

Abstract
Deletions of chromosome 1p36 affect approximately 1 in 5,000 newborns and are the most common terminal deletions in humans. Medical problems commonly caused by terminal deletions of 1p36 include developmental delay, intellectual disability, seizures, vision problems, hearing loss, short stature, distinctive facial features, brain anomalies, orofacial clefting, congenital heart defects, cardiomyopathy, and renal anomalies. Although 1p36 deletion syndrome is considered clinically recognizable, there is significant phenotypic variation among affected individuals. This variation is due, at least in part, to the genetic heterogeneity seen in 1p36 deletions which include terminal and interstitial deletions of varying lengths located throughout the 30 Mb of DNA that comprise chromosome 1p36. Array-based copy number variant analysis can easily identify genomic regions of 1p36 that are deleted in an affected individual. However, predicting the phenotype of an individual based solely on the location and extent of their 1p36 deletion remains a challenge since most of the genes that contribute to 1p36-related phenotypes have yet to be identified. In addition, haploinsufficiency of more than one gene may contribute to some phenotypes. In this article, we review recent successes in the effort to map and identify the genes and genomic regions that contribute to specific 1p36-related phenotypes. In particular, we highlight evidence implicating MMP23B, GABRD, SKI, PRDM16, KCNAB2, RERE, UBE4B, CASZ1, PDPN, SPEN, ECE1, HSPG2, and LUZP1 in various 1p36 deletion phenotypes.
Keywords: chromosome 1p36, chromosome deletion, 1p36 deletion syndrome, monosomy 1p36
Introduction
Deletions of chromosome 1p36 affect approximately 1 in 5,000 newborns and constitute the most common terminal chromosomal deletion in humans.1,2 The first reports of individuals with partial monosomy of chromosome 1p36 were published in the early 1980s, starting with a report by Hain et al.3 Many of the individuals described in these reports had unbalanced translocations in which their 1p36 deletions were accompanied by a gain of material from a nonhomologous chromosome.4 While the addition of chromosomal material to the long arm of chromosome 1 made it easier to identify these 1p36 deletions, it also made it more difficult to delineate with certainty the clinical effects of monosomy 1p36.
In 1987, Magenis et al5 published the first report of an individual with a de novo isolated 1p36 deletion. With the publication of additional case reports, and the clinical descriptions of large cohorts of individuals with 1p36 deletions, a pattern of characteristic functional deficits, congenital anomalies, and physical features associated with 1p36 deletions emerged.6–8 This pattern included developmental delay, intellectual disability, seizures, vision problems, hearing loss, short stature, brain anomalies, orofacial clefting, congenital heart defects, cardiomyopathy, renal anomalies, and distinctive facial features – straight eyebrows, deeply set eyes, midface retrusion, wide and depressed nasal bridge, long philtrum, pointed chin, large, late-closing anterior fontanel, microbrachycephaly, epicanthal folds, and posteriorly rotated, low-set, abnormal ears (Figures 1 and 2).7 Defining this pattern made the 1p36 deletion syndrome a clinically recognizable entity. At the same time, these reports also highlighted the significant phenotypic variability seen between patients.
Figure 1.

Facial features of a girl with a terminal 1p36 deletion (chr1:1–3,047,838; GRCh37/hg19).
Notes: Photos were taken at (A) 1 year 8 months, (B) 2 years 3 months, (C) 4 years, (D) 7 years, (E) 7 years 11 months, and (F) 10 years 3 months of age. These photos demonstrate several facial features that are typical of children with terminal 1p36 deletions, including straight eyebrows, a wide nasal bridge, and a pointed chin.
Figure 2.

Facial features of a woman with a large interstitial 1p36 deletion (chr1:3,313,081–12,530,129; GRCh37/hg19).
Notes: Photos were taken at (A) birth, (B) 16 months, (C) 5 years, (D) 7 years, (E) 9 years, (F) 14 years, (G) 25 years, (H) 31 years, and (I) 32 years of age. Her deletion partially overlaps the distal critical region and includes the entire proximal critical region of chromosome 1p36. Characteristic facial features that are evident in these photos include straight eyebrows, deeply set eyes and epicanthal folds. Other features that are not readily apparent in these photos include brachycephaly, small, low set ears, and facial hirsutism.
At first, it was suggested that the variable phenotypic expression of 1p36 deletions might be caused by a parent-of-origin effect in which deletions of the paternally-derived copy of 1p36 were not equivalent to deletions of the maternally-derived copy due to differences in imprinting. However, Shapira et al4 used DNA polymorphism analysis to show that there was no obvious parent-of-origin effect. Instead, they concluded that phenotypic variability was more likely to be caused by differences in the location and extent of the 1p36 deletions, which varied significantly in the patients they studied.
At the time Shapira et al4 came to this conclusion, 1p36 deletions were typically identified using G-banded chromosome analyses or telomere fluorescence in situ hybridization (FISH). As a result, most of the 1p36 deletion patients described in the literature had telomeric deletions. With the widespread clinical use of array-based copy number detection assays, an increasing number of small interstitial deletions are being identified throughout the 30 Mb of DNA that comprise chromosome 1p36.9,10 In many cases, the phenotypes of these individuals are distinct from those with terminal deletions since they are caused by haploinsufficiency of a discrete set of genes.9
The clinical and genetic heterogeneity seen among individuals with 1p36 deletions present a significant challenge to physicians who are called upon to provide prognostic information to families and to generate individualized care plans for their patients that include appropriate diagnostic and surveillance testing. This challenge arises, in part, because the genes that contribute to most 1p36-related phenotypes have yet to be identified, and many 1p36-related phenotypes may arise from haploinsufficiency for more than one gene within a particular genomic region.
In this article, we review recent successes in the effort to map and identify the genes and genomic regions that contribute to specific 1p36-related phenotypes. In particular, we highlight evidence implicating haploinsufficiency of MMP23B, GABRD, SKI, PRDM16, KCNAB2, RERE, UBE4B, CASZ1, PDPN, SPEN, ECE1, HSPG2, and LUZP1 in the development of various 1p36 deletion phenotypes. All of the coordinates referenced in the text and figures are based on human genome build GRCh37/hg19.
Identification of distal and proximal 1p36 critical regions
After the characteristic features of 1p36 deletion syndrome were described, efforts were made to determine the smallest terminal deletion that was required to cause individual 1p36 phenotypes. In a study of 30 individuals with 1p36 deletions, Wu et al11 determined that most genes contributing to the phenotypic features of 1p36 deletion syndrome were located distal to marker D1S2870 (chr1:6,289,764–6,289,973). This region was subsequently referred to as the distal or classical critical region (Figure 3).
Figure 3.
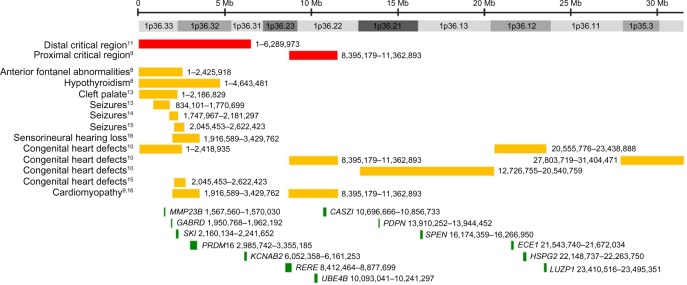
Critical regions and selected genes on chromosome 1p36.
Notes: Chromosome 1p36 spans approximately 30 Mb. Red bars represent the approximate locations of the distal and proximal critical regions. Orange bars represent the approximate locations of critical regions defined for various 1p36-related phenotypes. Green bars represent the approximate locations of selected genes whose haploinsufficiency is likely to contribute to phenotypes associated with 1p36 deletions. Coordinates are based on human genome build GRCh37/hg19.
Using array comparative genomic hybridization, Kang et al9 identified interstitial deletions affecting 1p36.23–1p36.11 in five individuals. This cohort included a 5-year-old male with a 2.97 Mb deletion (chr1:8,395,179–11,362,893) that did not overlap the distal critical region. Some of the facial features of this individual were distinct from those typically seen in 1p36 deletions and included frontal and parietal bossing, low-set, posteriorly rotated ears, epicanthal folds, anteverted nares, broad eyebrows, and hirsutism. However, this individual had many of the functional deficits and congenital anomalies commonly associated with deletion of the distal 1p36 critical region including developmental delay, speech delay, failure to thrive, hypotonia, congenital heart defects (a ventricular septal defect and an atrial septal defect), and dilated cardiomyopathy (CMD). Similar features were seen in other members of this cohort who had larger overlapping deletions.
Kang et al9 concluded that the features seen in these children might constitute a distinct proximal 1p36 deletion syndrome. Alternatively, they reasoned that the region could be considered a second contiguous gene deletion segment for 1p36 deletion syndrome. In either case, this data demonstrated that haploinsufficiency of two or more genes on 1p36 could affect somatic growth and the development and function of the brain and heart. These observations also helped to explain why large terminal deletions of 1p36 – which in some cases affected both critical regions – were associated with increased penetrance for individual phenotypes and a more severe clinical presentation.11,12
The identification of distal and proximal 1p36 critical regions also had important implications for future mapping studies. Since deletion of two or more 1p36-related genes or genomic regions could result in similar phenotypes, it would be unwise to automatically assume that the gene responsible for a specific phenotype must reside within the minimum region of overlap between two large deletions. Similarly, the incomplete penetrance seen for most, if not all, 1p36-related phenotypes suggested that the absence of a phenotype associated with a particular 1p36 deletion should not be used to conclude that the genes located within that region do not contribute to a specific phenotype.6,7
Mapping individual 1p36 deletion phenotypes
After publication of the distal 1p36 critical region, the critical regions for several individual phenotypes associated with terminal 1p36 deletions were refined further. Using large insert clones as markers, Heilstedt et al8 demonstrated that terminal deletions, including RP3-395M20 (chr1:2,424,876–2,425,918) were sufficient to cause large, late-closing anterior fontanels, while those including RP1-37J18 (chr1:4,631,608–4,643,481) could cause hypothyroidism, which is present in 15%–20% of individuals with 1p36 deletions.7 A critical region for cleft palate was defined by Shimida et al13 based on an individual with a terminal 1p36 deletion (chr1:1–2,186,829). Small, partially overlapping deletions that were sufficient to cause seizures were reported by Shimada et al13 (chr1:834,101–1,770,699), Rosenfeld et al14 (chr1:1,747,967–2,181,297), and Zhu et al15 (chr1:2,045,453–2,622,423). A critical region for sensorineural hearing loss was defined by Gajecka et al16 in two siblings, both of whom carried a 1.51 Mb interstitial deletion (chr1:1,916,589–3,429,762).
Rather than focusing on the distal critical region, Zaveri et al10 defined five critical regions for congenital heart defects along the entire length of chromosome 1p36 using a combination of published and previously unpublished individuals (chr1:1–2,418,935; chr1:8,395,179–11,362,893; chr1:12,726,755–20,540,759; chr1:20,555,776–23,438,888; chr1:27,803,719–31,404,471). The most distal of these critical regions overlaps partially with the deletion reported by Zhu et al15 (chr1:2,045,453–2,622,423), which was carried by an individual who had an atrial septal defect.
In contrast to the relatively large numbers of 1p36 critical regions for congenital heart defects identified by Zaveri et al,10 only two nonoverlapping critical regions for cardiomyopathy were identified by the same group. The first is the distal critical region for left ventricular noncompaction defined by Gajecka et al16 (chr1:1,916,589–3,429,762). The second is a proximally located critical region defined by the same deletion that was used by Kang et al9 to define the proximal critical region for 1p36 (chr1:8,395,179–11,362,893). This individual had CMD that appeared to be independent of his congenital heart defects.
Genes that may contribute to 1p36 deletion phenotypes
As critical regions for various phenotypes have been delineated, positional candidate genes that may contribute to these phenotypes have also been proposed based on their expression pattern, putative function, or the phenotypes seen in animal models and/or individuals who carry deleterious mutations in these genes. Some of the most strongly implicated 1p36 genes include MMP23B, GABRD, SKI, PRDM16, KCNAB2, RERE, UBE4B, CASZ1, PDPN, SPEN, ECE1, HSPG2, and LUZP1. The data supporting their potential role in 1p36-deletion phenotypes are summarized briefly in the following sections and in Table 1.
Table 1.
Summary of genes that may contribute to 1p36 deletion phenotypes
| Gene (OMIM#) | Phenotypes possibly associated with haploinsufficiency | Similar phenotypes seen in animal models | References |
|---|---|---|---|
| MMP23B (OMIM# 603321) | Large, late-closing anterior fontanel | Not reported | Gajecka et al18 |
| GABRD (OMIM# 137163) | Neurodevelopmental abnormalities, neuropsychiatric problems, seizures | Mice: depression-like and anxiety-like behaviors during the postpartum period | Mihalek et al,21 Maguire and Mody,22 Dibbens et al,23 Lenzen et al24 |
| SKI (OMIM# 64780) | Developmental delay, intellectual disability, seizures, orofacial clefting, congenital heart defects | Mice: neural tube defects, abnormal forebrain morphology, midline facial clefting, reduced skeletal muscle mass Zebrafish: craniofacial cartilage deficits, severe cardiac anomalies |
Colmenares et al,27 Doyle et al,28 Carmignac et al29 |
| PRDM16 (OMIM# 605557) | Left vertricular noncompaction, dilated cardiomyopathy | Mice: ventricular hypoplasia, abnormal ventricular morphology, cleft palate Zebrafish: bradycardia, reduced cardiac output, decreased cardiomyocyte number, partial uncoupling of cardiomyocytes, reduced impulse propagation velocity |
Arndt et al,33 Bjork et al35 |
| KCNAB2 (OMIM# 601142) | Developmental delay, intellectual disability, seizures | Mice: impaired associative learning and memory, sporadic seizures, cold swim-induced tremors | Perkowski and Murphy,37 McCormack et al38 |
| RERE (OMIM# 605226) | Short stature, developmental delay, intellectual disability, brain anomalies, vision problems, hearing loss, renal anomalies, congenital heart defects, cardiomyopathy | Mice: postnatal growth retardation, reduced brain size and weight, decreased numbers of NeuN-positive hippocampal neurons, cerebellar foliation defects, delayed Purkinje cell maturation and migration, microphthalmia, hearing loss, renal agenesis, congenital heart defects, cardiac fibrosis Zebrafish: microphthalmia, inconsistent startle response, decreased microphonic potentials |
Kim et al,40,41 Plaster et al,42 Schilling et al43 |
| UBE4B (OMIM# 613565) | Cardiomyopathy and neurodevelopmental phenotypes | Mice: reduced cardiac trabeculation, undeveloped and compact myocardial layer, high levels of cardiac-restricted apoptosis, defective assembly of myosin in cardiomyocytes, axonal dystrophy in the nucleus gracilis, degeneration of Purkinje cells, hindlimb gait abnormalities, impaired rotarod performance | Kaneko-Oshikawa et al45 |
| CASZ1 (OMIM# 609895) | Congenital heart defects and cardiomyopathy | Mice: abnormally shaped cardiac ventricular apices, ventricular septal defects, hypoplastic myocardium, abnormalities in ventricular cell alignment and fiber orientation | Liu et al47 |
| PDPN (OMIM# 608863) | Congenital heart defects, cardiomyopathy | Mice: congenital heart defects; hypoplasia of the sinoatrial node, primary atrial septum, and dorsal atrial wall; thin, perforated cardinal and pulmonary veins | Mahtab et al49,50 |
| SPEN (OMIM# 613484) | Congenital heart defects, cardiomyopathy, short stature, neurodevelopmental phenotypes | Mice: defective formation of the cardiac septum and muscle, postnatal growth retardation, reduced brain weight, hypoplastic cerebral cortex and hippocampus, enlarged lateral ventricles of the brain | Kuroda et al,52 Yabe et al53 |
| ECE1 (OMIM# 600423) | Congenital heart defects | Mice: congenital heart defects | Hofstra et al,55 Yanagisawa et al56 |
| HSPG2 (OMIM# 142461) | Cleft palate, congenital heart defects | Mice: cleft palate, congenital heart defects | Abdel-Aziz and Azab,60 Costell et al61,62 |
| LUZP1 (OMIM# 601422) | Congenital heart defects, cleft palate, brain anomalies | Mice: congenital heart defects, cleft palate, brain anomalies | Hsu et al63 |
MMP23B
The matrix metallopeptidase 23B gene (MMP23B; chr1:1,567,560–1,570,030; OMIM# 603321) is located in the critical region for large, late-closing anterior fontanel, delineated by Heilstedt et al,8 and encodes a metallopeptidase that is involved in bone matrix resorption and bone remodeling.17 Gajecka et al18 noted that individuals with deletions involving MMP23B had large, late-closing anterior fontanels, while individuals with duplications involving MMP23B developed craniosynostosis. After demonstrating that MMP23B is expressed in the cranial sutures, they concluded that haploinsufficiency of MMP23B was responsible for the large, late-closing anterior fontanels seen in children with 1p36 deletions.
GABRD
Haploinsufficiency of the gamma-aminobutyric acid (GABA) A receptor, delta gene (GABRD; chr1:1,950,768–1,962,192; OMIM# 137163) has been suggested as a possible contributor to the neurodevelopmental abnormalities, neuropsychiatric problems, and seizures seen in children with 1p36 deletions.19 GABRD encodes a subunit of a pentameric ligand-gated chloride channel that is activated by GABA,20 the major inhibitory neurotransmitter in the mammalian brain, and is located in the critical region for seizures defined by Rosenfeld et al.14 Gabrd-null mice exhibited depression-like and anxiety-like behaviors during the postpartum period. However, their learning and memory were normal when assessed using fear conditioning.21,22
By screening 72 unrelated patients with idiopathic generalized epilepsy, 65 patients with generalized epilepsy with febrile seizures plus (GEFS+), and 66 patients with febrile seizures for mutations in the GABRD gene, Dibbens et al23 identified two sequence variants in GABRD that lead to a significant reduction in maximal GABAA receptor current amplitude. The first of these heterozygous changes (c.530A>C, p.Glu177Ala [p.E177A]) was found in a small family with GEFS+. Since the unaffected mother also carried the mutation, it was thought that this change might represent a susceptibility allele.
The second change (c.659G>A, p.Arg220His [p.R220H]) was carried at similar frequencies in individuals with idiopathic generalized epilepsy (8.3%), GEFS+ (3.1%), febrile seizures (4.5%), juvenile myoclonic epilepsy (3.7%), and in controls (4.2%). Despite the fact that the carrier rates among the various epilepsy cohorts and controls were not statistically different, Dibbens et al23 suggested that this functional variant contributes to idiopathic generalized epilepsy and possibly other polygenic epilepsies. At the same time, they conceded that larger sample sizes would be required in order to explore the possibility of statistical associations. Lenzen et al,24 in a larger cohort of 562 German patients and 664 controls, found no association between the p.R220H change and idiopathic generalized epilepsy or juvenile myoclonic epilepsy.
SKI
The SKI proto-oncogene (SKI; chr1:2,160,134–2,241,652; OMIM# 164780) encodes a protein that acts as a transcriptional coregulator.25,26 Haploinsufficiency of SKI is thought to contribute to the developmental delay, intellectual disability, seizures, orofacial clefting, and congenital heart defects associated with distal/terminal 1p36 deletions. SKI is located within the critical region for seizures defined by Rosenfeld et al14 and Zhu et al,15 the critical region for cleft palate defined by Shimada et al,13 and the critical region for congenital heart defects defined by Zaveri et al10 and Zhu et al.15
Colmenares et al27 demonstrated that Ski-null mice have neural tube defects, abnormal forebrain morphology, midline facial clefting, and reduced skeletal muscle mass. Subsequently, Doyle et al28 showed that knockdown of the two paralogs of mammalian SKI in zebrafish (skia and skib) results in marked craniofacial cartilage deficits and severe cardiac anomalies.
Point mutations and small in-frame deletions of SKI have been shown to cause Shprintzen–Goldberg syndrome (OMIM# 182212).28,29 While some features of Shprintzen–Goldberg syndrome are similar to those seen with 1p36 deletions involving SKI – such as developmental delay, intellectual disability, and a high, narrow palate – other features of this disorder are unique, including craniosynostosis, a marfanoid habitus, mitral valve prolapse, aortic root dilatation, and distinct facial features.30,31 One possible explanation for these differences is that the SKI mutations seen in Shprintzen–Goldberg syndrome patients do not create typical loss-of-function alleles.
PRDM16
The PR domain containing 16 gene (PRDM16; chr1:2,985,742–3,355,185; OMIM# 605557) encodes a zinc finger transcription factor.32 Several lines of evidence suggest that haploinsufficiency of PRDM16 contributes to left vertricular noncompaction and CMD. PRDM16 is located in the critical region for cardiomyopathy defined by Gajecka et al,16 and PRDM16 protein is expressed in the nuclei of murine and human cardiomyocytes throughout development and into adulthood.33 Arndt et al33 resequenced PRDM16 in 75 non-syndromic individuals with left vertricular noncompaction and in a series of 131 cardiac biopsies from individuals with CMD. Eight of these samples were found to carry putatively deleterious sequence changes in PRDM16.
To confirm the role of PRDM16 in cardiomyopathy, Arndt et al33 performed knockdown of the zebrafish ortholog of PRDM16 using translation-blocking morpholinos. They observed dose-dependent bradycardia and reduced cardiac output in these morphant fish, which was rescued by wild-type human PRDM16 in a dose-dependent fashion. They also observed a decrease in total cardiomyocyte number in morphant fish compared to wild-type controls. This was associated with a decrease in cardiomyocyte proliferation. PRDM16 knockdown also caused partial uncoupling of cardiomyocytes with significant reductions in impulse propagation velocities seen in morphant hearts when compared with those of wild-type controls.
PRDM16 has been shown to interact physically with SKI to inhibit transforming growth factor-β signaling.34 Because most of the 1p36 deletions associated with cardiomyopathy include both PRDM16 and SKI – or include the proximal cardiomyopathy critical region defined by Kang et al9 – Arndt et al33 hypothesized that these genes may function together in the development of cardiomyopathy. By injecting subthreshold doses of morpholinos against PRDM16 and SKI independently and in combination, they demonstrated a synergistic genetic interaction between these genes in the development of decreased cardiac output in zebrafish.33 This suggests that depletion of SKI could have a modifying effect on the PRDM16 haploinsufficiency phenotype in cardiomyocytes and, potentially, in other tissues.
Studies in mice suggest that haploinsufficiency of PRDM16 may also play a role in the development of other 1p36-deletion phenotypes. Bjork et al35 showed that hearts of PRDM16-deficient mice exhibit ventricular hypoplasia and abnormal ventricular morphology with a cleft between ventricles. These mice also develop cleft palate as a result of micrognathia and failed palate shelf elevation due to physical obstruction by the tongue.35 However, PRDM16 is located outside of the currently reported critical regions for congenital heart defects and cleft palate defined by Zaveri et al10 and by Shimada et al,13 respectively.
KCNAB2
The potassium channel, voltage-gated subfamily A regulatory beta subunit 2 gene (KCNAB2; chr1:6,052,358–6,161,253; OMIM# 601142) encodes an auxiliary protein that alters the properties of functional potassium voltage-gated alpha subunits.36 Haploinsufficiency of this gene has been suggested to contribute to developmental delay, intellectual disability, and seizures.37 KCNAB2 is located within the distal 1p36 critical region defined by Wu et al,11 but not within the critical regions for seizures defined by Shimada et al,13 Rosenfeld et al,14 and Zhu et al.15
Individuals bearing deleterious mutations in KCNAB2 have not been reported. However, mice that are homozygous for a null mutation in Kcnab2 exhibit impaired associative learning and memory, sporadic seizures, and cold swim-induced tremors.37,38
RERE
The arginine-glutamic acid dipeptide (RE) repeats gene (RERE; chr1:8,412,464–8,877,699; OMIM# 605226) is located in the proximal 1p36 critical region and encodes a widely expressed nuclear receptor coregulator.39 Individuals carrying deleterious sequence changes in RERE have not been reported. However, mouse and zebrafish studies of RERE suggest that it may play a role in several 1p36 deletion-related phenotypes, including short stature, developmental delay, intellectual disability, brain anomalies, vision problems, hearing loss, renal anomalies, congenital heart defects, and cardiomyopathy.
Kim et al40 demonstrated that compound heterozygous mice carrying a null and a hypomorphic allele of Rere have postnatal growth retardation and reduced brain size and weight independent of somatic growth. Further studies revealed other abnormalities in brain development, including decreased numbers of NeuN-positive hippocampal neurons, cerebellar foliation defects, and delayed Purkinje cell maturation and migration.40,41 These mice also have microphthalmia, hearing loss, renal agenesis, and a variety of congenital heart defects, including aortic arch anomalies, double outlet right ventricle, transposition of the great arteries, and ventricular septal defects. On a different background, RERE-deficient mice of the same genotype did not have congenital heart defects. However, these mice spontaneously developed cardiac fibrosis.
Eye- and hearing loss-related phenotypes have also been documented in zebrafish carrying homozygous mutations in rerea (the zebrafish homologue of RERE). Specifically, these fish have microphthalmia, inconsistent startle response, and decreased microphonic potentials.42,43
UBE4B
The ubiquitination factor E4B gene (UBE4B; chr1:10,093,041–10,241,297; OMIM# 613565) encodes a ubiquitination factor that is involved in multiubiquitin chain assembly.44 UBE4B is located in the cardiomyopathy critical region defined by Kang et al.9 Kaneko-Oshikawa et al45 demonstrated that the hearts of Ube4b-null mice have reduced trabeculation and an undeveloped and compact myocardial layer. They also demonstrated high levels of cardiac-restricted apoptosis and defective assembly of myosin in cardiomyocytes.
The same authors then studied the effects of Ube4b haploinsufficiency on the brain.45 They found evidence of axonal dystrophy in the nucleus gracilis, as well as degeneration of Purkinje cells in adult Ube4b+/− mice. These mice also developed hind limb gait abnormalities and impaired performance in the rotarod test when compared to age-matched wild-type mice.45
These results suggest that haploinsufficiency of UBE4B may contribute to both cardiomyopathy and neurodevelopmental abnormalities.
CASZ1
The castor zinc finger 1 gene (CASZ1; chr1:10,696,666–10,856,733; OMIM# 609895) encodes a zinc finger transcription factor that is highly expressed in the heart.46 Although mutations in CASZ1 have not been reported in humans, this gene is located in one of the congenital heart defect critical regions defined by Zaveri et al10 and in the proximal cardiomyopathy critical region defined by Kang et al.9
Liu et al47 demonstrated that CASZ1-deficient mice had abnormal heart morphology with abnormally shaped ventricular apices and ventricular septal defects. The compact myocardium of their right and left ventricles was also found to be hypoplastic. Further investigations suggested that this was due to decreased cardiomyocyte proliferation. Abnormalities in cell alignment and fiber orientation were also observed in both ventricles. Thus, they concluded that haploinsufficiency of CASZ1 might contribute to the congenital heart defects and cardiomyopathy seen in individuals with 1p36 deletions.47
PDPN
The podoplanin gene (PDPN; chr1:13,910,252–13,944,452; OMIM# 608863) encodes an integral membrane glycoprotein, which is preferentially expressed in the vascular endothelium.48 PDPN is located inside one of the congenital heart defect critical regions defined by Zaveri et al,10 but outside those presently described for cardiomyopathy. Although no PDPN mutations have been described in humans, haploinsufficiency of this gene has been suggested as a possible contributor to 1p36 deletion-related cardiac phenotypes based on mouse studies.
Mahtab et al49 showed that Pdpn-null mice have high rates of in utero and perinatal death that may be attributable to cardiac-related abnormalities. Structural heart defects identified in these mice include atrioventricular valve anomalies and abnormal coronary artery morphology. Pdpn-null embryos were also found to have hypoplasia of the sinoatrial node, primary atrial septum, and dorsal atrial wall.50 The myocardium lining the wall of the cardinal and pulmonary veins of these mice was also found to be thin and perforated.
SPEN
The spen family transcriptional repressor gene (SPEN; chr1:16,174,359–16,266,950; OMIM# 613484) is located in one of the congenital heart defect critical regions defined by Zaveri et al,10 but outside of the currently defined critical regions for cardiomyopathy. SPEN is a transcriptional repressor that may function as a nuclear matrix platform that organizes and integrates transcriptional responses.51 Kuroda et al52 reported that Spen-null embryos die in utero and have defective formation of the cardiac septum and muscle. These embryos also had perturbed differentiation of pancreatic exocrine and endocrine cells, hypoplastic livers, and abnormal B-cell differentiation.
To analyze the function of SPEN in postnatal mice, Yabe et al53 developed Spen-conditional knockout mice. They subsequently ablated Spen throughout the nervous system using a transgenic nestin-Cre. Although the resulting mice were born at expected numbers and were morphologically indistinguishable from their littermates, they exhibited postnatal growth retardation. Their brain weight was reduced and histological analyses revealed severe reductions in cerebral cortex thickness and hippocampus size accompanied by enlargement of the lateral ventricles. Disruption of Spen in forebrain neurons using a transgenic CaMKIIα-Cre also resulted in brain hypoplasia. Thus, they concluded that Spen is required for survival of neurons in the postnatal mouse brain.
Taken together, these findings suggest that haploinsufficiency of SPEN may contribute to the congenital heart defects, cardiomyopathy, short stature, and neurodevelopmental phenotypes associated with 1p36 deletions.
ECE1
Haploinsufficiency of the endothelin-converting enzyme 1 gene (ECE1; chr1:21,543,740–21,672,034; OMIM# 600423) has been suggested as a possible contributor to congenital heart defects and is located in one of the congenital heart defect critical regions defined by Zaveri et al.10 ECE1 is a metalloprotease that is involved in the proteolytic processing of endothelin precursors to biologically active peptides.54 Hofstra et al55 identified a heterozygous missense mutation in the ECE1 gene of an individual with patent ductus arteriosus, a ventricular septal defect, an atrial septal defect, Hirschsprung disease, and autonomic dysfunction. This mutation is located in the vicinity of the ECE1 active site and severely compromises ECE1 activity.
Yanagisawa et al56 have shown that Ece1-null mice have a similar phenotype that includes cardiac defects – interrupted aortic arch, absent right subclavian artery, double outlet right ventricle, truncus arteriosus, double aortic arch, overriding aorta, and ventricular septal defects – and absence of the enteric neurons of the distal gut. These mice also display a variety of craniofacial anomalies, defects of the outer and middle ear, and absence of epidermal melanocytes.
HSPG2
The heparan sulfate proteoglycan 2 gene (HSPG2; chr1:22,148,737–22,263,750; OMIM# 142461) encodes perlecan, a large multidomain heparan sulfate proteoglycan of the extracellular matrix that binds to various basement membrane proteins.57,58 Recessive mutations in HSPG2 have been shown to underlie two skeletal dysplasia syndromes; dyssegmental dysplasia Silverman-Handmaker type (OMIM# 224410) and the less severe Schwartz–Jampel syndrome, type 1 (OMIM# 255800).58,59 Cleft palate can be seen in both of these syndromes.60 Similarly, Hspg2-null embryos also have skeletal abnormalities and cleft palates.61 Although congenital heart defects are not typical features associated with dyssegmental dysplasia Silverman-Handmaker type or Schwartz–Jampel syndrome, type 1, Hspg2-null embryos have cardiac anomalies, including transposition of the great arteries and malformations of the semilunar valves.62 HSPG2 is also located in one of the congenital heart defect critical regions defined by Zaveri et al.10 These findings suggest that haploinsufficiency of HSPG2 may contribute to the development of both cleft palate and congenital heart defects.
LUZP1
The leucine zipper protein 1 gene (LUZP1; chr1:23,410,516–23,495,351; OMIM# 601422) is located in one of the congenital heart defect critical regions defined by Zaveri et al.10 Although the phenotypic consequences of mutations in LUZP1 have not been described in humans, Hsu et al63 demonstrated that Luzp1-null mice have congenital heart defects, including double outlet right ventricle, transposition of the great arteries, and ventricular septal defects. Other features seen in these mice include cleft palate and abnormal brain development.63 This suggests that haploinsufficiency of LUZP1 may contribute to defects in cardiac, palatal, and brain development/function.
1p36 copy number gains
In contrast to the large numbers of 1p36 deletions that have been reported in the literature, relatively few cases of isolated 1p36 copy number gains have been published. Giannikou et al64 reported two individuals with relatively small, de novo, isolated terminal 1p36 duplications (chr1:1–1,565,789 and chr1:1–1,565,607): a 6-month-old male with hypotonia and severe psychomotor delay and a 17-year-old male with growth hormone deficiency, short stature, psychomotor delay, and left ventricular hypertrophy. Subsequently, Xu et al65 reported an 8-year-old female who carried four copies of approximately 5.28 Mb of the terminal region of 1p36. Her phenotypes included feeding difficulties in infancy, developmental delay, seizures, microcephaly, strabismus, hypertelorism, a low hairline, ear malformations, a broad nasal bridge, wide mouth, thick lips, and prominent incisors. Parental FISH was normal, suggesting that this tetrasomy was de novo.
Recently, Weaver et al66 reported the identification of 1p36.22p36.21 duplications and triplications in three individuals with focal facial dermal dysplasia 3, Setleis type (OMIM# 227260). This disorder is characterized by congenital bitemporal or preauricular atrophic skin lesions with variable facial findings, which may include thin and puckered periorbital skin, redundant facial skin, upslanting palpebral fissures, distichiasis and/or absent eyelashes, a flat nasal bridge with a broad nasal tip and large lips.67 The minimum overlapping region in these patients extended from nucleotides 11,696,993 to 12,920,040.
The majority of genes that contribute to the phenotypes associated with copy number gains on chromosome 1p36 have yet to be determined. One exception that we have previously mentioned is the matrix metallopeptidase 23B gene (MMP23B; chr1:1,567,560–1,570,030; OMIM # 603321) whose deletion contributes to large, late-closing anterior fontanels and whose duplication is associated with the development craniosynostosis.18
Conclusion
Most of the genes currently implicated in the development of 1p36 deletion-related phenotypes have been identified through a combination of molecular cytogenetic mapping, resequencing of positional candidate genes in humans, and/or the development of animal models. It is likely that the expanding use of array-based copy number variant detection assays will speed the identification of individuals with small 1p36 deletions that can be used to refine the 1p36 critical regions that have already been delineated and to identify new critical regions. At the same time, exome and genome sequencing efforts will help identify genes whose haploinsufficiency contributes significantly to an increased risk of developing specific 1p36-related phenotypes. Cellular and animal models will continue to provide important supportive evidence for the contribution of these genes and may also provide the initial evidence that a gene is involved in a particular phenotype. As a comprehensive deletion/phenotype map of the 1p36 region emerges from these efforts, physicians will find it easier to provide the prognostic information desired by families affected by 1p36 deletions and to generate individualized care plans for their patients who carry these deletions.
Acknowledgments
The authors would like to thank the families who contributed to this review by providing photographs and consenting to their publication.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet. 2000;34:297–329. doi: 10.1146/annurev.genet.34.1.297. [DOI] [PubMed] [Google Scholar]
- 2.Heilstedt HA, Ballif BC, Howard LA, Kashork CD, Shaffer LG. Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin Genet. 2003;64(4):310–316. doi: 10.1034/j.1399-0004.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- 3.Hain D, Leversha M, Campbell N, Daniel A, Barr PA, Rogers JG. The ascertainment and implications of an unbalanced translocation in the neonate. Familial 1:15 translocation. Aust Paediatr J. 1980;16(3):196–200. doi: 10.1111/j.1440-1754.1980.tb01296.x. [DOI] [PubMed] [Google Scholar]
- 4.Shapira SK, McCaskill C, Northrup H, et al. Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet. 1997;61(3):642–650. doi: 10.1086/515520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magenis RE, Shehy R, Lacey D, Brown MG, Litt M. Small terminal deletion of chromosome 1 short arm in an infant with multiple anomalies: confirmation by in situ hyridization of rove p1-79. Am J Hum Genet. 1987;41:A130. [Google Scholar]
- 6.Slavotinek A, Shaffer LG, Shapira SK. Monosomy 1p36. J Med Genet. 1999;36(9):657–663. [PMC free article] [PubMed] [Google Scholar]
- 7.Battaglia A, Hoyme HE, Dallapiccola B, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121(2):404–410. doi: 10.1542/peds.2007-0929. [DOI] [PubMed] [Google Scholar]
- 8.Heilstedt HA, Ballif BC, Howard LA, et al. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet. 2003;72(5):1200–1212. doi: 10.1086/375179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang SH, Scheffer A, Ou Z, et al. Identification of proximal 1p36 deletions using array-CGH: a possible new syndrome. Clin Genet. 2007;72(4):329–338. doi: 10.1111/j.1399-0004.2007.00876.x. [DOI] [PubMed] [Google Scholar]
- 10.Zaveri HP, Beck TF, Hernandez-Garcia A, et al. Identification of critical regions and candidate genes for cardiovascular malformations and cardiomyopathy associated with deletions of chromosome 1p36. PLoS One. 2014;9(1):e85600. doi: 10.1371/journal.pone.0085600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu YQ, Heilstedt HA, Bedell JA, et al. Molecular refinement of the 1p36 deletion syndrome reveals size diversity and a preponderance of maternally derived deletions. Hum Mol Genet. 1999;8(2):313–321. doi: 10.1093/hmg/8.2.313. [DOI] [PubMed] [Google Scholar]
- 12.Nicoulaz A, Rubi F, Lieder L, et al. Contiguous approximately 16 Mb 1p36 deletion: dominant features of classical distal 1p36 monosomy with haplo-lethality. Am J Med Genet A. 2011;155A(8):1964–1968. doi: 10.1002/ajmg.a.33210. [DOI] [PubMed] [Google Scholar]
- 13.Shimada S, Shimojima K, Okamoto N, et al. Microarray analysis of 50 patients reveals the critical chromosomal regions responsible for 1p36 deletion syndrome-related complications. Brain Dev. 2015;37(5):515–526. doi: 10.1016/j.braindev.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Rosenfeld JA, Crolla JA, Tomkins S, et al. Refinement of causative genes in monosomy 1p36 through clinical and molecular cytogenetic characterization of small interstitial deletions. Am J Med Genet A. 2010;152A(8):1951–1959. doi: 10.1002/ajmg.a.33516. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Zhang Y, Wang J, Yang JF, Yang YF, Tan ZP. 576 kb deletion in 1p36.33-p36.32 containing SKI is associated with limb malformation, congenital heart disease and epilepsy. Gene. 2013;528(2):352–355. doi: 10.1016/j.gene.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Gajecka M, Saitta SC, Gentles AJ, et al. Recurrent interstitial 1p36 deletions: evidence for germline mosaicism and complex rearrangement breakpoints. Am J Med Genet A. 2010;152A(12):3074–3083. doi: 10.1002/ajmg.a.33733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clancy BM, Johnson JD, Lambert AJ, et al. A gene expression profile for endochondral bone formation: oligonucleotide microarrays establish novel connections between known genes and BMP-2-induced bone formation in mouse quadriceps. Bone. 2003;33(1):46–63. doi: 10.1016/s8756-3282(03)00116-9. [DOI] [PubMed] [Google Scholar]
- 18.Gajecka M, Yu W, Ballif BC, et al. Delineation of mechanisms and regions of dosage imbalance in complex rearrangements of 1p36 leads to a putative gene for regulation of cranial suture closure. Eur J Hum Genet. 2005;13(2):139–149. doi: 10.1038/sj.ejhg.5201302. [DOI] [PubMed] [Google Scholar]
- 19.Windpassinger C, Kroisel PM, Wagner K, Petek E. The human gamma-aminobutyric acid A receptor delta (GABRD) gene: molecular characterisation and tissue-specific expression. Gene. 2002;292(1–2):25–31. doi: 10.1016/s0378-1119(02)00649-2. [DOI] [PubMed] [Google Scholar]
- 20.Mohler H. GABA(A) receptor diversity and pharmacology. Cell Tissue Res. 2006;326(2):505–516. doi: 10.1007/s00441-006-0284-3. [DOI] [PubMed] [Google Scholar]
- 21.Mihalek RM, Banerjee PK, Korpi ER, et al. Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proc Natl Acad Sci U S A. 1999;96(22):12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59(2):207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13(13):1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- 24.Lenzen KP, Heils A, Lorenz S, Hempelmann A, Sander T. Association analysis of the Arg220His variation of the human gene encoding the GABA delta subunit with idiopathic generalized epilepsy. Epilepsy Res. 2005;65(1–2):53–57. doi: 10.1016/j.eplepsyres.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Medrano EE. Repression of TGF-beta signaling by the oncogenic protein SKI in human melanomas: consequences for proliferation, survival, and metastasis. Oncogene. 2003;22(20):3123–3129. doi: 10.1038/sj.onc.1206452. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Xu W, Bales E, et al. SKI activates Wnt/beta-catenin signaling in human melanoma. Cancer Res. 2003;63(20):6626–6634. [PubMed] [Google Scholar]
- 27.Colmenares C, Heilstedt HA, Shaffer LG, et al. Loss of the SKI proto-oncogene in individuals affected with 1p36 deletion syndrome is predicted by strain-dependent defects in Ski−/− mice. Nat Genet. 2002;30(1):106–109. doi: 10.1038/ng770. [DOI] [PubMed] [Google Scholar]
- 28.Doyle AJ, Doyle JJ, Bessling SL, et al. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44(11):1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmignac V, Thevenon J, Ades L, et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am J Hum Genet. 2012;91(5):950–957. doi: 10.1016/j.ajhg.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shprintzen RJ, Goldberg RB. Dysmorphic facies, omphalocele, laryngeal and pharyngeal hypoplasia, spinal anomalies, and learning disabilities in a new dominant malformation syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):347–353. [PubMed] [Google Scholar]
- 31.Robinson PN, Neumann LM, Demuth S, et al. Shprintzen-Goldberg syndrome: fourteen new patients and a clinical analysis. Am J Med Genet A. 2005;135(3):251–262. doi: 10.1002/ajmg.a.30431. [DOI] [PubMed] [Google Scholar]
- 32.Mochizuki N, Shimizu S, Nagasawa T, et al. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000;96(9):3209–3214. [PubMed] [Google Scholar]
- 33.Arndt AK, Schafer S, Drenckhahn JD, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93(1):67–77. doi: 10.1016/j.ajhg.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahata M, Inoue Y, Tsuda H, et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J Biol Chem. 2009;284(5):3334–3344. doi: 10.1074/jbc.M808989200. [DOI] [PubMed] [Google Scholar]
- 35.Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010;19(5):774–789. doi: 10.1093/hmg/ddp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCormack K, McCormack T, Tanouye M, Rudy B, Stuhmer W. Alternative splicing of the human Shaker K+ channel beta 1 gene and functional expression of the beta 2 gene product. FEBS Lett. 1995;370(1–2):32–36. doi: 10.1016/0014-5793(95)00785-8. [DOI] [PubMed] [Google Scholar]
- 37.Perkowski JJ, Murphy GG. Deletion of the mouse homolog of KCNAB2, a gene linked to monosomy 1p36, results in associative memory impairments and amygdala hyperexcitability. J Neurosci. 2011;31(1):46–54. doi: 10.1523/JNEUROSCI.2634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormack K, Connor JX, Zhou L, et al. Genetic analysis of the mammalian K+ channel beta subunit Kvbeta 2 (Kcnab2) J Biol Chem. 2002;277(15):13219–13228. doi: 10.1074/jbc.M111465200. [DOI] [PubMed] [Google Scholar]
- 39.Zoltewicz JS, Stewart NJ, Leung R, Peterson AS. Atrophin 2 recruits histone deacetylase and is required for the function of multiple signaling centers during mouse embryogenesis. Development. 2004;131(1):3–14. doi: 10.1242/dev.00908. [DOI] [PubMed] [Google Scholar]
- 40.Kim BJ, Zaveri HP, Shchelochkov OA, et al. An allelic series of mice reveals a role for RERE in the development of multiple organs affected in chromosome 1p36 deletions. PLoS One. 2013;8(2):e57460. doi: 10.1371/journal.pone.0057460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim BJ, Scott DA. Mouse model reveals the role of RERE in cerebellar foliation and the migration and maturation of Purkinje cells. PLoS One. 2014;9(1):e87518. doi: 10.1371/journal.pone.0087518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plaster N, Sonntag C, Schilling TF, Hammerschmidt M. REREa/Atrophin-2 interacts with histone deacetylase and Fgf8 signaling to regulate multiple processes of zebrafish development. Dev Dyn. 2007;236(7):1891–1904. doi: 10.1002/dvdy.21196. [DOI] [PubMed] [Google Scholar]
- 43.Schilling TF, Piotrowski T, Grandel H, et al. Jaw and branchial arch mutants in zebrafish I: branchial arches. Development. 1996;123:329–344. doi: 10.1242/dev.123.1.329. [DOI] [PubMed] [Google Scholar]
- 44.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96(5):635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 45.Kaneko-Oshikawa C, Nakagawa T, Yamada M, et al. Mammalian E4 is required for cardiac development and maintenance of the nervous system. Mol Cell Biol. 2005;25(24):10953–10964. doi: 10.1128/MCB.25.24.10953-10964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vacalla CM, Theil T. Cst, a novel mouse gene related to Drosophila Castor, exhibits dynamic expression patterns during neurogenesis and heart development. Mech Dev. 2002;118(1–2):265–268. doi: 10.1016/s0925-4773(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Li W, Ma X, et al. Essential role of the zinc finger transcription factor Casz1 for mammalian cardiac morphogenesis and development. J Biol Chem. 2014;289(43):29801–29816. doi: 10.1074/jbc.M114.570416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmer G, Oeffner F, Von Messling V, et al. Cloning and characterization of gp36, a human mucin-type glycoprotein preferentially expressed in vascular endothelium. Biochem J. 1999;341(Pt 2):277–284. [PMC free article] [PubMed] [Google Scholar]
- 49.Mahtab EA, Wijffels MC, Van Den Akker NM, et al. Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: correlation with abnormal epicardial development. Dev Dyn. 2008;237(3):847–857. doi: 10.1002/dvdy.21463. [DOI] [PubMed] [Google Scholar]
- 50.Mahtab EA, Vicente-Steijn R, Hahurij ND, et al. Podoplanin deficient mice show a RhoA-related hypoplasia of the sinus venosus myocardium including the sinoatrial node. Dev Dyn. 2009;238(1):183–193. doi: 10.1002/dvdy.21819. [DOI] [PubMed] [Google Scholar]
- 51.Sierra OL, Cheng SL, Loewy AP, Charlton-Kachigian N, Towler DA. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J Biol Chem. 2004;279(31):32913–32923. doi: 10.1074/jbc.M314098200. [DOI] [PubMed] [Google Scholar]
- 52.Kuroda K, Han H, Tani S, et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 2003;18(2):301–312. doi: 10.1016/s1074-7613(03)00029-3. [DOI] [PubMed] [Google Scholar]
- 53.Yabe D, Fukuda H, Aoki M, et al. Generation of a conditional knockout allele for mammalian Spen protein Mint/SHARP. Genesis. 2007;45(5):300–306. doi: 10.1002/dvg.20296. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt M, Kroger B, Jacob E, et al. Molecular characterization of human and bovine endothelin converting enzyme (ECE-1) FEBS Lett. 1994;356(2–3):238–243. doi: 10.1016/0014-5793(94)01277-6. [DOI] [PubMed] [Google Scholar]
- 55.Hofstra RM, Valdenaire O, Arch E, et al. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am J Hum Genet. 1999;64(1):304–308. doi: 10.1086/302184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yanagisawa H, Yanagisawa M, Kapur RP, et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125(5):825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 57.Farach-Carson MC, Carson DD. Perlecan – a multifunctional extracellular proteoglycan scaffold. Glycobiology. 2007;17(9):897–905. doi: 10.1093/glycob/cwm043. [DOI] [PubMed] [Google Scholar]
- 58.Nicole S, Davoine CS, Topaloglu H, et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia) Nat Genet. 2000;26(4):480–483. doi: 10.1038/82638. [DOI] [PubMed] [Google Scholar]
- 59.Arikawa-Hirasawa E, Wilcox WR, Le AH, et al. Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat Genet. 2001;27(4):431–434. doi: 10.1038/86941. [DOI] [PubMed] [Google Scholar]
- 60.Abdel-Aziz M, Azab NA. A case of Schwartz-Jampel syndrome with cleft palate. Int J Pediatr Otorhinolaryngol. 2009;73(11):1601–1603. doi: 10.1016/j.ijporl.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 61.Costell M, Gustafsson E, Aszodi A, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147(5):1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91(2):158–164. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- 63.Hsu CY, Chang NC, Lee MW, et al. LUZP deficiency affects neural tube closure during brain development. Biochem Biophys Res Commun. 2008;376(3):466–471. doi: 10.1016/j.bbrc.2008.08.170. [DOI] [PubMed] [Google Scholar]
- 64.Giannikou K, Fryssira H, Oikonomakis V, et al. Further delineation of novel 1p36 rearrangements by array-CGH analysis: narrowing the breakpoints and clarifying the “extended” phenotype. Gene. 2012;506(2):360–368. doi: 10.1016/j.gene.2012.06.060. [DOI] [PubMed] [Google Scholar]
- 65.Xu F, Zhang YN, Cheng DH, et al. The first patient with a pure 1p36 microtriplication associated with severe clinical phenotypes. Mol Cytogenet. 2014;7(1):64. doi: 10.1186/s13039-014-0064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weaver DD, Norby AR, Rosenfeld JA, et al. Chromosome 1p36.22p36.21 duplications/triplication causes Setleis syndrome (focal facial dermal dysplasia type III) Am J Med Genet A. 2015;167(5):1061–1070. doi: 10.1002/ajmg.a.36973. [DOI] [PubMed] [Google Scholar]
- 67.Slavotinek AM, Mehrotra P, Nazarenko I, et al. Focal facial dermal dysplasia, type IV, is caused by mutations in CYP26C1. Hum Mol Genet. 2013;22(4):696–703. doi: 10.1093/hmg/dds477. [DOI] [PMC free article] [PubMed] [Google Scholar]