sinapinic acid was dissolved at 10 mg ml−1 in 0.1% TFA and 50% acetonitrile. PFD was diluted to 10 pmol ml−1 and mixed with the sinapinic acid solution. The mixed solution was spotted onto a MALDI plate and the solvents were vaporized. Mass calibration was carried out using apo myoglobin (16.95 kDa) and cytochrome c (12.36 kDa). The mass spectrum showed peaks corresponding to all six subunits of PFD, suggesting the correct assembly of PFD. The molecular masses of PFD1, PFD5 and PFD6 were ∼131 Da lower than those calculated from the sequences, respectively. We interpret these results as the deletion of the N-terminal methionines during expression in E. coli.
2.6. Crystallization
Initial screening for crystallization condition was carried out at 293 K by the sitting-drop vapour-diffusion method using several commercial screening kits. Each crystallization drop was prepared by mixing equal volumes (0.3 µl each) of protein solution (10 mg ml−1 in 20 mM Tris–HCl pH 7.5, 1 mM DTT) and reservoir solution and equilibrating against 60 µl reservoir solution. Crystals were obtained in one condition with a reservoir consisting of 100 mM Tris–HCl pH 8.5, 9.9%(v/v) 2-propanol, 9.9%(w/v) polyethylene glycol (PEG) 3350. After optimizing the condition, crystals suitable for diffraction experiments were obtained by the hanging-drop vapour-diffusion method. Equal volumes (1.5 µl each) of protein solution (7.5 mg ml−1 in 10 mM Tris–HCl pH 7.5, 1 mM DTT) and reservoir solution [100 mM Tris–HCl pH 8.6, 11%(w/v) PEG 3350] were mixed and equilibrated against 500 µl reservoir solution at 293 K. Crystals with approximate dimensions of 300 × 100 × 20 µm were obtained within one week (Fig. 2 ▸).
Figure 2.

Crystals of human PFD. The black bar represents 0.5 mm.
2.7. X-ray data collection
The crystals were soaked in reservoir solution supplemented with 20%(w/v) PEG 4000 and flash-cooled in a nitrogen-gas stream at 95 K. X-ray diffraction data were collected on beamline BL1A at the Photon Factory, Tsukuba, Japan using a Pilatus 2M-F detector (Dectris, Baden, Switzerland; Fig. 3 ▸) and on BL41XU at SPring-8, Harima, Japan using a MX225HE detector (Rayonix, Illinois, USA). A data set consisting of 180 frames was collected with an oscillation angle of 1° per frame. The data set was processed and scaled with the HKL-2000 package (Otwinowski & Minor, 1997 ▸). The data-collection statistics are summarized in Table 2 ▸.
Figure 3.

X-ray diffraction pattern from a crystal of human PFD. The circle represents 4.7 Å resolution.
Table 2. Data-collection statistics for a crystal of human PFD.
Values in parentheses are for the highest resolution shell.
| Diffraction source | BL1A, Photon Factory |
| Wavelength () | 1.1 |
| Temperature (K) | 95 |
| Detector | Pilatus 2M-F |
| Crystal-to-detector distance (mm) | 440 |
| Rotation range per image () | 1 |
| Total rotation range () | 180 |
| Exposure time per image (s) | 0.5 |
| Space group | P21212 |
| a, b, c () | 123.2, 152.4, 105.9 |
| Mosaicity () | 0.873.40 |
| Resolution range () | 504.7 (4.874.70) |
| Total No. of reflections | 52378 |
| No. of unique reflections | 10172 |
| Completeness (%) | 92.9 (88.1) |
| Multiplicity | 5.2 (4.0) |
| I/(I) | 22.7 (2.3) |
| R p.i.m. † (%) | 3.0 (30.5) |
R
p.i.m. = 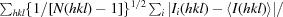
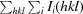 , where Ii(hkl) is the ith intensity measurement of reflection hkl, I(hkl) is the mean intensity for this reflection and N(hkl) is the multiplicity.
, where Ii(hkl) is the ith intensity measurement of reflection hkl, I(hkl) is the mean intensity for this reflection and N(hkl) is the multiplicity.
3. Results and discussion
Recombinant human PFD was reconstituted and purified from an E. coli expression system. SDS–PAGE, SEC-MALS and MALDI-TOF MS analyses suggested successful reconstitution and purification of heterohexameric PFD. A CD spectroscopic study suggested that human PFD has a jellyfish-like structure similar to those of its archaeal homologues. Crystals of human PFD were obtained using PEG 3350 as the main precipitant. X-ray diffraction data were indexed and scaled at 4.7 Å resolution. The crystals belonged to space group P21212, with unit-cell parameters a = 123.2, b = 152.4, c = 105.9 Å. Assuming the presence of two PFD hexamers in the asymmetric unit, the Matthews coefficient (V M) and solvent content were calculated to be 2.5 Å3 Da−1 and 50.2%, respectively (Matthews, 1968 ▸).
Acknowledgments
The authors are grateful to the beamline staff at Photon Factory and SPring-8 for their assistance during data collection. The authors are also grateful to Dr Masafumi Yohda for providing purified PhPFD. The diffraction experiment was performed on beamlines BL1A at the Photon Factory with the approval of KEK (Proposal No. 2013G659) and BL41XU at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; Proposal Nos. 2007A1829, 2009B1694, 2011A1602, 2011B1672 and 2012A1704). This work was supported by a grant from the National Project on Protein Structural and Functional Analyses (to KM) and from the Global COE Program ‘Integrated Material Science’ (No. B-024) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (to YA and HK).
References
- Hansen, W. J., Cowan, N. J. & Welch, W. J. (1999). J. Cell Biol. 145, 265–277. [DOI] [PMC free article] [PubMed]
- Hartl, F. U. & Hayer-Hartl, M. (2002). Science, 295, 1852–1858. [DOI] [PubMed]
- Kida, H., Sugano, Y., Iizuka, R., Fujihashi, M., Yohda, M. & Miki, K. (2008). J. Mol. Biol. 383, 465–474. [DOI] [PubMed]
- Leroux, M. R., Fändrich, M., Klunker, D., Siegers, K., Lupas, A. N., Brown, J. R., Schiebel, E., Dobson, C. M. & Hartl, F. U. (1999). EMBO J. 18, 6730–6743. [DOI] [PMC free article] [PubMed]
- Martín-Benito, J., Boskovic, J., Gómez-Puertas, P., Carrascosa, J. L., Simons, C. T., Lewis, S. A., Bartolini, F., Cowan, N. J. & Valpuesta, J. M. (2002). EMBO J. 21, 6377–6386. [DOI] [PMC free article] [PubMed]
- Martín-Benito, J., Gómez-Reino, J., Stirling, P. C., Lundin, V. F., Gómez-Puertas, P., Boskovic, J., Chacón, P., Fernández, J. J., Berenguer, J., Leroux, M. R. & Valpuesta, J. M. (2007). Structure, 15, 101–110. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Ohtaki, A., Kida, H., Miyata, Y., Ide, N., Yonezawa, A., Arakawa, T., Iizuka, R., Noguchi, K., Kita, A., Odaka, M., Miki, K. & Yohda, M. (2008). J. Mol. Biol. 376, 1130–1141. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Siegert, R., Leroux, M. R., Scheufler, C., Hartl, F. U. & Moarefi, I. (2000). Cell, 103, 621–632. [DOI] [PubMed]
- Vainberg, I. E., Lewis, S. A., Rommelaere, H., Ampe, C., Vandekerckhove, J., Klein, H. L. & Cowan, N. J. (1998). Cell, 93, 863–873. [DOI] [PubMed]